
Delineation of Pathogenomic Insights of Breast Cancer in Young Women


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of Age Groups’ Specific Patterns of Gene Expression
2.2. MRNA–Protein Correlation Analysis
2.3. Prognostic Significance
2.4. Gene Ontology Analysis
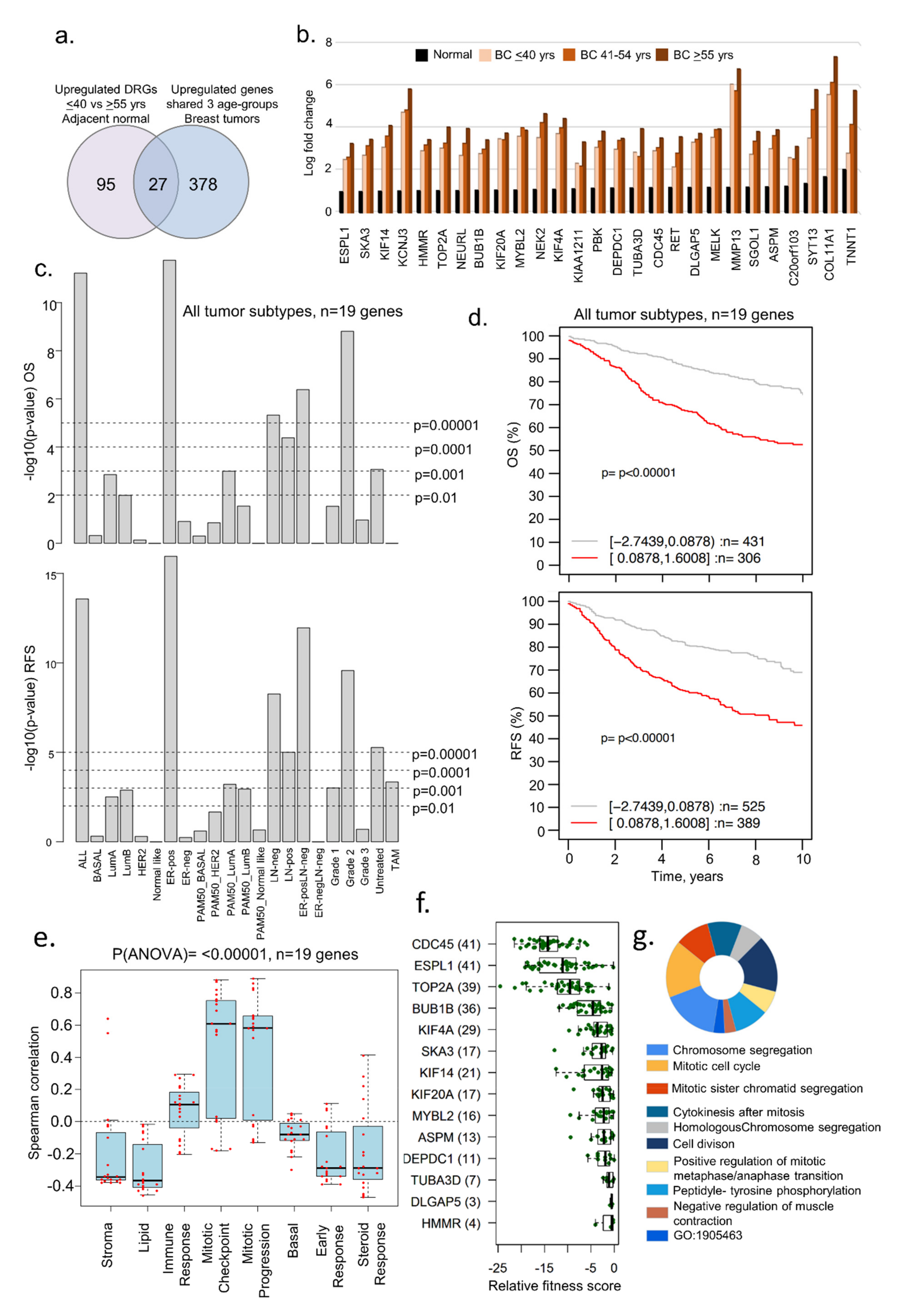
2.5. Fitness Analysis
2.6. Secreted Proteins
3. Results
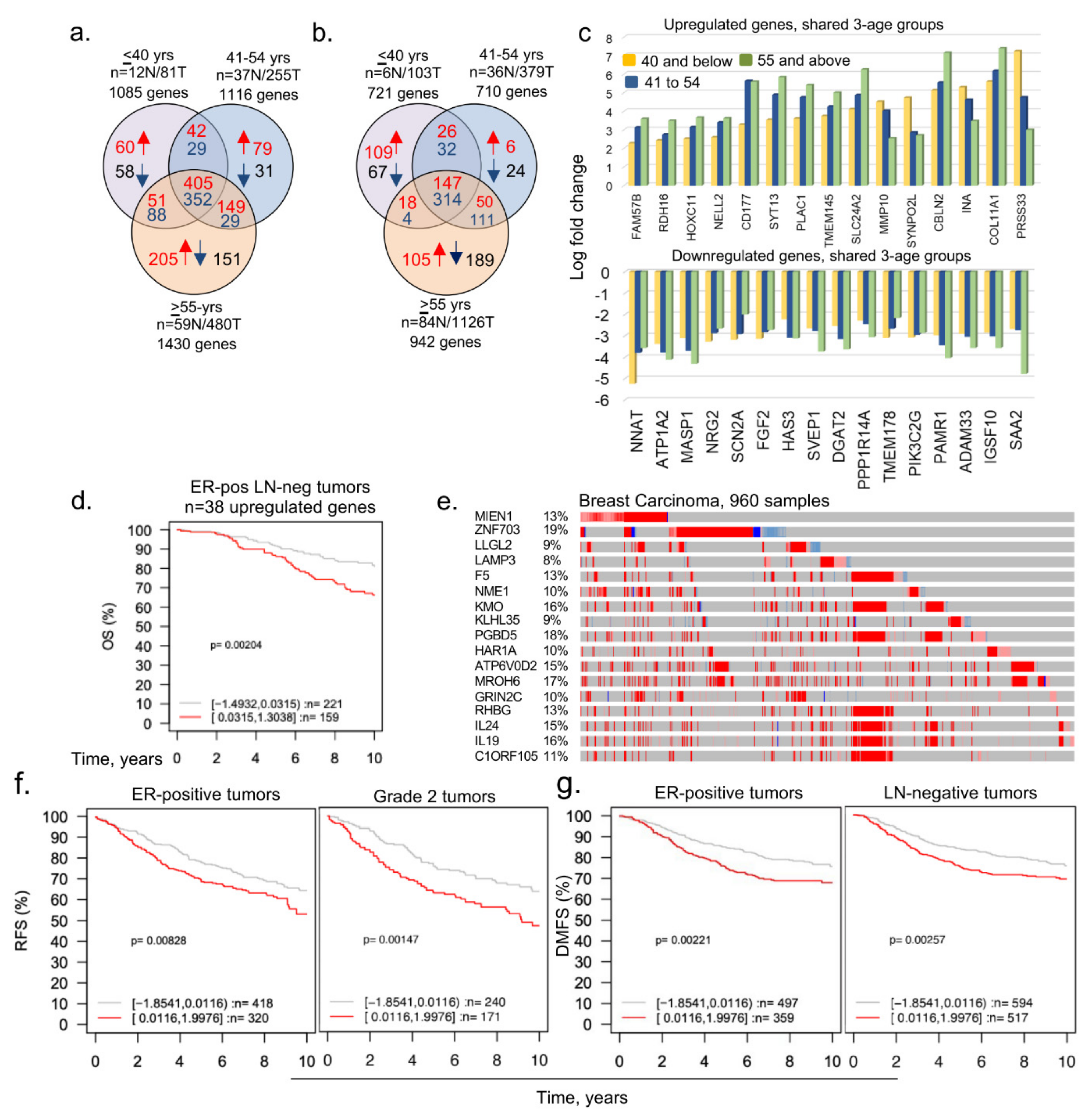
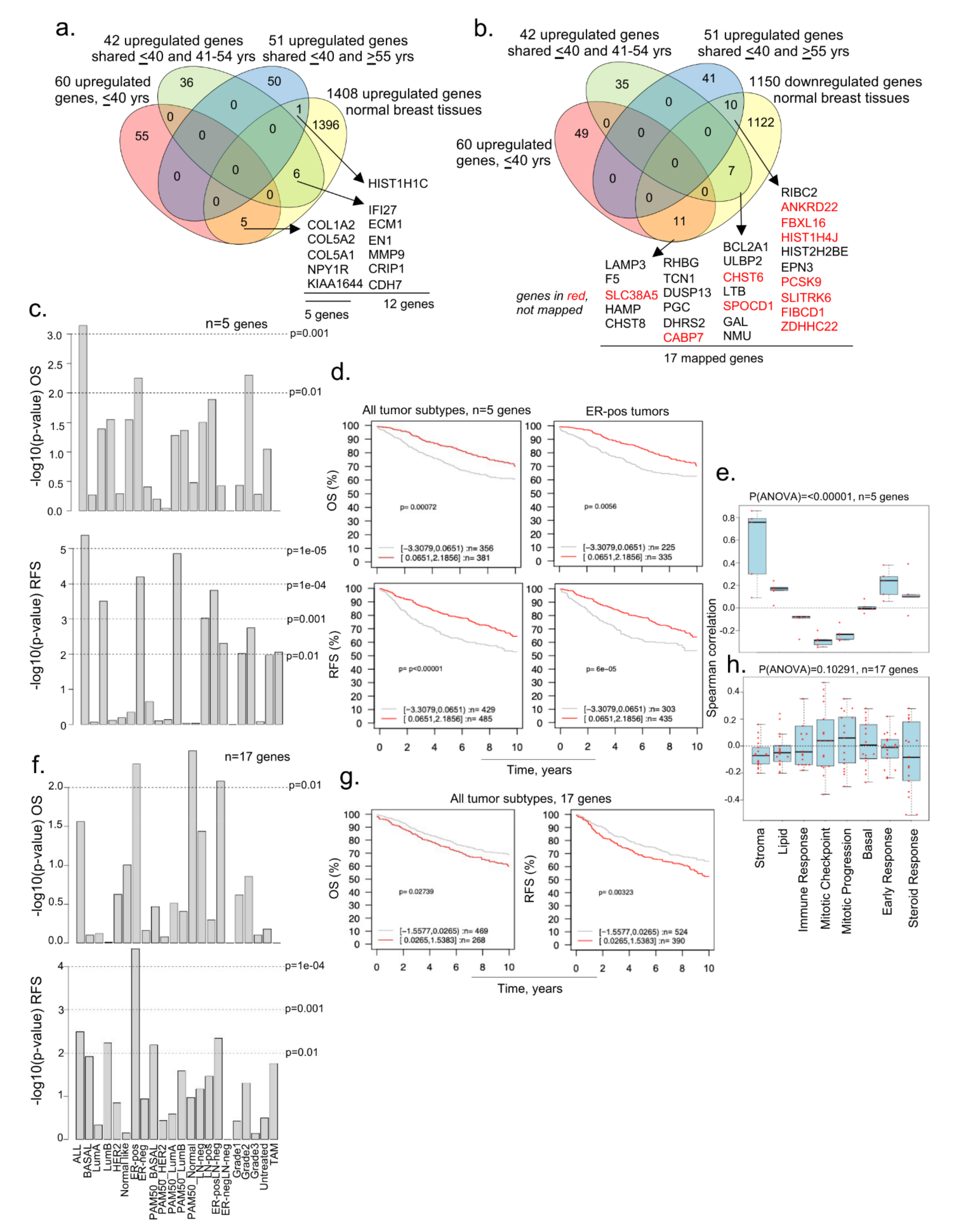
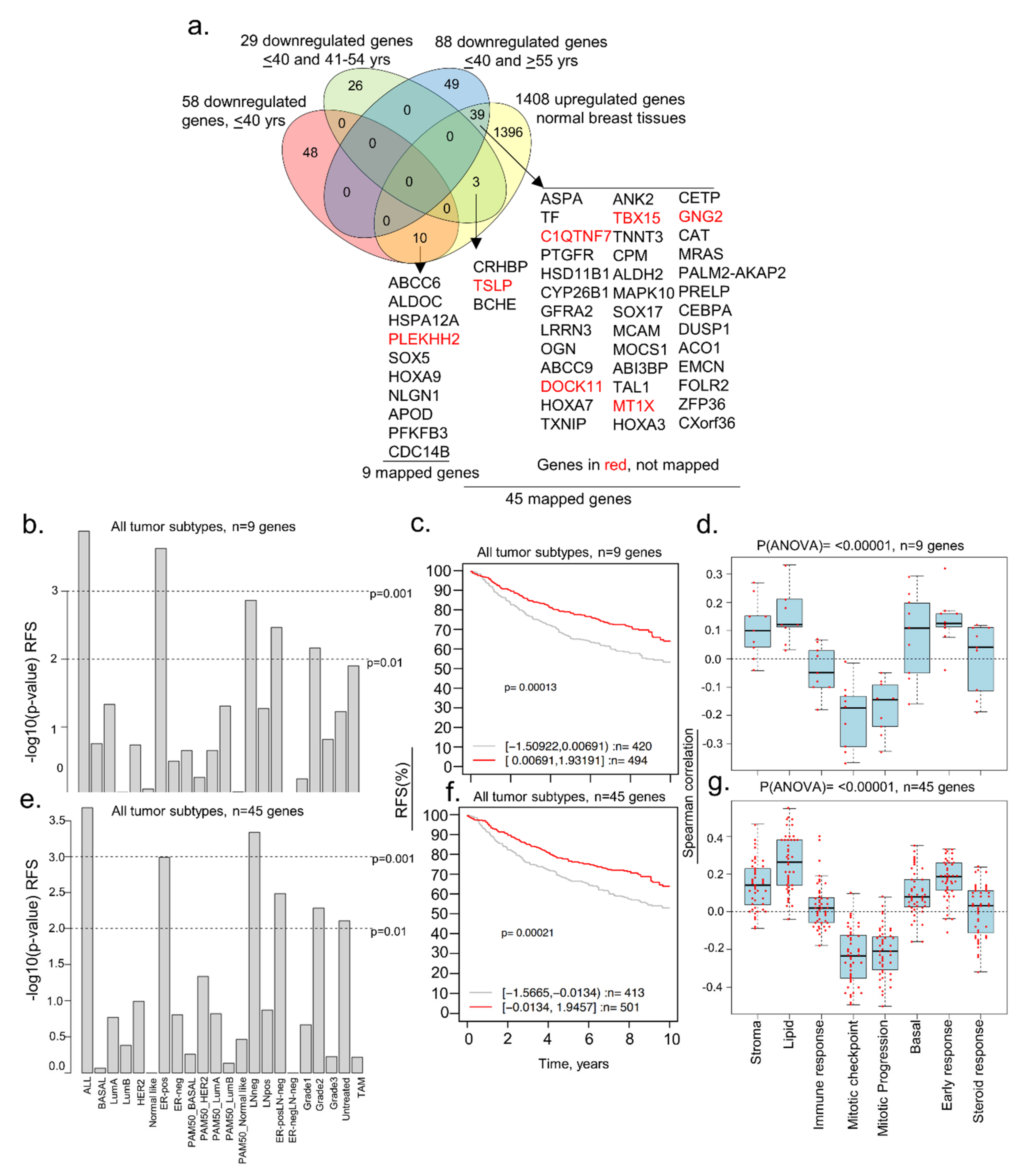
3.1. Identification of Dysregulated Genes in BCYW Aged ≤40 Years
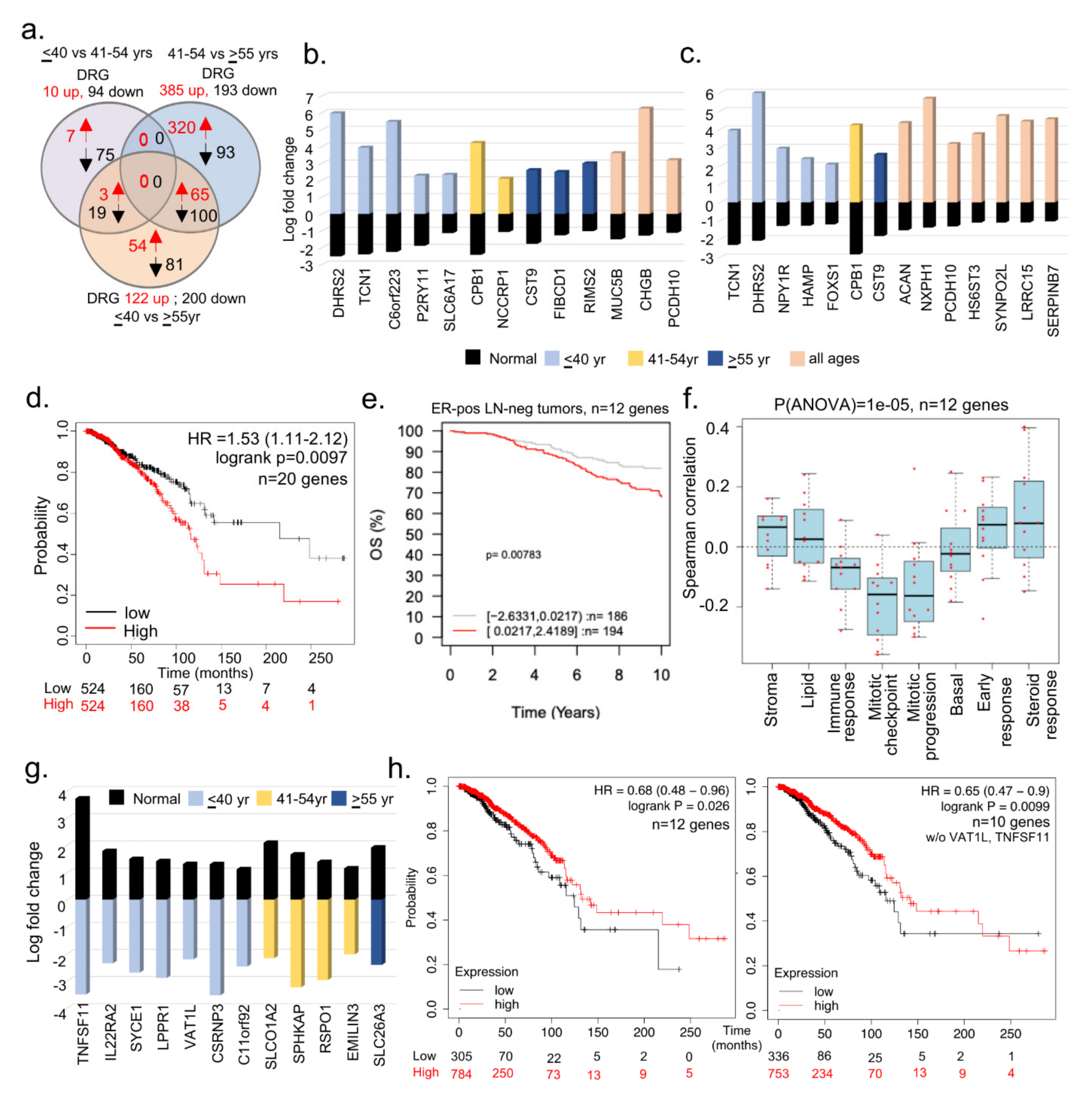
3.2. Subset of Differentially Dysregulated and Clinically Significant Genes in BCYW
3.3. Evidence of Dysregulated Cancer Genes in BCYW and Normal Breast Tissues
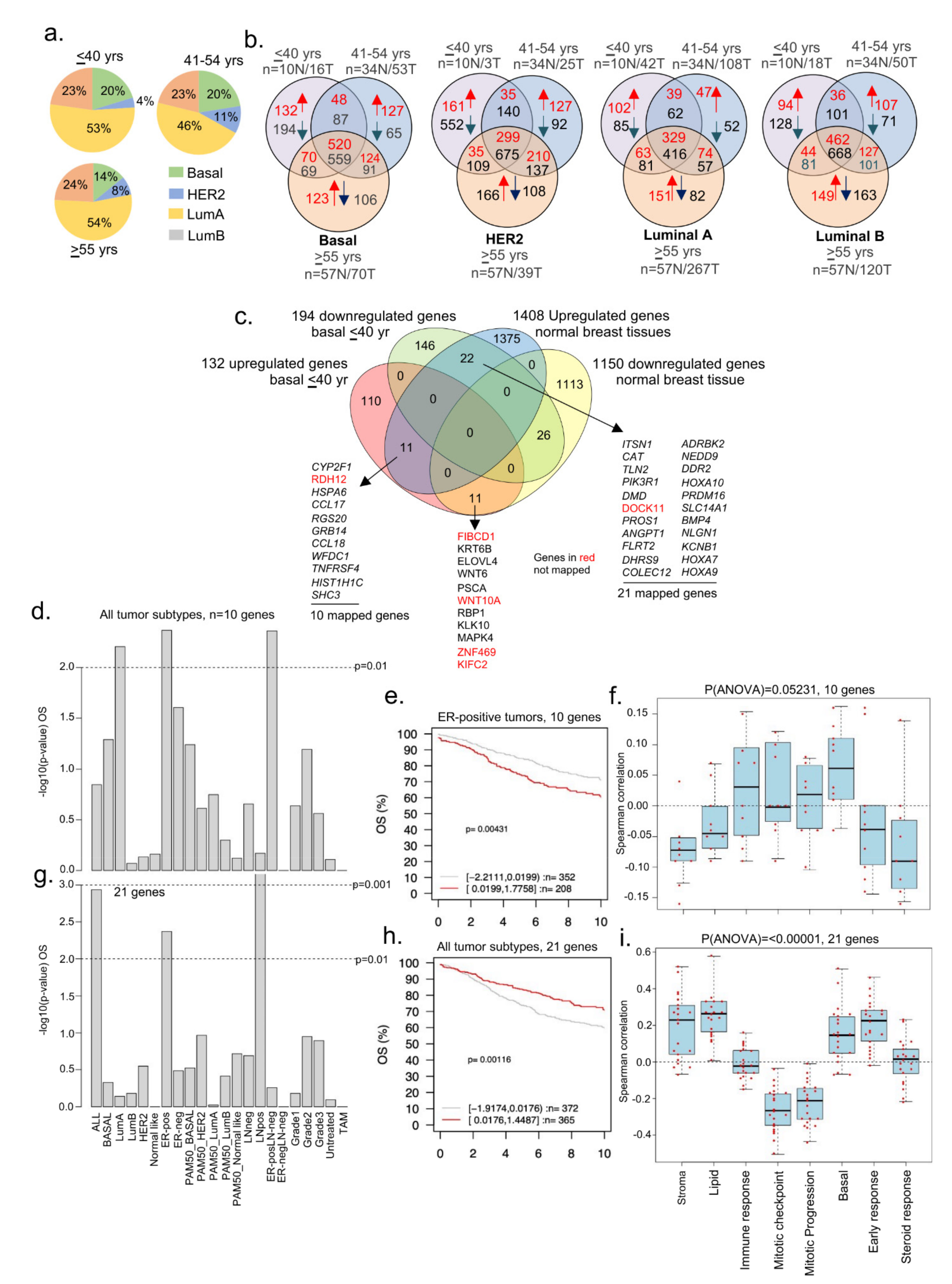
3.4. Dysregulated Genes in BCYW Vary with the Intrinsic Tumor Subtype
3.5. Tumor Laterality and Age-Associated Dysregulated Genes
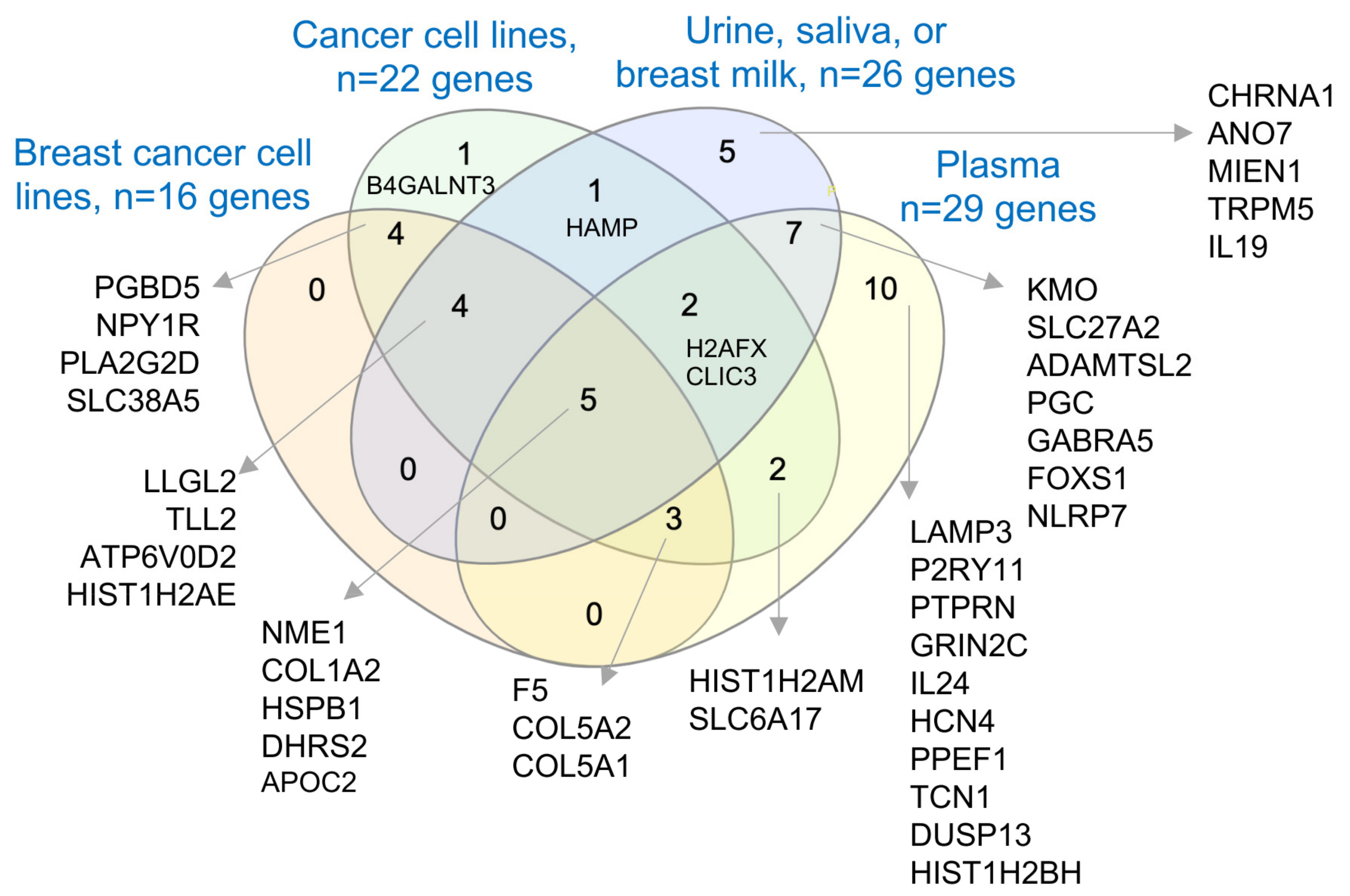
3.6. Secretory Nature of Dysregulated Gene Products in Young Women’s Breast Tumors
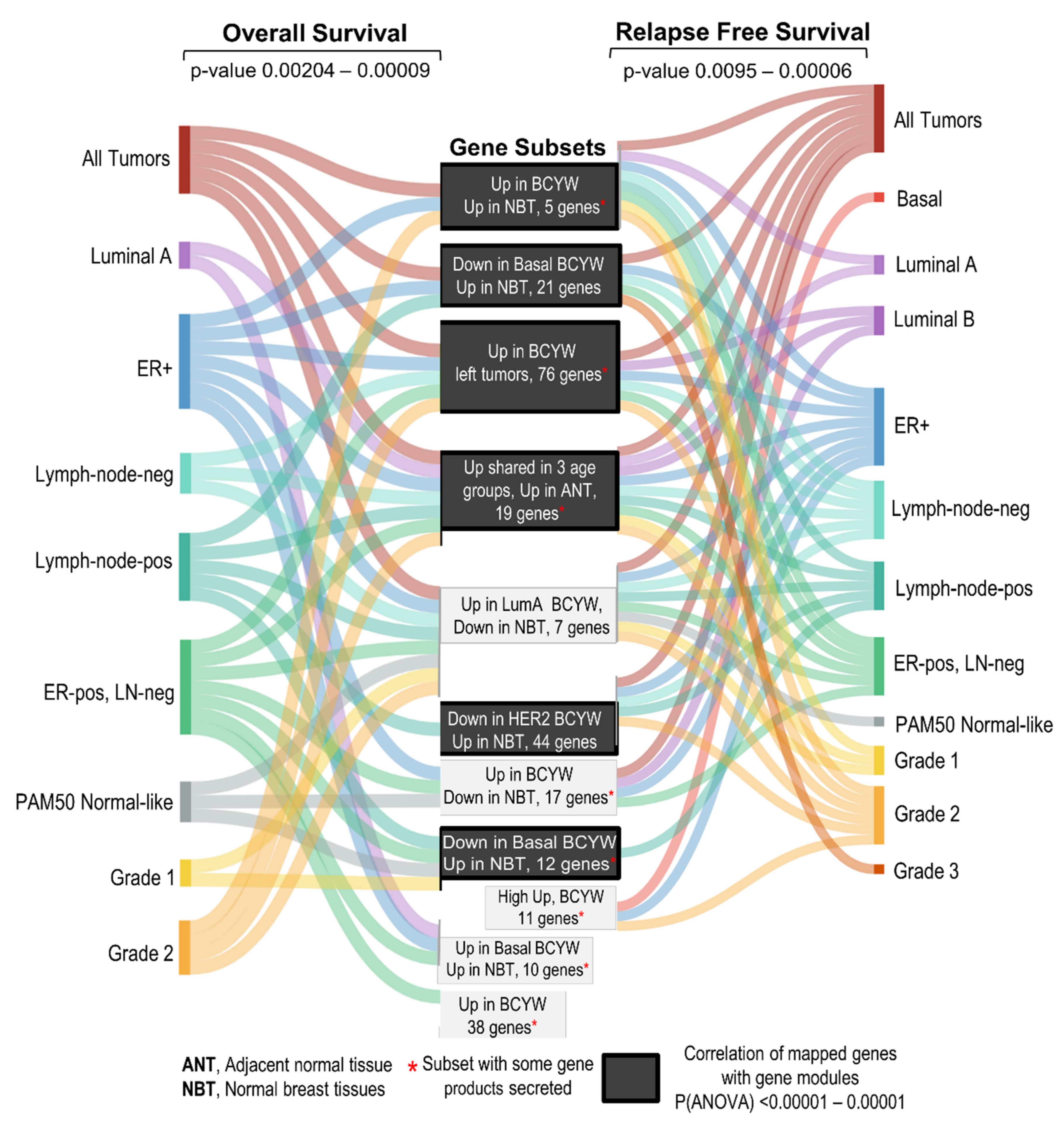
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hendrick, R.E.; Helvie, M.A.; Monticciolo, D.L. Breast Cancer Mortality Rates Have Stopped Declining in U.S. Women Younger than 40 Years. Radiology 2021, 299, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Abreu, C.; Toi, M.; Saini, S.; Casimiro, S.; Arora, A.; Paul, A.M.; Velaga, R.; Rameshwar, P.; Lipton, A.; et al. Oncobiology and Treatment of Breast Cancer in Young Women. Cancer Metastasis Rev. 2022, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, S.; Freedman, R.A.; Partridge, A.H. The Impact of Young Age at Diagnosis (Age <40 Years) on Prognosis Varies by Breast Cancer Subtype: A U.S. SEER Database Analysis. Breast 2022, 61, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Paluch-Shimon, S.; Cardoso, F.; Partridge, A.H.; Abulkhair, O.; Azim, H.A.J.; Bianchi-Micheli, G.; Cardoso, M.-J.; Curigliano, G.; Gelmon, K.A.; Harbeck, N.; et al. ESO-ESMO 4th International Consensus Guidelines for Breast Cancer in Young Women (BCY4). Ann. Oncol. 2020, 31, 674–696. [Google Scholar] [CrossRef]
- Bajpai, J.; Ventrapati, P.; Joshi, S.; Wadasadawala, T.; Rath, S.; Pathak, R.; Nandhana, R.; Mohanty, S.; Chougle, Q.; Engineer, M.; et al. Unique Challenges and Outcomes of Young Women with Breast Cancers from a Tertiary Care Cancer Centre in India. Breast 2021, 60, 177–184. [Google Scholar] [CrossRef]
- Breast Cancer Incidence (Invasive) Statistics|Cancer Research UK. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/breast-cancer/incidence-invasive#heading-One (accessed on 11 February 2022).
- Okazaki, M.; Bando, H.; Tohno, E.; Kujiraoka, Y.; Iguchi-Manaka, A.; Ichioka, E.; Tsushima, Y.; Watanabe, H.; Hara, H. Investigation of the Significance of Population-Based Breast Cancer Screening among Women Aged under 40 Years. Breast Cancer 2021, 28, 75–81. [Google Scholar] [CrossRef]
- Nakata, K.; Hiyama, E.; Katanoda, K.; Matsuda, T.; Tada, Y.; Inoue, M.; Kawa, K.; Maru, M.; Shimizu, C.; Horibe, K.; et al. Cancer in Adolescents and Young Adults in Japan: Epidemiology and Cancer Strategy. Int. J. Clin. Oncol. 2022, 27, 7–15. [Google Scholar] [CrossRef]
- Hayashi, N.; Kumamaru, H.; Isozumi, U.; Aogi, K.; Asaga, S.; Iijima, K.; Kadoya, T.; Kojima, Y.; Kubo, M.; Miyashita, M.; et al. Annual Report of the Japanese Breast Cancer Registry for 2017. Breast Cancer 2020, 27, 803–809. [Google Scholar] [CrossRef]
- Alvarez-Bañuelos, M.T.; Segura-Jaramillo, K.A.; Gómez-Rivera, E.D.C.; Alarcón-Rojas, C.A.; Morales-Romero, J.; Sampieri, C.L.; Guzmán-García, R.E. Age Under 30 Years As a Predictor of Poor Survival in a Cohort of Mexican Women With Breast Cancer. Cancer Control 2021, 28, 10732748211047408. [Google Scholar] [CrossRef]
- Ntirenganya, F.; Twagirumukiza, J.D.; Bucyibaruta, G.; Rugwizangoga, B.; Rulisa, S. Premenopausal Breast Cancer Risk Factors and Associations with Molecular Subtypes: A Case-Control Study. Int. J. Breast Cancer 2021, 2021, 5560559. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-J.; Chang, Y.-J.; Chang, Y.-J. Treatment and Long-Term Outcome of Breast Cancer in Very Young Women: Nationwide Population-Based Study. BJS Open 2021, 5, zrab087. [Google Scholar] [CrossRef] [PubMed]
- Conte, B.; Soldato, D.; Razeti, M.G.; Fregatti, P.; de Azambuja, E.; Schettini, F.; Prat, A.; del Mastro, L.; Lambertini, M. De Novo Metastatic Breast Cancer Arising in Young Women: Review of the Current Evidence. Clin. Breast Cancer 2022, 22, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Szollár, A.; Újhelyi, M.; Polgár, C.; Oláh, E.; Pukancsik, D.; Rubovszky, G.; Udvarhelyi, N.; Kovács, T.; Sávolt, Á.; Kenessey, I.; et al. A Long-Term Retrospective Comparative Study of the Oncological Outcomes of 598 Very Young (≤35 Years) and Young (36–45 Years) Breast Cancer Patients. Eur. J. Surg. Oncol. 2019, 45, 2009–2015. [Google Scholar] [CrossRef]
- Suter, M.B.; Pagani, O. Should Age Impact Breast Cancer Management in Young Women? Fine Tuning of Treatment Guidelines. Ther. Adv. Med. Oncol. 2018, 10, 1758835918776923. [Google Scholar] [CrossRef]
- Azim, H.A.J.; Partridge, A.H. Biology of Breast Cancer in Young Women. Breast Cancer Res. 2014, 16, 427. [Google Scholar] [CrossRef]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer Statistics, 2010. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Tan, L.; Jiang, W.G.; Chen, K.; You, N.; Sanders, A.J.; Liang, G.; Liu, Z.; Ling, Y.; Gong, C. Effect of Younger Age on Survival Outcomes in T1N0M0 Breast Cancer: A Propensity Score Matching Analysis. J. Surg. Oncol. 2019, 119, 1039–1046. [Google Scholar] [CrossRef]
- Park, C.; Yoon, K.-A.; Kim, J.; Park, I.H.; Park, S.J.; Kim, M.K.; Jang, W.; Cho, S.Y.; Park, B.; Kong, S.-Y.; et al. Integrative Molecular Profiling Identifies a Novel Cluster of Estrogen Receptor-Positive Breast Cancer in Very Young Women. Cancer Sci. 2019, 110, 1760–1770. [Google Scholar] [CrossRef] [PubMed]
- Yau, C.; Fedele, V.; Roydasgupta, R.; Fridlyand, J.; Hubbard, A.; Gray, J.W.; Chew, K.; Dairkee, S.H.; Moore, D.H.; Schittulli, F.; et al. Aging Impacts Transcriptomes but Not Genomes of Hormone-Dependent Breast Cancers. Breast Cancer Res. 2007, 9, R59. [Google Scholar] [CrossRef]
- Azim, H.A.J.; Nguyen, B.; Brohée, S.; Zoppoli, G.; Sotiriou, C. Genomic Aberrations in Young and Elderly Breast Cancer Patients. BMC Med. 2015, 13, 266. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Kim, D.; Jain, E.; Snow, C.; Kirkner, G.J.; Rosenberg, S.M.; Oh, C.; Poorvu, P.D.; Ruddy, K.J.; Tamimi, R.M.; et al. Somatic and Germline Genomic Alterations in Very Young Women with Breast Cancer. Clin. Cancer Res. 2022, 28, 2339–2348. [Google Scholar] [CrossRef] [PubMed]
- Azim, H.A., Jr.; Peccatori, F.A.; Brohée, S.; Branstetter, D.; Loi, S.; Viale, G.; Piccart, M.; Dougall, W.C.; Pruneri, G.; Sotiriou, C. RANK-ligand (RANKL) expression in young breast cancer patients and during pregnancy. Breast Cancer Res. 2015, 17, 24. [Google Scholar] [CrossRef]
- Gu, X.; Wang, B.; Zhu, H.; Zhou, Y.; Horning, A.M.; Huang, T.H.-M.; Chen, Y.; Houghton, P.; Lai, Z.; Michalek, J.E.; et al. Age-Associated Genes in Human Mammary Gland Drive Human Breast Cancer Progression. Breast Cancer Res. 2020, 22, 64. [Google Scholar] [CrossRef] [PubMed]
- Colak, D.; Nofal, A.; Albakheet, A.; Nirmal, M.; Jeprel, H.; Eldali, A.; Al-Tweigeri, T.; Tulbah, A.; Ajarim, D.; Malik, O.; et al. Age-Specific Gene Expression Signatures for Breast Tumors and Cross-Species Conserved Potential Cancer Progression Markers in Young Women. PLoS ONE 2013, 8, e63204. [Google Scholar] [CrossRef]
- Anders, C.K.; Hsu, D.S.; Broadwater, G.; Acharya, C.R.; Foekens, J.A.; Zhang, Y.; Blackwell, K.L. Young age at diagnosis correlates with worse prognosis and defines a subset of breast cancers with shared patterns of gene expression. J. Clin. Oncol. 2008, 26, 3324–3330. [Google Scholar] [CrossRef]
- Kan, Z.; Ding, Y.; Kim, J.; Jung, H.H.; Chung, W.; Lal, S.; Cho, S.; Fernandez-Banet, J.; Lee, S.K.; Kim, S.W.; et al. Multi-Omics Profiling of Younger Asian Breast Cancers Reveals Distinctive Molecular Signatures. Nat. Commun. 2018, 9, 1725. [Google Scholar] [CrossRef]
- Eiro, N.; Gonzalez, L.O.; Fraile, M.; Cid, S.; Schneider, J.; Vizoso, F.J. Breast Cancer Tumor Stroma: Cellular Components, Phenotypic Heterogeneity, Intercellular Communication, Prognostic Implications and Therapeutic Opportunities. Cancers 2019, 11, 664. [Google Scholar] [CrossRef] [PubMed]
- Pirone, J.R.; D’Arcy, M.; Stewart, D.A.; Hines, W.C.; Johnson, M.; Gould, M.N.; Yaswen, P.; Jerry, D.J.; Smith Schneider, S.; Troester, M.A. Age-Associated Gene Expression in Normal Breast Tissue Mirrors Qualitative Age-at-Incidence Patterns for Breast Cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.; Yau, C.; Wong, C.K.; Sanborn, J.Z.; Newton, Y.; Vaske, C.; Benz, S.C.; Krings, G.; Camarda, R.; Henry, J.E.; et al. A Risk-Associated Active Transcriptome Phenotype Expressed by Histologically Normal Human Breast Tissue and Linked to a pro-Tumorigenic Adipocyte Population. Breast Cancer Res. 2020, 22, 81. [Google Scholar] [CrossRef]
- Hinck, L.; Silberstein, G.B. Key Stages in Mammary Gland Development: The Mammary End Bud as a Motile Organ. Breast Cancer Res. 2005, 7, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Woodward, T.L.; Xie, J.W.; Haslam, S.Z. The Role of Mammary Stroma in Modulating the Proliferative Response to Ovarian Hormones in the Normal Mammary Gland. J. Mammary Gland Biol. Neoplasia 1998, 3, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Pardo, I.; Lillemoe, H.A.; Blosser, R.J.; Choi, M.; Sauder, C.A.M.; Doxey, D.K.; Mathieson, T.; Hancock, B.A.; Baptiste, D.; Atale, R.; et al. Next-Generation Transcriptome Sequencing of the Premenopausal Breast Epithelium Using Specimens from a Normal Human Breast Tissue Bank. Breast Cancer Res. 2014, 16, R26. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor Package for Integrative Analysis of TCGA Data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The Genomic and Transcriptomic Architecture of 2,000 Breast Tumours Reveals Novel Subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byearsne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics DataThe CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Lánczky, A.; Győrffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Ringnér, M.; Fredlund, E.; Häkkinen, J.; Borg, Å.; Staaf, J. GOBO: Gene Expression-Based Outcome for Breast Cancer Online. PLoS ONE 2011, 6, e17911. [Google Scholar] [CrossRef] [PubMed]
- Fredlund, E.; Staaf, J.; Rantala, J.K.; Kallioniemi, O.; Borg, Å.; Ringnér, M. The gene expression landscape of breast cancer is shaped by tumor protein p53 status and epithelial-mesenchymal transition. Breast Cancer Res. 2012, 14, R113. [Google Scholar] [CrossRef] [PubMed]
- Fonseka, P.; Pathan, M.; Chitti, S.V.; Kang, T.; Mathivanan, S. FunRich Enables Enrichment Analysis of OMICs Datasets. J. Mol. Biol. 2021, 433, 166747. [Google Scholar] [CrossRef] [PubMed]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of Cancer Therapeutic Targets Using CRISPR-Cas9 Screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- George, B.; Pillai, P.M.; Paul, A.M.; Amjesh, R.; Leitzel, K.; Ali, S.M.; Sandiford, O.; Lipton, A.; Rameshwar, P.; Hortobagyi, G.N.; et al. Cellular Fitness Phenotypes of Cancer Target Genes from Oncobiology to Cancer Therapeutics. Cells 2021, 10, 433. [Google Scholar] [CrossRef]
- Kalra, H.; Drummen, G.P.C.; Mathivanan, S. Focus on Extracellular Vesicles: Introducing the Next Small Big Thing. Int. J. Mol. Sci. 2016, 17, 170. [Google Scholar] [CrossRef]
- Nanjappa, V.; Thomas, J.K.; Marimuthu, A.; Muthusamy, B.; Radhakrishnan, A.; Sharma, R.; Ahmad Khan, A.; Balakrishnan, L.; Sahasrabuddhe, N.A.; Kumar, S.; et al. Plasma Proteome Database as a Resource for Proteomics Research: 2014 Update. Nucleic Acids Res. 2014, 42, D959–D965. [Google Scholar] [CrossRef]
- Muthusamy, B.; Hanumanthu, G.; Suresh, S.; Rekha, B.; Srinivas, D.; Karthick, L.; Vrushabendra, B.M.; Sharma, S.; Mishra, G.; Chatterjee, P.; et al. Plasma Proteome Database as a Resource for Proteomics Research. Proteomics 2005, 5, 3531–3536. [Google Scholar] [CrossRef]
- Azim, H.A.J.; Michiels, S.; Bedard, P.L.; Singhal, S.K.; Criscitiello, C.; Ignatiadis, M.; Haibe-Kains, B.; Piccart, M.J.; Sotiriou, C.; Loi, S. Elucidating Prognosis and Biology of Breast Cancer Arising in Young Women Using Gene Expression Profiling. Clin. Cancer Res. 2012, 18, 1341–1351. [Google Scholar] [CrossRef]
- Deng, G.; Lu, Y.; Zlotnikov, G.; Thor, A.D.; Smith, H.S. Loss of Heterozygosity in Normal Tissue Adjacent to Breast Carcinomas. Science 1996, 274, 2057–2059. [Google Scholar] [CrossRef]
- Aran, D.; Camarda, R.; Odegaard, J.; Paik, H.; Oskotsky, B.; Krings, G.; Goga, A.; Sirota, M.; Butte, A.J. Comprehensive Analysis of Normal Adjacent to Tumor Transcriptomes. Nat. Commun. 2017, 8, 1077. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, L.; Yang, H.; Li, C.; Fang, C. Identification of Potential Genes Related to Breast Cancer Brain Metastasis in Breast Cancer Patients. Biosci. Rep. 2021, 41, BSR20211615. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zheng, X.; Zheng, Y.; Chen, Y.; Fei, W.; Wang, F.; Zheng, C. Extracellular Matrix: Emerging Roles and Potential Therapeutic Targets for Breast Cancer. Front. Oncol. 2021, 11, 650453. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, P.J.; Pascetta, S.A.; Kirsh, S.M.; Al-Khazraji, B.K.; Uniacke, J. Expression of Hypoxia Inducible Factor-Dependent Neuropeptide Y Receptors Y1 and Y5 Sensitizes Hypoxic Cells to NPY Stimulation. J. Biol. Chem. 2022, 298, 101645. [Google Scholar] [CrossRef]
- Bhat, R.; Thangavel, H.; Abdulkareem, N.M.; Vasaikar, S.; de Angelis, C.; Bae, L.; Cataldo, M.L.; Nanda, S.; Fu, X.; Zhang, B.; et al. NPY1R Exerts Inhibitory Action on Estradiol-Stimulated Growth and Predicts Endocrine Sensitivity and Better Survival in ER-Positive Breast Cancer. Sci. Rep. 2022, 12, 1972. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xu, Q.; Cheng, L.; Ma, C.; Xiao, L.; Xu, D.; Gao, Y.; Wang, J.; Song, H. NPY1R Is a Novel Peripheral Blood Marker Predictive of Metastasis and Prognosis in Breast Cancer Patients. Oncol. Lett. 2015, 9, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, M.A.H.; Maier, C.M.; Falzoni, R.; Quadros, L.G.D.A.; Lima, G.R.; Baracat, E.C.; Nazário, A.C.P. Assessment of the Proliferative, Apoptotic and Cellular Renovation Indices of the Human Mammary Epithelium during the Follicular and Luteal Phases of the Menstrual Cycle. Breast Cancer Res. 2005, 7, R306. [Google Scholar] [CrossRef]
- Olsson, H.L.; Olsson, M.L. The Menstrual Cycle and Risk of Breast Cancer: A Review. Front. Oncol. 2020, 10, 21. [Google Scholar] [CrossRef]
- Liang, H.; Xiao, J.; Zhou, Z.; Wu, J.; Ge, F.; Li, Z.; Zhang, H.; Sun, J.; Li, F.; Liu, R.; et al. Hypoxia Induces MiR-153 through the IRE1α-XBP1 Pathway to Fine Tune the HIF1α/VEGFA Axis in Breast Cancer Angiogenesis. Oncogene 2018, 37, 1961–1975. [Google Scholar] [CrossRef]
- Li, X.; Tian, R.; Gao, H.; Yang, Y.; Williams, B.R.G.; Gantier, M.P.; McMillan, N.A.J.; Xu, D.; Hu, Y.; Gao, Y. Identification of a Histone Family Gene Signature for Predicting the Prognosis of Cervical Cancer Patients. Sci. Rep. 2017, 7, 16495. [Google Scholar] [CrossRef]
- Xie, W.; Zhang, J.; Zhong, P.; Qin, S.; Zhang, H.; Fan, X.; Yin, Y.; Liang, R.; Han, Y.; Liao, Y.; et al. Expression and Potential Prognostic Value of Histone Family Gene Signature in Breast Cancer. Exp. Ther. Med. 2019, 18, 4893–4903. [Google Scholar] [CrossRef] [PubMed]
- Senie, R.T.; Rosen, P.P.; Lesser, M.L.; Snyder, R.E.; Schottenfeld, D.; Duthie, K. Epidemiology of Breast Carcinoma II: Factors Related to the Predominance of Left-sided Disease. Cancer 1980, 46, 1705–1713. [Google Scholar] [CrossRef]
- Perkins, C.I.; Hotes, J.; Kohler, B.A.; Howe, H.L. Association between Breast Cancer Laterality and Tumor Location, United States, 1994–1998. Cancer Causes Control 2004, 15, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Wilting, J.; Hagedorn, M. Left-Right Asymmetry in Embryonic Development and Breast Cancer: Common Molecular Determinants? Curr. Med. Chem. 2011, 18, 5519–5527. [Google Scholar] [CrossRef]
- Veltmaat, J.M.; Ramsdell, A.F.; Sterneck, E. Positional Variations in Mammary Gland Development and Cancer. J. Mammary Gland Biol. Neoplasia 2013, 18, 179–188. [Google Scholar] [CrossRef]
- Barbara, R.C.; Piotr, R.; Kornel, B.; Elżbieta, Z.; Danuta, R.; Eduardo, N. Divergent Impact of Breast Cancer Laterality on Clinicopathological, Angiogenic, and Hemostatic Profiles: A Potential Role of Tumor Localization in Future Outcomes. J. Clin. Med. 2020, 9, 1708. [Google Scholar] [CrossRef]
- Robichaux, J.P.; Hallett, R.M.; Fuseler, J.W.; Hassell, J.A.; Ramsdell, A.F. Mammary Glands Exhibit Molecular Laterality and Undergo Left-Right Asymmetric Ductal Epithelial Growth in MMTV-CNeu Mice. Oncogene 2015, 34, 2003–2010. [Google Scholar] [CrossRef]
- Campoy, E.M.; Laurito, S.R.; Branham, M.T.; Urrutia, G.; Mathison, A.; Gago, F.; Orozco, J.; Urrutia, R.; Mayorga, L.S.; Roqué, M. Asymmetric Cancer Hallmarks in Breast Tumors on Different Sides of the Body. PLoS ONE 2016, 11, e0157416. [Google Scholar] [CrossRef][Green Version]
- Newton, Y.; Szeto, C.; Vaske, C.; Reddy, L.; Reddy, S. Abstract 2533: The Genomic and Transcriptomic Landscape of Left versus Right Sided Breast Cancer in 410 Cases. Cancer Res. 2019, 79, 2533. [Google Scholar] [CrossRef]
- Sandiford, O.A.; Donnelly, R.J.; El-Far, M.H.; Burgmeyer, L.M.; Sinha, G.; Pamarthi, S.H.; Sherman, L.S.; Ferrer, A.I.; DeVore, D.E.; Patel, S.A.; et al. Mesenchymal Stem Cell-Secreted Extracellular Vesicles Instruct Stepwise Dedifferentiation of Breast Cancer Cells into Dormancy at the Bone Marrow Perivascular Region. Cancer Res. 2021, 81, 1567–1582. [Google Scholar] [CrossRef]
- Eswaran, J.; Cyanam, D.; Mudvari, P.; Divijendra, S.; Reddy, N.; Pakala, S.B.; Nair, S.S.; Florea, L.; Fuqua, S.A.; Godbole, S.; et al. Transcriptomic landscape of breast cancers through mRNA sequencing. Sci. Rep. 2012, 2, 264. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.S.; Kumar, R. Chromatin remodeling in cancer: A gateway to regulate gene transcription. Mol. Oncol. 2012, 6, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.S.; Mishra, S.K.; Yang, Z.; Balasenthil, S.; Kumar, R.; Vadlamudi, R.K. Potential role of a novel transcriptional coactivator PELP1 in histone H1 displacement in cancer cells. Cancer Res. 2004, 64, 6416–6423. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Manavathi, B.; Acconcia, F.; Rayala, S.K.; Kumar, R. An inherent role of microtubule network in the action of nuclear receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 15981–15986. [Google Scholar] [CrossRef] [PubMed]
- Jordan, K.R.; Hall, J.K.; Schedin, T.; Borakove, M.; Xian, J.J.; Dzieciatkowska, M.; Borges, V.F. Extracellular vesicles from young women’s breast cancer patients drive increased invasion of non-malignant cells via the focal adhesion kinase pathway: A proteomic approach. Breast Cancer Res. 2020, 22, 128. [Google Scholar] [CrossRef] [PubMed]
- Stevic, I.; Müller, V.; Weber, K.; Fasching, P.A.; Karn, T.; Marmé, F.; Schem, C.; Schwarzenbach, H. Specific microRNA signatures in exosomes of triple-negative and HER2-positive breast cancer patients undergoing neoadjuvant therapy within the GeparSixto trial. BMC Med. 2018, 16, 179. [Google Scholar] [CrossRef]
- Tkach, M.; Thalmensi, J.; Timperi, E.; Gueguen, P.; Névo, N.; Grisard, E.; Sirven, P. Extracellular vesicles from triple negative breast cancer promote pro-inflammatory macrophages associated with better clinical outcome. Proc. Natl. Acad. Sci. USA 2022, 119, e2107394119. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Yu, G.; Sun, Z.; Wang, T.; Tian, X.; Duan, X.; Zhang, C. Exosomal miR-1246 and miR-155 as predictive and prognostic biomarkers for trastuzumab-based therapy resistance in HER2-positive breast cancer. Cancer Chemother. Pharmacol. 2020, 86, 761–772. [Google Scholar] [CrossRef]
- Knower, K.C.; To, S.Q.; Leung, Y.K.; Ho, S.M.; Clyne, C.D. Endocrine disruption of the epigenome: A breast cancer link. Endocr. Relat. Cancer 2014, 21, T33. [Google Scholar] [CrossRef]
- Remely, M.; Stefanska, B.; Lovrecic, L.; Magnet, U.; Haslberger, A.G. Nutriepigenomics: The role of nutrition in epigenetic control of human diseases. Curr. Opin. Clin. Nutr. Metab. Care. 2015, 18, 328–333. [Google Scholar] [CrossRef]
- Paris, A.; Tardif, N.; Galibert, M.D.; Corre, S. AhR and cancer: From gene profiling to targeted therapy. Int. J. Mol. Sci. 2021, 22, 752. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, A.M.; George, B.; Saini, S.; Pillai, M.R.; Toi, M.; Costa, L.; Kumar, R. Delineation of Pathogenomic Insights of Breast Cancer in Young Women. Cells 2022, 11, 1927. https://doi.org/10.3390/cells11121927
Paul AM, George B, Saini S, Pillai MR, Toi M, Costa L, Kumar R. Delineation of Pathogenomic Insights of Breast Cancer in Young Women. Cells. 2022; 11(12):1927. https://doi.org/10.3390/cells11121927
Chicago/Turabian StylePaul, Aswathy Mary, Bijesh George, Sunil Saini, Madhavan Radhakrishna Pillai, Masakazu Toi, Luis Costa, and Rakesh Kumar. 2022. "Delineation of Pathogenomic Insights of Breast Cancer in Young Women" Cells 11, no. 12: 1927. https://doi.org/10.3390/cells11121927
APA StylePaul, A. M., George, B., Saini, S., Pillai, M. R., Toi, M., Costa, L., & Kumar, R. (2022). Delineation of Pathogenomic Insights of Breast Cancer in Young Women. Cells, 11(12), 1927. https://doi.org/10.3390/cells11121927