
Biomarkers in Nephropathic Cystinosis: Current and Future Perspectives

 , , ,
, , ,
Abstract
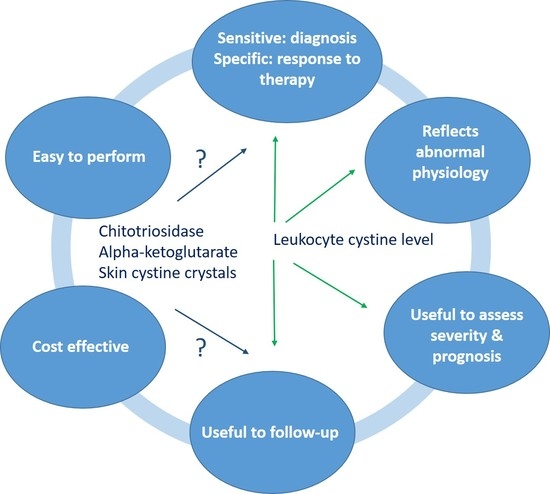
:
1. Introduction
2. Methods for Measuring Intracellular Leucocyte Cystine
2.1. Cell Isolation
2.2. Cystine and Protein Determination
3. Prenatal and Neonatal Diagnosis
3.1. Prenatal Diagnosis
3.2. Neonatal Diagnosis in at Risk Siblings
3.3. Newborn Screening in Unaffected Population
4. Treatment Monitoring: Importance of Target Cystine Values
5. Limitations of Current Monitoring
6. Novel Biomarkers
6.1. Biochemical Biomarkers
6.1.1. Chitotriosidase
6.1.2. Alpha-Ketoglutarate
6.2. Quantification of Cystine Crystals
6.2.1. Intestinal Cystine Crystals
6.2.2. Intradermal Cystine Crystal Determination and Skin Aging
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thoene, J.; Lemons, R.; Anikster, Y.; Mullet, J.; Paelicke, K.; Lucero, C.; Gahl, W.; Schneider, J.; Shu, S.G.; Campbell, H.T. Mutations of CTNS causing intermediate cystinosis. Mol. Genet. Metab. 1999, 67, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Town, M.; Jean, G.; Cherqui, S.; Attard, M.; Forestier, L.; Whitmore, S.A.; Callen, D.F.; Gribouval, O.; Broyer, M.; Bates, G.P.; et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat. Genet. 1998, 18, 319–324. [Google Scholar] [CrossRef] [PubMed]
- The Cystinosis Collaborative Research Group. Linkage of the gene for cystinosis to markers on the short arm of chromosome 17. Nat. Genet. 1995, 10, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Veys, K.R.; Elmonem, M.A.; Arcolino, F.O.; van den Heuvel, L.; Levtchenko, E. Nephropathic cystinosis: An update. Curr. Opin. Pediatr. 2017, 29, 168–178. [Google Scholar] [CrossRef]
- Besouw, M.T.; Van Dyck, M.; Cassiman, D.; Claes, K.J.; Levtchenko, E.N. Management dilemmas in pediatric nephrology: Cystinosis. Pediatr. Nephrol. 2015, 30, 1349–1360. [Google Scholar] [CrossRef]
- Gultekingil Keser, A.; Topaloglu, R.; Bilginer, Y.; Besbas, N. Long-term endocrinologic complications of cystinosis. Minerva. Pediatr. 2014, 66, 123–130. [Google Scholar]
- Emma, F.; Nesterova, G.; Langman, C.; Labbe, A.; Cherqui, S.; Goodyer, P.; Janssen, M.C.; Greco, M.; Topaloglu, R.; Elenberg, E.; et al. Nephropathic cystinosis: An international consensus document. Nephrol. Dial. Transpl. 2014, 29 (Suppl. 4), iv87–iv94. [Google Scholar] [CrossRef] [Green Version]
- Gahl, W.A.; Thoene, J.G.; Schneider, J.A. Cystinosis. N. Engl. J. Med. 2002, 347, 111–121. [Google Scholar] [CrossRef]
- Topaloglu, R. Nephropathic cystinosis: An update on genetic conditioning. Pediatr. Nephrol. 2021, 36, 1347–1352. [Google Scholar] [CrossRef]
- David, D.; Princiero Berlingerio, S.; Elmonem, M.A.; Oliveira Arcolino, F.; Soliman, N.; van den Heuvel, B.; Gijsbers, R.; Levtchenko, E. Molecular Basis of Cystinosis: Geographic Distribution, Functional Consequences of Mutations in the CTNS Gene, and Potential for Repair. Nephron. 2019, 141, 133–146. [Google Scholar] [CrossRef]
- Ivanova, E.A.; Arcolino, F.O.; Elmonem, M.A.; Rastaldi, M.P.; Giardino, L.; Cornelissen, E.M.; van den Heuvel, L.P.; Levtchenko, E.N. Cystinosin deficiency causes podocyte damage and loss associated with increased cell motility. Kidney Int. 2016, 89, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Levtchenko, E.; Monnens, L. Development of Fanconi syndrome during infancy in a patient with cystinosis. Acta. Paediatr. 2006, 95, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Long, W.S.; Seashore, M.R.; Siegel, N.J.; Bia, M.J. Idiopathic Fanconi syndrome with progressive renal failure: A case report and discussion. Yale. J. Biol. Med. 1990, 63, 15–28. [Google Scholar] [PubMed]
- Brodin-Sartorius, A.; Tete, M.J.; Niaudet, P.; Antignac, C.; Guest, G.; Ottolenghi, C.; Charbit, M.; Moyse, D.; Legendre, C.; Lesavre, P.; et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 2012, 81, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Nesterova, G.; Gahl, W.A. Cystinosis: Tthe evolution of a treatable disease. Pediatr. Nephrol. 2013, 28, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Gahl, W.A.; Balog, J.Z.; Kleta, R. Nephropathic cystinosis in adults: Natural history and effects of oral cysteamine therapy. Ann. Intern. Med. 2007, 147, 242–250. [Google Scholar] [CrossRef]
- Schiefer, J.; Zenker, M.; Grone, H.J.; Chatzikyrkou, C.; Mertens, P.R.; Liakopoulos, V. Unrecognized juvenile nephropathic cystinosis. Kidney. Int. 2018, 94, 1027. [Google Scholar] [CrossRef]
- Manz, F.; Gretz, N. Cystinosis in the Federal Republic of Germany. Coordination and analysis of the data. J. Inherit. Metab. Dis. 1985, 8, 2–4. [Google Scholar] [CrossRef]
- Cherqui, S.; Courtoy, P.J. The renal Fanconi syndrome in cystinosis: Pathogenic insights and therapeutic perspectives. Nat. Rev. Nephrol. 2017, 13, 115–131. [Google Scholar] [CrossRef]
- Greco, M.; Brugnara, M.; Zaffanello, M.; Taranta, A.; Pastore, A.; Emma, F. Long-term outcome of nephropathic cystinosis: A 20-year single-center experience. Pediatr. Nephrol. 2010, 25, 2459–2467. [Google Scholar] [CrossRef]
- Schulman, J.D.; Wong, V.G.; Kuwabara, T.; Bradley, K.H.; Seegmiller, J.E. Intracellular cystine content of leukocyte populations in cystinosis. Arch. Intern. Med. 1970, 125, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Bradley, K.; Seegmiller, J.E. Increased cystine in leukocytes from individuals homozygous and heterozygous for cystinosis. Science 1967, 157, 1321–1322. [Google Scholar] [CrossRef]
- Oshima, R.G.; Willis, R.C.; Furlong, C.E.; Schneider, J.A. Binding assays for amino acids. The utilization of a cystine binding protein from Escherichia coli for the determination of acid-soluble cystine in small physiological samples. J. Biol. Chem. 1974, 249, 6033–6039. [Google Scholar] [CrossRef]
- Smith, M.; Furlong, C.E.; Greene, A.A.; Schneider, J.A. Cystine: Binding protein assay. Methods Enzymol. 1987, 143, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Chabli, A.; Aupetit, J.; Raehm, M.; Ricquier, D.; Chadefaux-Vekemans, B. Measurement of cystine in granulocytes using liquid chromatography-tandem mass spectrometry. Clin. Biochem. 2007, 40, 692–698. [Google Scholar] [CrossRef]
- de Graaf-Hess, A.; Trijbels, F.; Blom, H. New method for determining cystine in leukocytes and fibroblasts. Clin. Chem. 1999, 45, 2224–2228. [Google Scholar] [CrossRef]
- Elmonem, M.A.; Veys, K.R.; Soliman, N.A.; van Dyck, M.; van den Heuvel, L.P.; Levtchenko, E. Cystinosis: A review. Orphanet. J. Rare. Dis. 2016, 11, 47. [Google Scholar] [CrossRef] [Green Version]
- Smolin, L.A.; Clark, K.F.; Schneider, J.A. An improved method for heterozygote detection of cystinosis, using polymorphonuclear leukocytes. Am. J. Hum. Genet. 1987, 41, 266–275. [Google Scholar]
- Levtchenko, E.; de Graaf-Hess, A.; Wilmer, M.; van den Heuvel, L.; Monnens, L.; Blom, H. Comparison of cystine determination in mixed leukocytes vs polymorphonuclear leukocytes for diagnosis of cystinosis and monitoring of cysteamine therapy. Clin. Chem. 2004, 50, 1686–1688. [Google Scholar] [CrossRef] [Green Version]
- Fowler, B.; Bielsky, M.C.; Farrington, Z. White Cell Cystine Group; BIMDG Bulletin Spring 2001, WCYS Compiled.doc.; BIMDG: Cambridge, UK, 2001. [Google Scholar]
- Chadefaux-Vekemans, B. White Cell Cystine Group: Guideline no. 2. Polymorphonuclear Leucocyte Preparation; BIMDG Bulletin Spring 2001; BIMDG: Cambridge, UK, 2001. [Google Scholar]
- Gertsman, I.; Johnson, W.S.; Nishikawa, C.; Gangoiti, J.A.; Holmes, B.; Barshop, B.A. Diagnosis and Monitoring of Cystinosis Using Immunomagnetically Purified Granulocytes. Clin. Chem. 2016, 62, 766–772. [Google Scholar] [CrossRef] [Green Version]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Powell, K.L.; Langman, C.B. An unexpected problem in the clinical assessment of cystinosis. Pediatr. Nephrol. 2012, 27, 687–688. [Google Scholar] [CrossRef] [Green Version]
- Langman, C.B.; Barshop, B.A.; Deschenes, G.; Emma, F.; Goodyer, P.; Lipkin, G.; Midgley, J.P.; Ottolenghi, C.; Servais, A.; Soliman, N.A.; et al. Controversies and research agenda in nephropathic cystinosis: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney. Int. 2016, 89, 1192–1203. [Google Scholar] [CrossRef]
- Emma, F.; Hoff, W.V.; Hohenfellner, K.; Topaloglu, R.; Greco, M.; Ariceta, G.; Bettini, C.; Bockenhauer, D.; Veys, K.; Pape, L.; et al. An international cohort study spanning five decades assessed outcomes of nephropathic cystinosis. Kidney. Int. 2021, 100, 1112–1123. [Google Scholar] [CrossRef]
- Jackson, M.; Young, E. Prenatal diagnosis of cystinosis by quantitative measurement of cystine in chorionic villi and cultured cells. Prenat. Diagn. 2005, 25, 1045–1047. [Google Scholar] [CrossRef]
- da Silva, V.A.; Zurbrugg, R.P.; Lavanchy, P.; Blumberg, A.; Suter, H.; Wyss, S.R.; Luthy, C.M.; Oetliker, O.H. Long-term treatment of infantile nephropathic cystinosis with cysteamine. N. Engl. J. Med. 1985, 313, 1460–1463. [Google Scholar] [CrossRef]
- Gahl, W.A.; Thoene, J.G. Cystinosis: A disorder of lysosomal membrane transport. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Sly, W.S., Childs, B., Beaudet, A.L., Valle, D., Kinzler, K.W., Vogelstein, B., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 5085–5108. [Google Scholar]
- Surmeli Doven, S.; Delibas, A.; Kayacan, U.R.; Unal, S. Short-cut diagnostic tool in cystinosis: Bone marrow aspiration. Pediatr. Int. 2017, 59, 1178–1182. [Google Scholar] [CrossRef]
- Wamelink, M.M.; Struys, E.A.; Jansen, E.E.; Blom, H.J.; Vilboux, T.; Gahl, W.A.; Komhoff, M.; Jakobs, C.; Levtchenko, E.N. Elevated concentrations of sedoheptulose in bloodspots of patients with cystinosis caused by the 57-kb deletion: Implications for diagnostics and neonatal screening. Mol. Genet. Metab. 2011, 102, 339–342. [Google Scholar] [CrossRef]
- Fleige, T.; Burggraf, S.; Czibere, L.; Haring, J.; Gluck, B.; Keitel, L.M.; Landt, O.; Harms, E.; Hohenfellner, K.; Durner, J.; et al. Next generation sequencing as second-tier test in high-throughput newborn screening for nephropathic cystinosis. Eur. J. Hum. Genet. 2020, 28, 193–201. [Google Scholar] [CrossRef]
- Hohenfellner, K.; Bergmann, C.; Fleige, T.; Janzen, N.; Burggraf, S.; Olgemoller, B.; Gahl, W.A.; Czibere, L.; Froschauer, S.; Roschinger, W.; et al. Molecular based newborn screening in Germany: Follow-up for cystinosis. Mol. Genet. Metab. Rep. 2019, 21, 100514. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, G.; Williams, C.; Bernardini, I.; Gahl, W.A. Cystinosis: Renal glomerular and renal tubular function in relation to compliance with cystine-depleting therapy. Pediatr. Nephrol. 2015, 30, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Servais, A.; Saitovitch, A.; Hummel, A.; Boisgontier, J.; Scemla, A.; Sberro-Soussan, R.; Snanoudj, R.; Lemaitre, H.; Legendre, C.; Pontoizeau, C.; et al. Central nervous system complications in adult cystinosis patients. J. Inherit. Metab. Dis. 2020, 43, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Wilmer, M.J.; Schoeber, J.P.; van den Heuvel, L.P.; Levtchenko, E.N. Cystinosis: Practical tools for diagnosis and treatment. Pediatr. Nephrol. 2011, 26, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Levtchenko, E.N.; van Dael, C.M.; de Graaf-Hess, A.C.; Wilmer, M.J.; van den Heuvel, L.P.; Monnens, L.A.; Blom, H.J. Strict cysteamine dose regimen is required to prevent nocturnal cystine accumulation in cystinosis. Pediatr. Nephrol. 2006, 21, 110–113. [Google Scholar] [CrossRef]
- Linden, S.; Klank, S.; Harms, E.; Gruneberg, M.; Park, J.H.; Marquardt, T. Cystinosis: Therapy adherence and metabolic monitoring in patients treated with immediate-release cysteamine. Mol. Genet. Metab. Rep. 2020, 24, 100620. [Google Scholar] [CrossRef]
- Ariceta, G.; Giordano, V.; Santos, F. Effects of long-term cysteamine treatment in patients with cystinosis. Pediatr. Nephrol. 2019, 34, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Kleta, R.; Kaskel, F.; Dohil, R.; Goodyer, P.; Guay-Woodford, L.M.; Harms, E.; Ingelfinger, J.R.; Koch, V.H.; Langman, C.B.; Leonard, M.B.; et al. Diseases, N.I.H.O.o.R. First NIH/Office of Rare Diseases Conference on Cystinosis: Past, present, and future. Pediatr. Nephrol. 2005, 20, 452–454. [Google Scholar] [CrossRef]
- Markello, T.C.; Bernardini, I.M.; Gahl, W.A. Improved renal function in children with cystinosis treated with cysteamine. N. Engl. J. Med. 1993, 328, 1157–1162. [Google Scholar] [CrossRef]
- Gahl, W.A.; Reed, G.F.; Thoene, J.G.; Schulman, J.D.; Rizzo, W.B.; Jonas, A.J.; Denman, D.W.; Schlesselman, J.J.; Corden, B.J.; Schneider, J.A. Cysteamine therapy for children with nephropathic cystinosis. N. Engl. J. Med. 1987, 316, 971–977. [Google Scholar] [CrossRef]
- Labbe, A.; Niaudet, P.; Loirat, C.; Charbit, M.; Guest, G.; Baudouin, C. In vivo confocal microscopy and anterior segment optical coherence tomography analysis of the cornea in nephropathic cystinosis. Ophthalmology 2009, 116, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Chiaverini, C.; Kang, H.Y.; Sillard, L.; Berard, E.; Niaudet, P.; Guest, G.; Cailliez, M.; Bahadoran, P.; Lacour, J.P.; Ballotti, R.; et al. In vivo reflectance confocal microscopy of the skin: A noninvasive means of assessing body cystine accumulation in infantile cystinosis. J. Am. Acad. Dermatol. 2013, 68, e111–e116. [Google Scholar] [CrossRef] [PubMed]
- Elmonem, M.A.; Makar, S.H.; van den Heuvel, L.; Abdelaziz, H.; Abdelrahman, S.M.; Bossuyt, X.; Janssen, M.C.; Cornelissen, E.A.; Lefeber, D.J.; Joosten, L.A.; et al. Clinical utility of chitotriosidase enzyme activity in nephropathic cystinosis. Orphanet. J. Rare. Dis. 2014, 9, 155. [Google Scholar] [CrossRef] [Green Version]
- Prencipe, G.; Caiello, I.; Cherqui, S.; Whisenant, T.; Petrini, S.; Emma, F.; De Benedetti, F. Inflammasome activation by cystine crystals: Implications for the pathogenesis of cystinosis. J. Am. Soc. Nephrol. 2014, 25, 1163–1169. [Google Scholar] [CrossRef] [Green Version]
- Veys, K.R.P.; Elmonem, M.A.; Van Dyck, M.; Janssen, M.C.; Cornelissen, E.A.M.; Hohenfellner, K.; Prencipe, G.; van den Heuvel, L.P.; Levtchenko, E. Chitotriosidase as a Novel Biomarker for Therapeutic Monitoring of Nephropathic Cystinosis. J. Am. Soc. Nephrol. 2020, 31, 1092–1106. [Google Scholar] [CrossRef]
- Lobry, T.; Miller, R.; Nevo, N.; Rocca, C.J.; Zhang, J.; Catz, S.D.; Moore, F.; Thomas, L.; Pouly, D.; Bailleux, A.; et al. Interaction between galectin-3 and cystinosin uncovers a pathogenic role of inflammation in kidney involvement of cystinosis. Kidney Int. 2019, 96, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.J. A clinico-pathological study of cystinosis in two siblings. Arch. Dis. Child. 1952, 27, 428–433. [Google Scholar] [CrossRef] [Green Version]
- DiDomenico, P.; Berry, G.; Bass, D.; Fridge, J.; Sarwal, M. Noncirrhotic portal hypertension in association with juvenile nephropathic cystinosis: Case presentation and review of the literature. J. Inherit. Metab. Dis. 2004, 27, 693–699. [Google Scholar] [CrossRef]
- Dohil, R.; Carrigg, A.; Newbury, R. A potential new method to estimate tissue cystine content in nephropathic cystinosis. J. Pediatr. 2012, 161, 531–535.e1. [Google Scholar] [CrossRef]
- Guillet, G.; Sassolas, B.; Fromentoux, S.; Gobin, E.; Leroy, J.P. Skin storage of cystine and premature skin ageing in cystinosis. Lancet 1998, 352, 1444–1445. [Google Scholar] [CrossRef]
- Monier, L.; Mauvieux, L. Cystine crystals in bone marrow aspirate. Blood 2015, 126, 1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmonem, M.A.; Ramadan, D.I.; Issac, M.S.; Selim, L.A.; Elkateb, S.M. Blood spot versus plasma chitotriosidase: A systematic clinical comparison. Clin. Biochem. 2014, 47, 38–43. [Google Scholar] [CrossRef]
- Guo, Y.; He, W.; Boer, A.M.; Wevers, R.A.; de Bruijn, A.M.; Groener, J.E.; Hollak, C.E.; Aerts, J.M.; Galjaard, H.; van Diggelen, O.P. Elevated plasma chitotriosidase activity in various lysosomal storage disorders. J. Inherit. Metab. Dis. 1995, 18, 717–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamalpoor, A.; van Gelder, C.A.; Yousef Yengej, F.A.; Zaal, E.A.; Berlingerio, S.P.; Veys, K.R.; Pou Casellas, C.; Voskuil, K.; Essa, K.; Ammerlaan, C.M.; et al. Cysteamine-bicalutamide combination therapy corrects proximal tubule phenotype in cystinosis. EMBO Mol. Med. 2021, 13, e13067. [Google Scholar] [CrossRef] [PubMed]
- Elmonem, M.A.; Veys, K.; Oliveira Arcolino, F.; Van Dyck, M.; Benedetti, M.C.; Diomedi-Camassei, F.; De Hertogh, G.; van den Heuvel, L.P.; Renard, M.; Levtchenko, E. Allogeneic HSCT transfers wild-type cystinosin to nonhematological epithelial cells in cystinosis: First human report. Am. J. Transplant. 2018, 18, 2823–2828. [Google Scholar] [CrossRef] [Green Version]
- Veys, K.R.P.; Elmonem, M.A.; Dhaenens, F.; Van Dyck, M.; Janssen, M.; Cornelissen, E.A.M.; Hohenfellner, K.; Reda, A.; Quatresooz, P.; van den Heuvel, B.; et al. Enhanced Intrinsic Skin Aging in Nephropathic Cystinosis Assessed by High-Definition Optical Coherence Tomography. J. Investig. Dermatol. 2019, 139, 2242–2245. [Google Scholar] [CrossRef]
- Bengali, M.; Goodman, S.; Sun, X.; Dohil, M.A.; Dohil, R.; Newbury, R.; Lobry, T.; Hernandez, L.; Antignac, C.; Jain, S.; et al. Non-invasive intradermal imaging of cystine crystals in cystinosis. PLoS ONE 2021, 16, e0247846. [Google Scholar] [CrossRef]
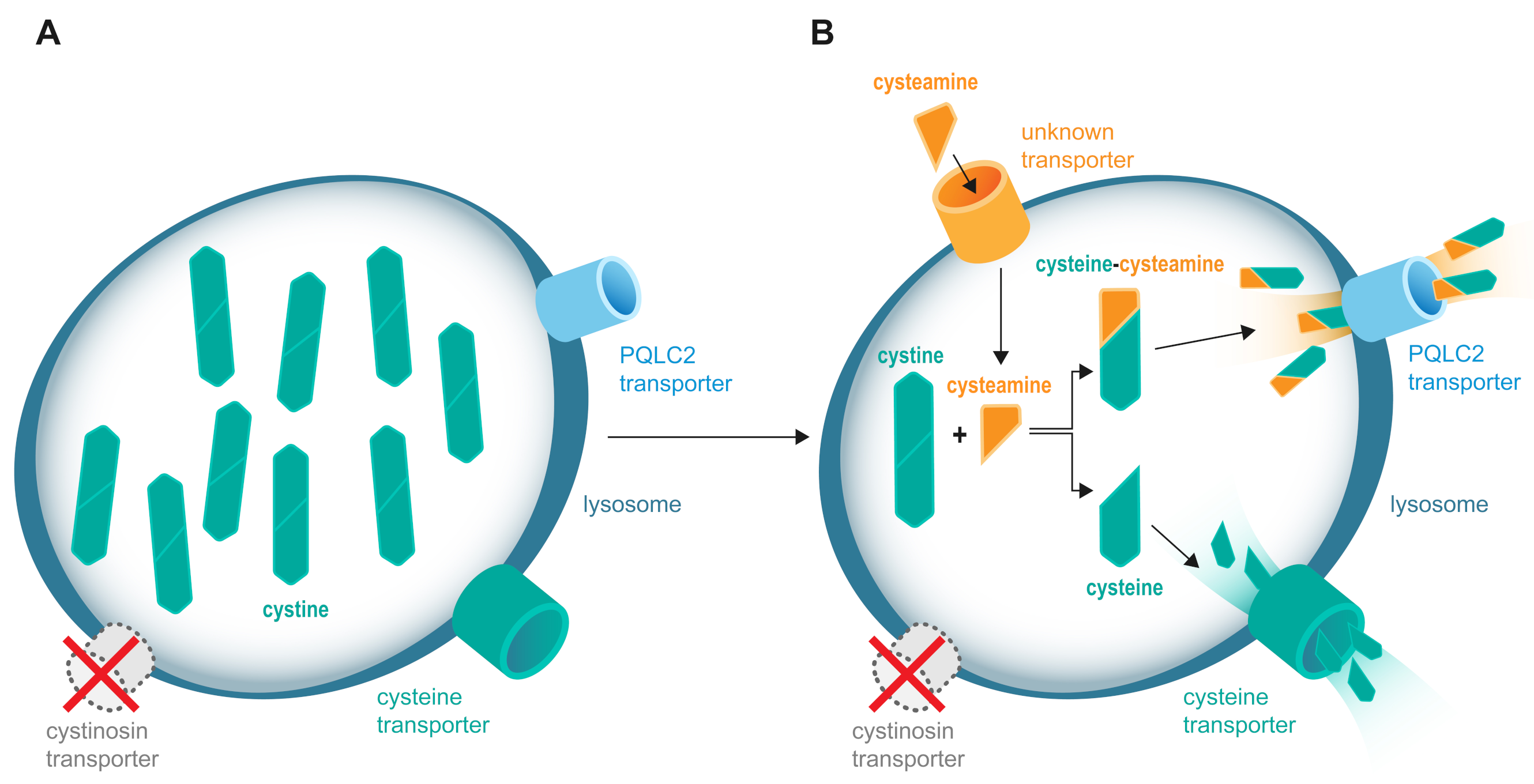

{kind=link}
{kind=link}
| Study | Kidney Outcome | Extra-Renal Outcome | Reference |
|---|---|---|---|
| US study; children treated with cysteamine up to 73 months (n = 93) | Mean creatinine clearance reduced in patients not adequately depleted (< 1 vs. > 3 nmol half-cystine/mg protein: 50.5 vs. 29.7 mL per min per 1.73 m2) | [53] | |
| US single center study; patients analyzed between 1960 and 1992 (n = 76) | Non-adequate treatment as shown by leucocyte cystine levels > 2 nmol half-cystine/mg protein. Treatment started at > 2 y with reduced creatinine clearance and early onset of renal impairment (mean creatinine clearance predicted = 0 ml at the age of 20 vs. 74 y for children receiving adequate treatment compared to those with only partial treatment) | [52] | |
| US database; age 18–45 y analyzed between January 1985 and May 2006 (n = 100) | Non-adequate treatment, highlighted by leucocyte cystine levels above the cut-off, need for kidney transplantation at least 3.8 y earlier | Non-adequate treatment associated with an increased incidence of hypothyroidism (87% vs. 56%) and death (49% vs. 8%) | [16] |
| French study; age ≥ 15 y, diagnosis time: 1961–1995, mean follow-up 24.6 y (n = 86) | Patients with leucocyte cystine levels > 3 nmol half-cystine/mg protein show early onset ESRD compared to adequately depleted patients (p < 0.0001) | [14] | |
| Turkish study; single center retrospective study, age 0.5–29 y, median follow-up 8 y (n = 21) | Non-adequate treatment associated with an increased incidence of complications; < 2 vs. ≥2 nmol half-cystine/mg protein with deficit of growth (66.6% vs. 90.9%), pubertal delay (0% vs 66.6%), hypothyroidism (33.3% vs. 54.5%), and diabetes (0% vs 18.1%) | [6] | |
| US database; age 11–48 y analyzed between 1975 and 2005 (n = 147) | Adequate treatment associated with delayed onset ESRD (R2 = 0.997) Every year of sub-optimal treatment corresponded to a loss of 0.9 y of renal glomerular function 21 patients (born in the 1960s and 1970s) reached ESKD in the first 8 years of life | Mean leucocyte cystine levels (p = 0.01), and earlier initiation of cysteamine therapy (p = 0.03) significantly associated with improved growth Children reaching CKD stage 5 before 15 years of age grew on average 0.55 height standard deviation scores worse than children that reached dialysis after their 15th birthday (95% CI: 0.09–1.01; p = 0.03) | [45] |
| International European cohort of patients born between 1964 and 2016 (n = 453) | Earlier age at start of cysteamine and lower mean leucocyte cystine levels are associated with delayed development of CKD stage 5 | [37] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emma, F.; Montini, G.; Pennesi, M.; Peruzzi, L.; Verrina, E.; Goffredo, B.M.; Canalini, F.; Cassiman, D.; Rossi, S.; Levtchenko, E. Biomarkers in Nephropathic Cystinosis: Current and Future Perspectives. Cells 2022, 11, 1839. https://doi.org/10.3390/cells11111839
Emma F, Montini G, Pennesi M, Peruzzi L, Verrina E, Goffredo BM, Canalini F, Cassiman D, Rossi S, Levtchenko E. Biomarkers in Nephropathic Cystinosis: Current and Future Perspectives. Cells. 2022; 11(11):1839. https://doi.org/10.3390/cells11111839
Chicago/Turabian StyleEmma, Francesco, Giovanni Montini, Marco Pennesi, Licia Peruzzi, Enrico Verrina, Bianca Maria Goffredo, Fabrizio Canalini, David Cassiman, Silvia Rossi, and Elena Levtchenko. 2022. "Biomarkers in Nephropathic Cystinosis: Current and Future Perspectives" Cells 11, no. 11: 1839. https://doi.org/10.3390/cells11111839
APA StyleEmma, F., Montini, G., Pennesi, M., Peruzzi, L., Verrina, E., Goffredo, B. M., Canalini, F., Cassiman, D., Rossi, S., & Levtchenko, E. (2022). Biomarkers in Nephropathic Cystinosis: Current and Future Perspectives. Cells, 11(11), 1839. https://doi.org/10.3390/cells11111839