
The Actin Cytoskeleton Responds to Inflammatory Cues and Alters Macrophage Activation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
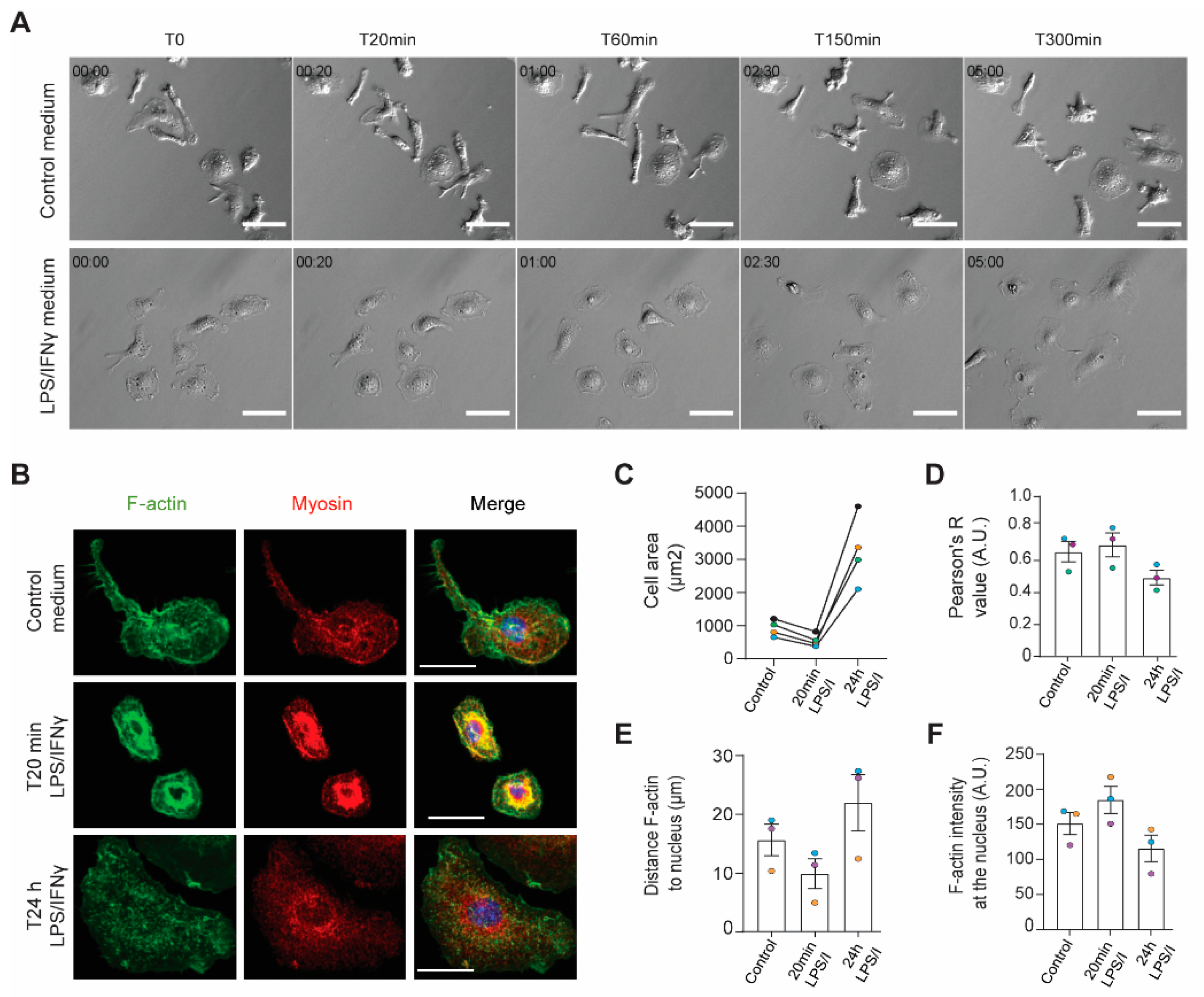
2.1. The Actin Cytoskeleton Responds to Inflammatory Stimulation in Two Temporal Phases
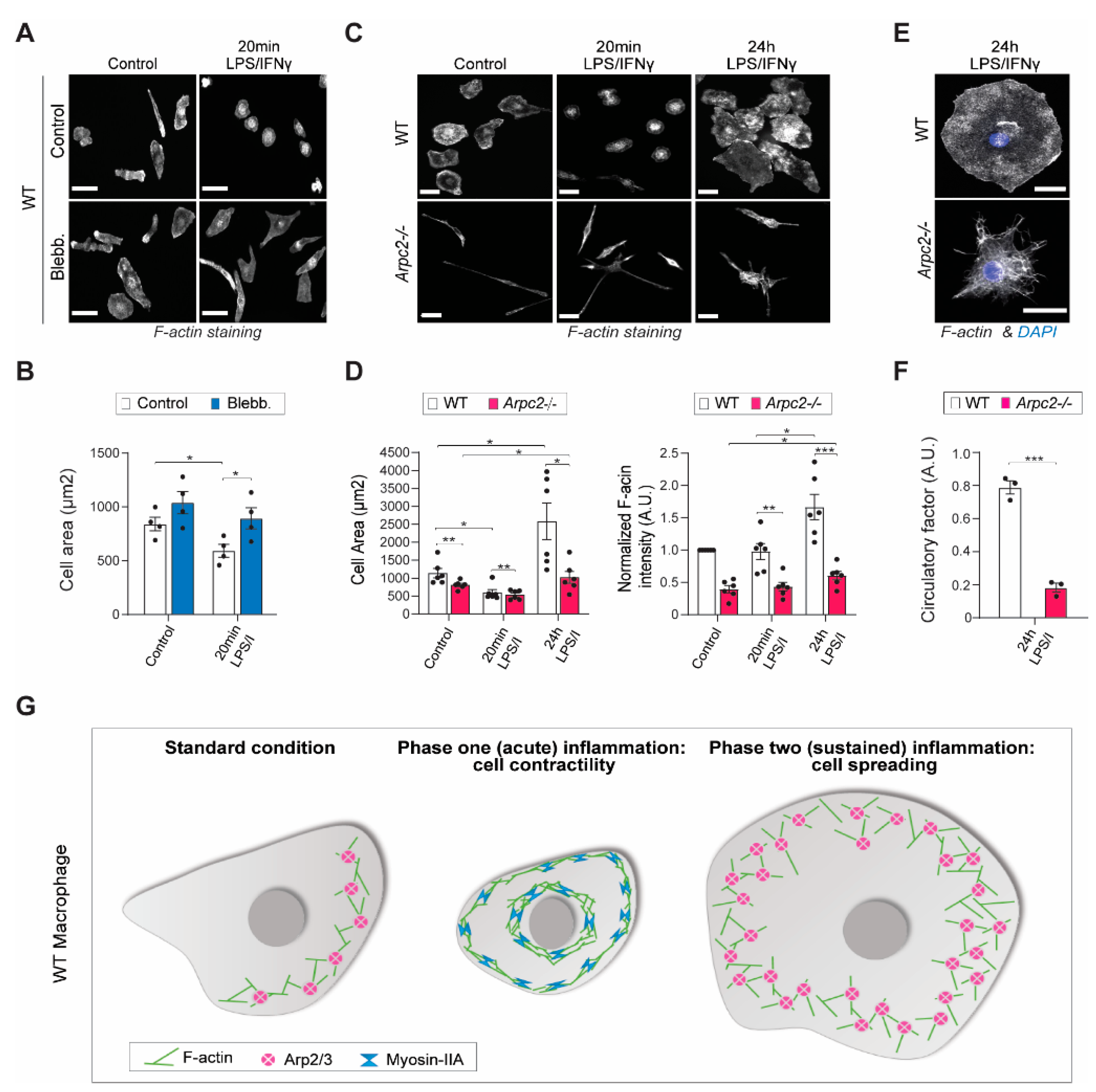
2.2. Arp2/3 Complex and Myosin II Reciprocally Regulate iNOS Activity
2.3. Antigen Presentation and Th-Skewing Cytokine Secretion Are Altered by Loss of Arp2/3 Complex
3. Discussion
4. Materials and Methods
4.1. Mouse Lines Used to Derive Macrophages
4.2. Bone Marrow-Derived Macrophage Preparation and Cell Culture
4.3. Antibodies
4.4. Cell Treatments
4.4.1. Arpc2 Allele Recombination with 4-Hydroxytamoxifen (4-OHT) Treatment
4.4.2. LPS and IFN-g co-Stimulation
4.4.3. Myosin II Inhibition via Blebbistatin
4.5. Western Blot
4.6. Immunofluorescence Staining
4.7. Imaging
4.7.1. Fixed Cell Imaging
4.7.2. Live Cell Imaging
4.7.3. Quantitative Image Analysis
4.8. Elisa
4.9. Chemokine Array
4.10. RT-qPCR
4.11. Nitrite Concentration Measurement
4.12. Proteasome Inhibitor Treatment
4.13. FACS Analysis
4.14. Statistics
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buracco, S.; Claydon, S.; Insall, R. Control of actin dynamics during cell motility. F1000Research 2019, 8, 1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlier, M.-F.; Pantaloni, D. Control of actin assembly dynamics in cell motility. J. Biol. Chem. 2007, 282, 23005–23009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotty, J.D.; Wu, C.; Bear, J.E. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Dos Remedios, C.G.; Chhabra, D.; Kekic, M.; Dedova, I.V.; Tsubakihara, M.; Berry, D.A.; Nosworthy, N.J. Actin binding proteins: Regulation of cytoskeletal microfilaments. Physiol. Rev. 2003, 83, 433–473. [Google Scholar] [CrossRef]
- Frieden, B.R.; Gatenby, R.A. Signal transmission through elements of the cytoskeleton form an optimized information network in eukaryotic cells. Sci. Rep. 2019, 9, 6110. [Google Scholar] [CrossRef]
- Rouiller, I.; Xu, X.P.; Amann, K.J.; Egile, C.; Nickell, S.; Nicastro, D.; Li, R.; Pollard, T.D.; Volkmann, N.; Hanein, D. The structural basis of actin filament branching by the Arp2/3 complex. J. Cell Biol. 2008, 180, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Machesky, L.; Atkinson, S.J.; Ampe, C.; Vandekerckhove, J.; Pollard, T.D. Purification of a cortical complex containing two unconventional actins from Acanthamoeba by affinity chromatography on profilin-agarose. J. Cell Biol. 1994, 127, 107–115. [Google Scholar] [CrossRef]
- Mullins, R.D.; Stafford, W.F.; Pollard, T.D. Structure, subunit topology, and actin-binding activity of the Arp2/3 complex from Acanthamoeba. J. Cell Biol. 1997, 136, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Welch, M.D.; Iwamatsu, A.; Mitchison, T.J. Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature 1997, 385, 265–269. [Google Scholar] [CrossRef]
- Goley, E.D.; Rammohan, A.; Znameroski, E.A.; Firat-Karalar, E.N.; Sept, D.; Welch, M.D. An actin-filament-binding interface on the Arp2/3 complex is critical for nucleation and branch stability. Proc. Natl. Acad. Sci. USA 2010, 107, 8159–8164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayel, M.J.; Holleran, E.A.; Mullins, R.D. Arp2/3 complex requires hydrolyzable ATP for nucleation of new actin filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 14871–14876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Asokan, S.B.; Berginski, M.E.; Haynes, E.M.; Sharpless, N.E.; Griffith, J.D.; Gomez, S.M.; Bear, J.E. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell 2012, 148, 973–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suraneni, P.; Rubinstein, B.; Unruh, J.R.; Durnin, M.; Hanein, D.; Li, R. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J. Cell Biol. 2012, 197, 239–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotty, J.D.; Wu, C.; Haynes, E.M.; Suarez, C.; Winkelman, J.D.; Johnson, H.E.; Haugh, J.M.; Kovar, D.R.; Bear, J.E. Profilin-1 serves as a gatekeeper for actin assembly by Arp2/3-dependent and -independent pathways. Dev. Cell 2015, 32, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Rotty, J.D.; Brighton, H.E.; Craig, S.L.; Asokan, S.B.; Cheng, N.; Ting, J.P.; Bear, J.E. Arp2/3 Complex Is Required for Macrophage Integrin Functions but Is Dispensable for FcR Phagocytosis and In Vivo Motility. Dev. Cell 2017, 42, 498–513.e6. [Google Scholar] [CrossRef] [Green Version]
- Insall, R.H.; Machesky, L.M. Actin dynamics at the leading edge: From simple machinery to complex networks. Dev. Cell 2009, 17, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Haynes, E.M.; Asokan, S.B.; Simon, J.M.; Sharpless, N.E.; Baldwin, A.S.; Davis, I.J.; Johnson, G.L.; Bear, J.E. Loss of Arp2/3 induces an NF-kappaB-dependent, nonautonomous effect on chemotactic signaling. J. Cell Biol. 2013, 203, 907–916. [Google Scholar] [CrossRef]
- Pedersen, E.; Wang, Z.; Stanley, A.; Peyrollier, K.; Rösner, L.M.; Werfel, T.; Quondamatteo, F.; Brakebusch, C. RAC1 in keratinocytes regulates crosstalk to immune cells by Arp2/3-dependent control of STAT1. J Cell Sci 2012, 125 Pt 22, 5379–5390. [Google Scholar] [CrossRef] [Green Version]
- Ambruso, D.R.; Knall, C.; Abell, A.N.; Panepinto, J.; Kurkchubasche, A.; Thurman, G.; Gonzalez-Aller, C.; Hiester, A.; Deboer, M.; Harbeck, R.J.; et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 4654–4659. [Google Scholar] [CrossRef] [Green Version]
- Kashani, P.; Marwaha, A.; Feanny, S.; Kim, V.H.-D.; Atkinson, A.R.; Leon-Ponte, M.; Mendoza-Londono, R.; Grunebaum, E. Progressive decline of T and B cell numbers and function in a patient with CDC42 deficiency. Immunol. Res. 2021, 69, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Takenouchi, T.; Kosaki, R.; Niizuma, T.; Hata, K.; Kosaki, K. Macrothrombocytopenia and developmental delay with a de novo CDC42 mutation: Yet another locus for thrombocytopenia and developmental delay. Am. J. Med. Genet. A 2015, 167A, 2822–2825. [Google Scholar] [CrossRef] [PubMed]
- Lanzi, G.; Moratto, D.; Vairo, D.; Masneri, S.; Delmonte, O.; Paganini, T.; Parolini, S.; Tabellini, G.; Mazza, C.; Savoldi, G.; et al. A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. J. Exp. Med. 2012, 209, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, K.R.; McGhee, S.; Winkler, S.; Sassi, A.; Woellner, C.; Lopez-Herrera, G.; Chen, A.; Kim, H.S.; Lloret, M.G.; Schulze, I.; et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J. Allergy Clin. Immunol. 2009, 124, 1289–1302.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Davis, J.C.; Lamborn, I.T.; Freeman, A.F.; Jing, H.; Favreau, A.J.; Matthews, H.F.; Davis, J.; Turner, M.L.; Uzel, G.; et al. Combined immunodeficiency associated with DOCK8 mutations. N. Engl. J. Med. 2009, 361, 2046–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbs, K.; Conde, C.D.; Zhang, S.-Y.; Parolini, S.; Audry, M.; Chou, J.; Haapaniemi, E.; Keles, S.; Bilic, I.; Okada, S.; et al. Inherited DOCK2 Deficiency in Patients with Early-Onset Invasive Infections. N. Engl. J. Med. 2015, 372, 2409–2422. [Google Scholar] [CrossRef]
- A Aldrich, R.; Steinberg, A.G.; Campbell, D.C. Pedigree demonstrating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis and bloody diarrhea. Pediatrics 1954, 13, 133–139. [Google Scholar] [CrossRef]
- Derry, J.M.; Ochs, H.D.; Francke, U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell 1994, 78, 635–644. [Google Scholar] [CrossRef]
- Linder, S.; Nelson, D.; Weiss, M.; Aepfelbacher, M. Wiskott-Aldrich syndrome protein regulates podosomes in primary human macrophages. Proc. Natl. Acad. Sci. USA 1999, 96, 9648–9653. [Google Scholar] [CrossRef] [Green Version]
- Piperno, G.M.; Naseem, A.; Silvestrelli, G.; Amadio, R.; Caronni, N.; Cervantes-Luevano, K.E.; Liv, N.; Klumperman, J.; Colliva, A.; Ali, H.; et al. Wiskott-Aldrich syndrome protein restricts cGAS/STING activation by dsDNA immune complexes. JCI Insight 2020, 5, e132857. [Google Scholar] [CrossRef]
- Prete, F.; Catucci, M.; Labrada, M.; Gobessi, S.; Castiello, M.C.; Bonomi, E.; Aiuti, A.; Vermi, W.; Cancrini, C.; Metin, A.; et al. Wiskott-Aldrich syndrome protein–mediated actin dynamics control type-I interferon production in plasmacytoid dendritic cells. J. Exp. Med. 2013, 210, 355–374. [Google Scholar] [CrossRef]
- Cook, S.A.; Comrie, W.A.; Poli, M.C.; Similuk, M.; Oler, A.J.; Faruqi, A.J.; Kuhns, D.B.; Yang, S.; Vargas-Hernández, A.; Carisey, A.F.; et al. HEM1 deficiency disrupts mTORC2 and F-actin control in inherited immunodysregulatory disease. Science 2020, 369, 202–207. [Google Scholar] [CrossRef]
- Kahr, W.H.; Pluthero, F.G.; Elkadri, A.; Warner, N.; Drobac, M.; Chen, C.H.; Lo, R.W.; Li, L.; Li, R.; Li, Q.; et al. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat. Commun. 2017, 8, 14816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musial, A.; Eissa, N.T. Inducible nitric-oxide synthase is regulated by the proteasome degradation pathway. J. Biol. Chem. 2001, 276, 24268–24273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, N.; Vogel, V. Spatial confinement downsizes the inflammatory response of macrophages. Nat. Mater. 2018, 17, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Iellem, A.; Mariani, M.; Lang, R.; Recalde, H.; Panina-Bordignon, P.; Sinigaglia, F.; D’Ambrosio, D. Unique chemotactic response profile and specific expression of chemokine receptors Ccr4 and Ccr8 by Cd4+Cd25+ regulatory T cells. J. Exp. Med. 2001, 194, 847–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Rapp, M.; Wintergerst, M.W.; Kunz, W.G.; Vetter, V.K.; Knott, M.M.; Lisowski, D.; Haubner, S.; Moder, S.; Thaler, R.; Eiber, S.; et al. CCL22 controls immunity by promoting regulatory T cell communication with dendritic cells in lymph nodes. J. Exp. Med. 2019, 216, 1170–1181. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; McKeon, D.; Sao, K.; Yang, C.; Naranjo, N.M.; Svitkina, T.M.; Petrie, R.J. Myosin II and Arp2/3 cross-talk governs intracellular hydraulic pressure and lamellipodia formation. Mol. Biol. Cell 2021, 32, 579–589. [Google Scholar] [CrossRef]
- Leeuwen, F.N.; Kain, H.E.; Kammen, R.A.; Michiels, F.; Kranenburg, O.W.; Collard, J.G. The guanine nucleotide exchange factor Tiam1 affects neuronal morphology; opposing roles for the small GTPases Rac and Rho. J. Cell Biol. 1997, 139, 797–807. [Google Scholar] [CrossRef] [Green Version]
- Nimnual, A.S.; Taylor, L.; Bar-Sagi, D. Redox-dependent downregulation of Rho by Rac. Nat. Cell Biol. 2003, 5, 236–241. [Google Scholar] [CrossRef]
- Sander, E.E.; ten Klooster, J.P.; van Delft, S.; van der Kammen, R.A.; Collard, J.G. Rac downregulates Rho activity: Reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 1999, 147, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Lomakin, A.J.; Lee, K.-C.; Han, S.J.; Bui, D.A.; Davidson, M.; Mogilner, A.; Danuser, G. Competition for actin between two distinct F-actin networks defines a bistable switch for cell polarization. Nat. Cell Biol. 2015, 17, 1435–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, P.; Maiuri, P.; Bretou, M.; Sáez, P.J.; Pierobon, P.; Maurin, M.; Chabaud, M.; Lankar, D.; Obino, D.; Terriac, E.; et al. Innate control of actin nucleation determines two distinct migration behaviours in dendritic cells. Nat. Cell Biol. 2015, 18, 43–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wales, P.; E Schuberth, C.; Aufschnaiter, R.; Fels, J.; García-Aguilar, I.; Janning, A.; Dlugos, C.P.; Schäfer-Herte, M.; Klingner, C.; Wälte, M.; et al. Calcium-mediated actin reset (CaAR) mediates acute cell adaptations. eLife 2016, 5, e19850. [Google Scholar] [CrossRef]
- Chauhan, A.; Sun, Y.; Sukumaran, P.; Zangbede, F.O.Q.; Jondle, C.N.; Sharma, A.; Evans, D.L.; Chauhan, P.; Szlabick, R.E.; Aaland, M.O.; et al. M1 Macrophage Polarization Is Dependent on TRPC1-Mediated Calcium Entry. iScience 2018, 8, 85–102. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.Y.; Veckman, V.; Limmer, K.; David, M. Phospholipase Cgamma-2 and intracellular calcium are required for lipopolysaccharide-induced Toll-like receptor 4 (TLR4) endocytosis and interferon regulatory factor 3 (IRF3) activation. J. Biol. Chem. 2012, 287, 3704–3709. [Google Scholar] [CrossRef] [Green Version]
- Atcha, H.; Jairaman, A.; Holt, J.R.; Meli, V.S.; Nagalla, R.R.; Veerasubramanian, P.K.; Brumm, K.T.; Lim, H.E.; Othy, S.; Cahalan, M.D.; et al. Mechanically activated ion channel Piezo1 modulates macrophage polarization and stiffness sensing. Nat. Commun. 2021, 12, 3256. [Google Scholar] [CrossRef]
- McWhorter, F.Y.; Wang, T.; Nguyen, P.; Chung, T.; Liu, W.F. Modulation of macrophage phenotype by cell shape. Proc. Natl. Acad. Sci. USA 2013, 110, 17253–17258. [Google Scholar] [CrossRef] [Green Version]
- Pugin, J.; Dunn, I.; Jolliet, P.; Tassaux, D.; Magnenat, J.-L.; Nicod, L.P.; Chevrolet, J.-C. Activation of human macrophages by mechanical ventilation in vitro. Am. J. Physiol. 1998, 275, L1040–L1050. [Google Scholar] [CrossRef]
- Wehner, S.; Buchholz, B.M.; Schuchtrup, S.; Rocke, A.; Schaefer, N.; Lysson, M.; Hirner, A.; Kalff, J.C. Mechanical strain and TLR4 synergistically induce cell-specific inflammatory gene expression in intestinal smooth muscle cells and peritoneal macrophages. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G1187–G1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamori, Y.; Emoto, M.; Fukuda, N.; Taguchi, A.; Okuya, S.; Tajiri, M.; Miyagishi, M.; Taira, K.; Wada, Y.; Tanizawa, Y. Myosin motor Myo1c and its receptor NEMO/IKK-gamma promote TNF-alpha-induced serine307 phosphorylation of IRS-1. J. Cell Biol. 2006, 173, 665–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, J.L.; Harvey, M.W.; Holden, D.W.; Evans, T.J. Macrophage nitric oxide synthase associates with cortical actin but is not recruited to phagosomes. Infect. Immun. 2001, 69, 6391–6400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuai, K.; Schindler, C.; Prezioso, V.R.; Darnell, J.E., Jr. Activation of transcription by IFN-gamma: Tyrosine phosphorylation of a 91-kD DNA binding protein. Science 1992, 258, 1808–1812. [Google Scholar] [CrossRef]
- Xie, Q.W.; Whisnant, R.; Nathan, C. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J. Exp. Med. 1993, 177, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, U.; Kuroda, E. Regulation of macrophage-derived chemokine (MDC/CCL22) production. Crit. Rev. Immunol. 2002, 22, 105–114. [Google Scholar] [CrossRef]
- Steimle, V.; Siegrist, C.A.; Mottet, A.; Lisowska-Grospierre, B.; Mach, B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science 1994, 265, 106–109. [Google Scholar] [CrossRef]
- Tyryshkin, A.; Gorgun, F.; Fattah, E.A.; Mazumdar, T.; Pandit, L.; Zeng, S.; Eissa, N. Src kinase-mediated phosphorylation stabilizes inducible nitric-oxide synthase in normal cells and cancer cells. J. Biol. Chem. 2010, 285, 784–792. [Google Scholar] [CrossRef] [Green Version]
- Bergeron, M.; Olivier, M. Trypanosoma cruzi-mediated IFN-gamma-inducible nitric oxide output in macrophages is regulated by iNOS mRNA stability. J. Immunol. 2006, 177, 6271–6280. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Brovkovych, V.; Brovkovych, S.; Tan, F.; Lee, B.-S.; Sharma, T.; Skidgel, R.A. Dynamic receptor-dependent activation of inducible nitric-oxide synthase by ERK-mediated phosphorylation of Ser745. J. Biol. Chem. 2007, 282, 32453–32461. [Google Scholar] [CrossRef] [Green Version]
- Quang, B.A.T.; Peters, R.; Cassani, D.A.D.; Chugh, P.; Clark, A.G.; Agnew, M.; Charras, G.; Paluch, E.K. Extent of myosin penetration within the actin cortex regulates cell surface mechanics. Nat. Commun. 2021, 12, 6511. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, F.; Reis-Rodrigues, P.; de Vries, I.; Hons, M.; Aguilera, J.; Riedl, M.; Leithner, A.; Tasciyan, S.; Kopf, A.; Merrin, J.; et al. WASp triggers mechanosensitive actin patches to facilitate immune cell migration in dense tissues. Dev. Cell 2021, 57, 47–62.e9. [Google Scholar] [CrossRef]
- Leithner, A.; Eichner, A.; Müller, J.; Reversat, A.; Brown, M.; Schwarz, J.; Merrin, J.; De Gorter, D.J.J.; Schur, F.; Bayerl, J.; et al. Diversified actin protrusions promote environmental exploration but are dispensable for locomotion of leukocytes. Nat. Cell Biol. 2016, 18, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Stahnke, S.; Döring, H.; Kusch, C.; de Gorter, D.J.; Dütting, S.; Guledani, A.; Pleines, I.; Schnoor, M.; Sixt, M.; Geffers, R.; et al. Loss of Hem1 disrupts macrophage function and impacts migration, phagocytosis, and integrin-mediated adhesion. Curr. Biol. 2021, 31, 2051–2064.e8. [Google Scholar] [CrossRef] [PubMed]
- Badour, K.; McGavin, M.K.H.; Zhang, J.; Freeman, S.; Vieira, C.; Filipp, D.; Julius, M.; Mills, G.B.; Siminovitch, K.A. Interaction of the Wiskott–Aldrich syndrome protein with sorting nexin 9 is required for CD28 endocytosis and cosignaling in T cells. Proc. Natl. Acad. Sci. USA 2007, 104, 1593–1598. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, R.M.; Kockelkoren, G.; Raghavan, P.; Bell, G.R.; Britain, D.; Puri, N.; Collins, S.R.; Leonetti, M.D.; Stamou, D.; Weiner, O.D. WASP integrates substrate topology and cell polarity to guide neutrophil migration. J. Cell Biol. 2021, 221, e202104046. [Google Scholar] [CrossRef]
- Fritz-Laylin, L.K.; Lord, S.J.; Mullins, R.D. WASP and SCAR are evolutionarily conserved in actin-filled pseudopod-based motility. J. Cell Biol. 2017, 216, 1673–1688. [Google Scholar] [CrossRef] [Green Version]
- Salzer, E.; Zoghi, S.; Kiss, M.G.; Kage, F.; Rashkova, C.; Stahnke, S.; Haimel, M.; Platzer, R.; Caldera, M.; Ardy, R.C.; et al. The cytoskeletal regulator HEM1 governs B cell development and prevents autoimmunity. Sci. Immunol. 2020, 5, eabc3979. [Google Scholar] [CrossRef]
- Pulecio, J.; Tagliani, E.; Scholer, A.; Prete, F.; Fetler, L.; Burrone, O.R.; Benvenuti, F. Expression of Wiskott-Aldrich syndrome protein in dendritic cells regulates synapse formation and activation of naive CD8+ T cells. J. Immunol. 2008, 181, 1135–1142. [Google Scholar] [CrossRef] [Green Version]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric oxide and the regulation of gene expression. Trends Cell Biol. 2001, 11, 66–75. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Weber, S.E.; Harbertson, J.; Godebu, E.; Mros, G.A.; Padrick, R.C.; Carson, B.D.; Ziegler, S.F.; Bradley, L.M. Adaptive islet-specific regulatory CD4 T cells control autoimmune diabetes and mediate the disappearance of pathogenic Th1 cells in vivo. J. Immunol. 2006, 176, 4730–4739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proto, J.D.; Doran, A.C.; Gusarova, G.; Yurdagul, A.; Sozen, E.; Subramanian, M.; Islam, M.N.; Rymond, C.C.; Du, J.; Hook, J.; et al. Regulatory T Cells Promote Macrophage Efferocytosis during Inflammation Resolution. Immunity 2018, 49, 666–677.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okeke, E.B.; Okwor, I.; Uzonna, J.E. Regulatory T cells restrain CD4+ T cells from causing unregulated immune activation and hypersensitivity to lipopolysaccharide challenge. J. Immunol. 2014, 193, 655–662. [Google Scholar] [CrossRef] [Green Version]
- Tur-Gracia, S.; Martinez-Quiles, N. Emerging functions of cytoskeletal proteins in immune diseases. J. Cell Sci. 2021, 134, jcs253534. [Google Scholar] [CrossRef]
- Thrasher, A.; Burns, S. WASP: A key immunological multitasker. Nat. Rev. Immunol. 2010, 10, 182–192. [Google Scholar] [CrossRef]
- Kim, J.H.; Wang, A.; Conti, M.A.; Adelstein, R. Nonmuscle myosin II is required for internalization of the epidermal growth factor receptor and modulation of downstream signaling. J. Biol. Chem. 2012, 287, 27345–27358. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ronzier, E.; Laurenson, A.J.; Manickam, R.; Liu, S.; Saintilma, I.M.; Schrock, D.C.; Hammer, J.A.; Rotty, J.D. The Actin Cytoskeleton Responds to Inflammatory Cues and Alters Macrophage Activation. Cells 2022, 11, 1806. https://doi.org/10.3390/cells11111806
Ronzier E, Laurenson AJ, Manickam R, Liu S, Saintilma IM, Schrock DC, Hammer JA, Rotty JD. The Actin Cytoskeleton Responds to Inflammatory Cues and Alters Macrophage Activation. Cells. 2022; 11(11):1806. https://doi.org/10.3390/cells11111806
Chicago/Turabian StyleRonzier, Elsa, Alexander J. Laurenson, Rohini Manickam, Sophia Liu, Imelda M. Saintilma, Dillon C. Schrock, John A. Hammer, and Jeremy D. Rotty. 2022. "The Actin Cytoskeleton Responds to Inflammatory Cues and Alters Macrophage Activation" Cells 11, no. 11: 1806. https://doi.org/10.3390/cells11111806
APA StyleRonzier, E., Laurenson, A. J., Manickam, R., Liu, S., Saintilma, I. M., Schrock, D. C., Hammer, J. A., & Rotty, J. D. (2022). The Actin Cytoskeleton Responds to Inflammatory Cues and Alters Macrophage Activation. Cells, 11(11), 1806. https://doi.org/10.3390/cells11111806