
Claudin-17 Deficiency in Mice Results in Kidney Injury Due to Electrolyte Imbalance and Oxidative Stress

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
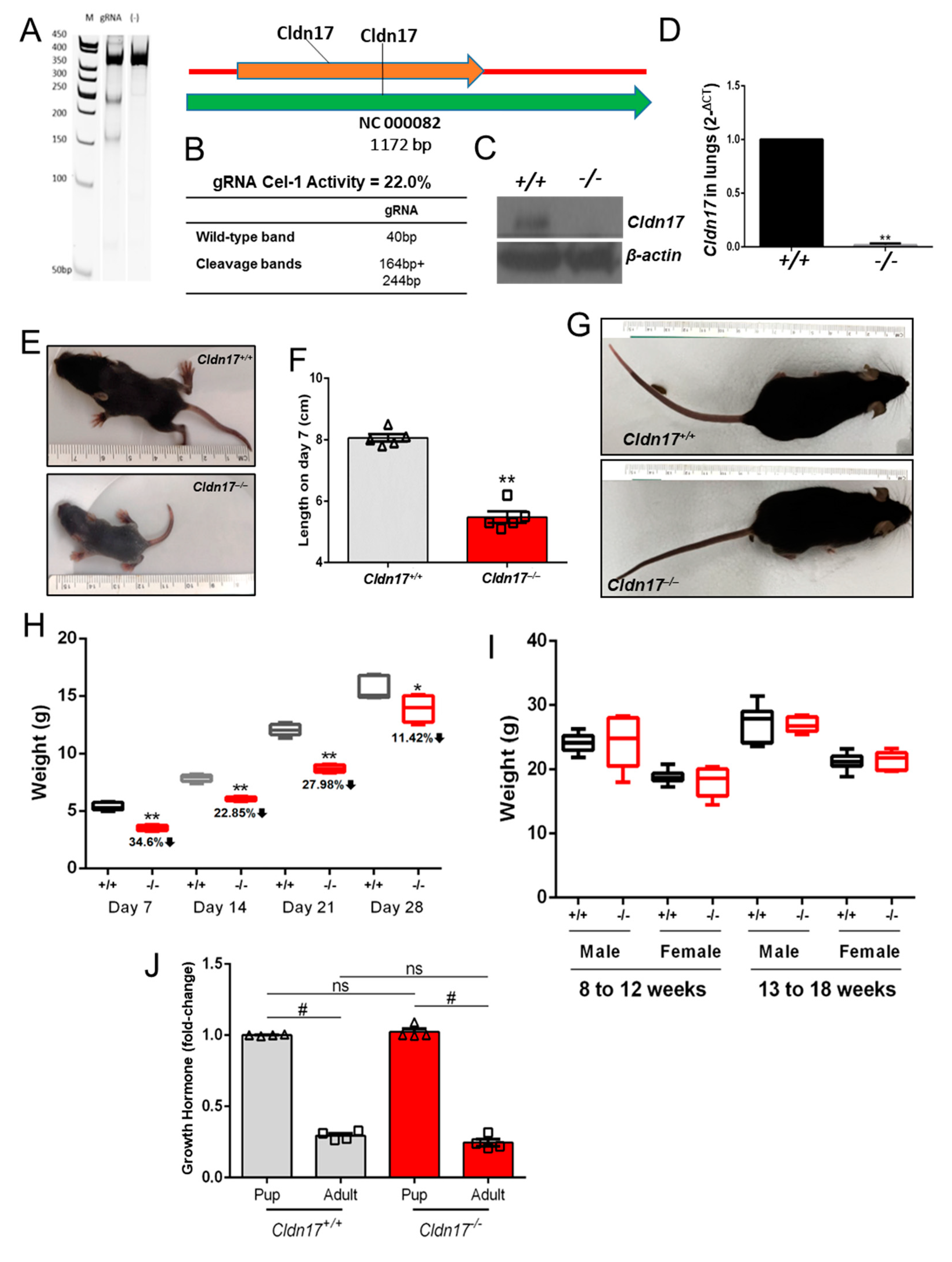
2.1. Generation and Maintenance of Cldn17−/− Mice
2.2. RNA Isolation, cDNA Preparation, and qRT-PCR
2.3. Western Blot Analysis
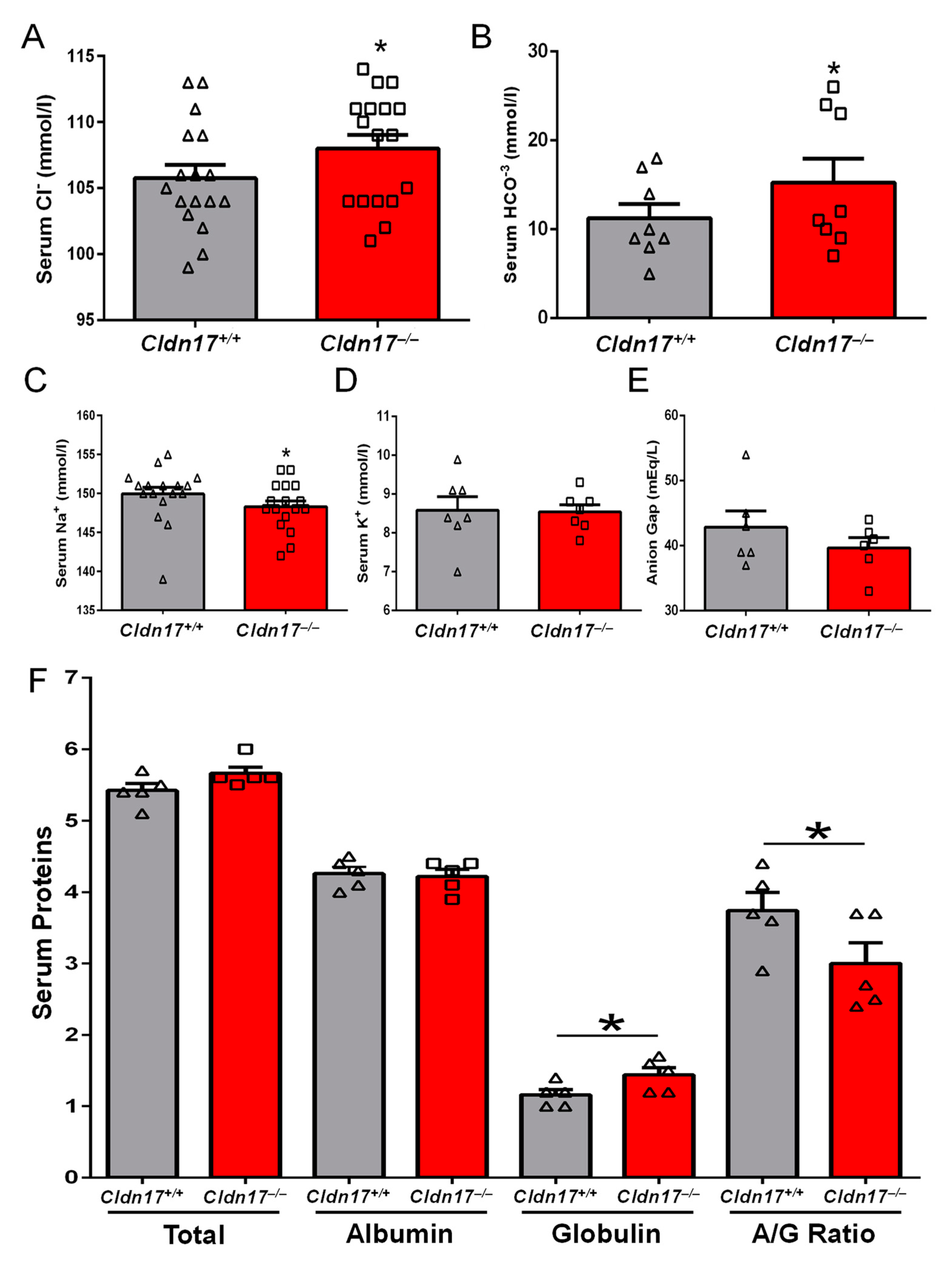
2.4. Analysis of Serum Electrolytes and Urine Parameters
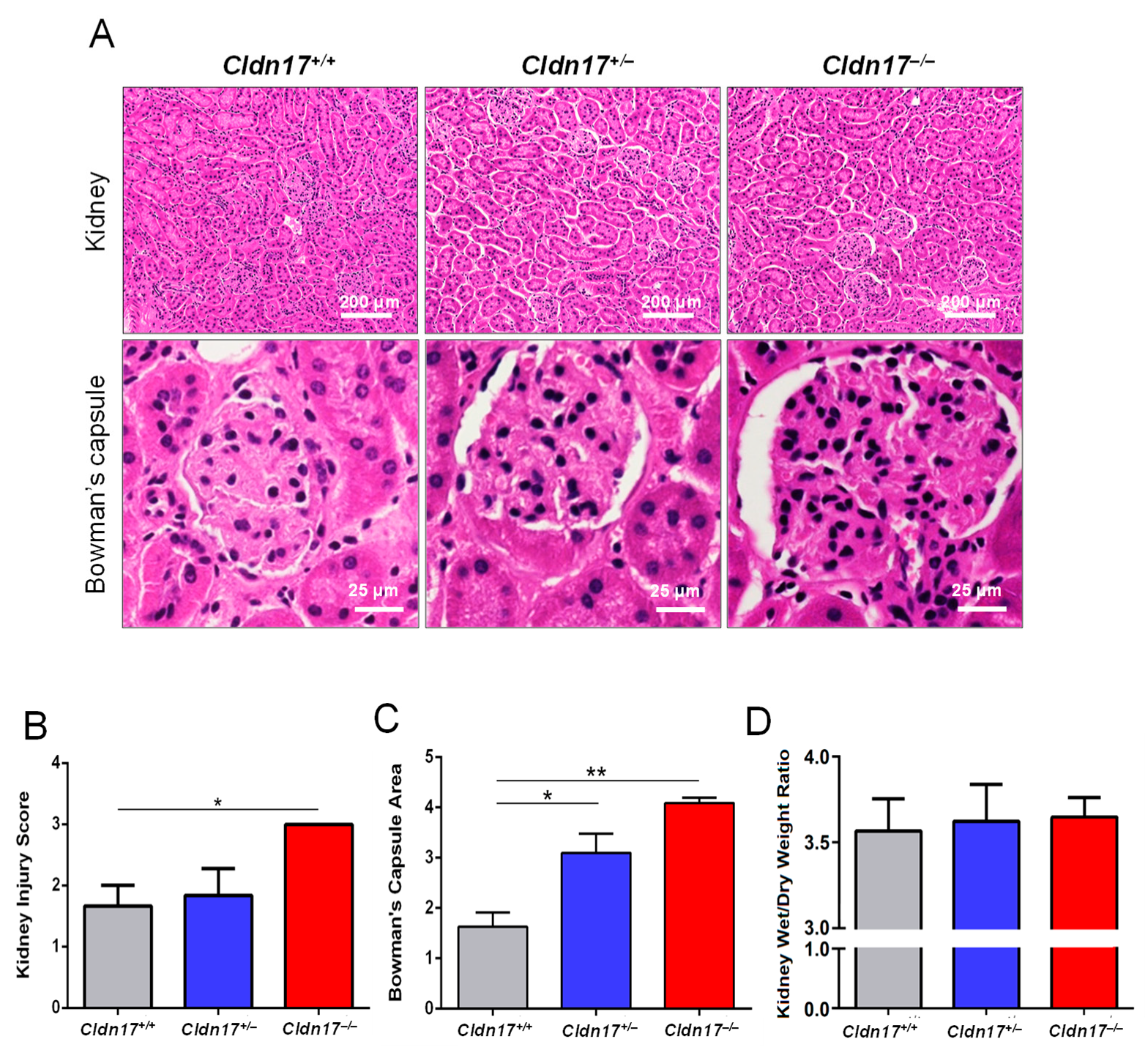
2.5. Kidney Injury and Wet/Dry Ratio Analysis
2.6. Genomics Data Co-Expression Analyses
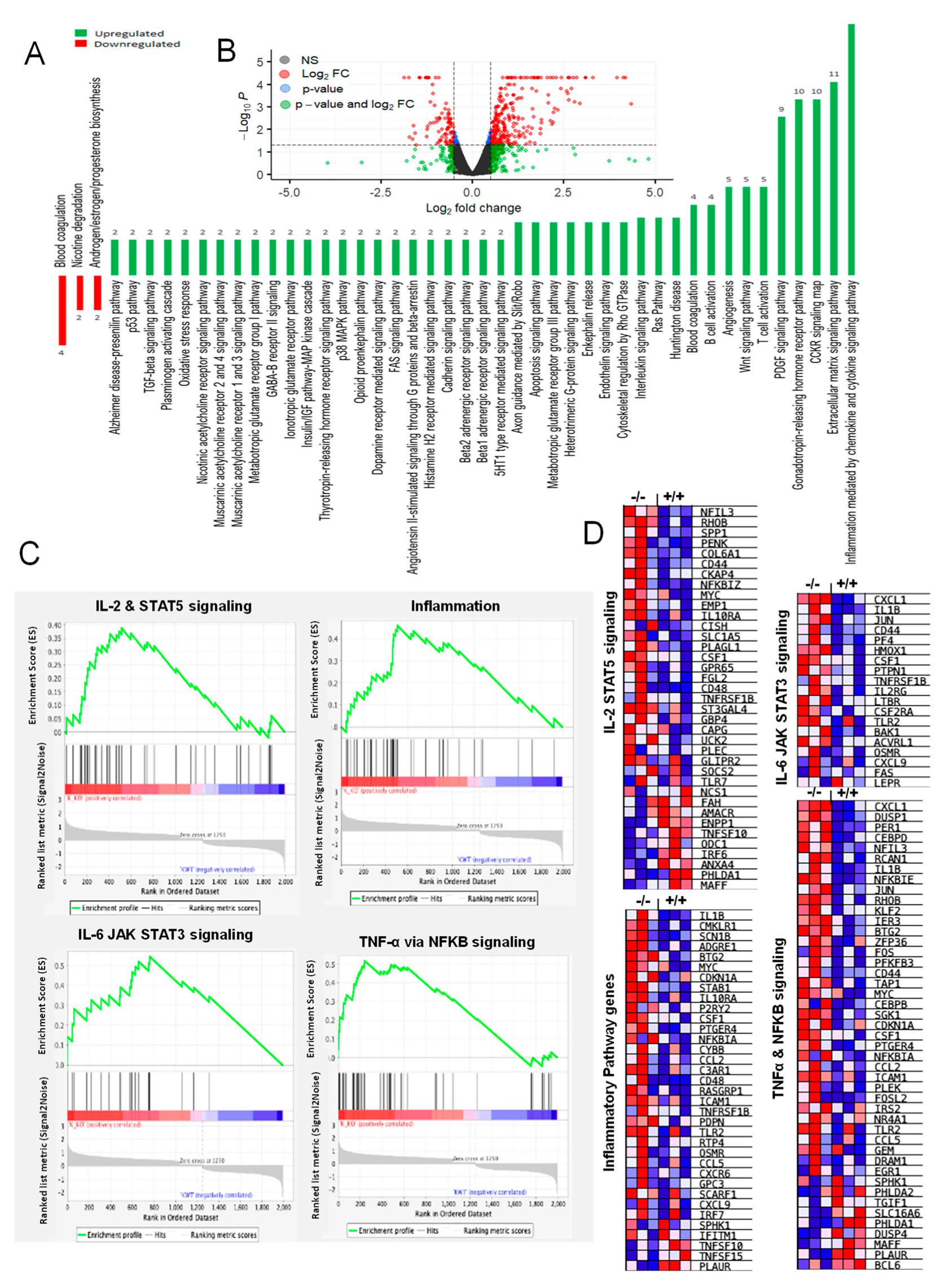
2.7. RNA-Seq and Bioinformatics Analysis
2.8. Ingenuity Pathway Analysis (IPA)
2.9. ROS Analysis
2.10. Analysis of Growth Hormone Expression
2.11. Statistical Analysis
3. Results
3.1. Cldn17−/− Mice Exhibit Delayed Growth
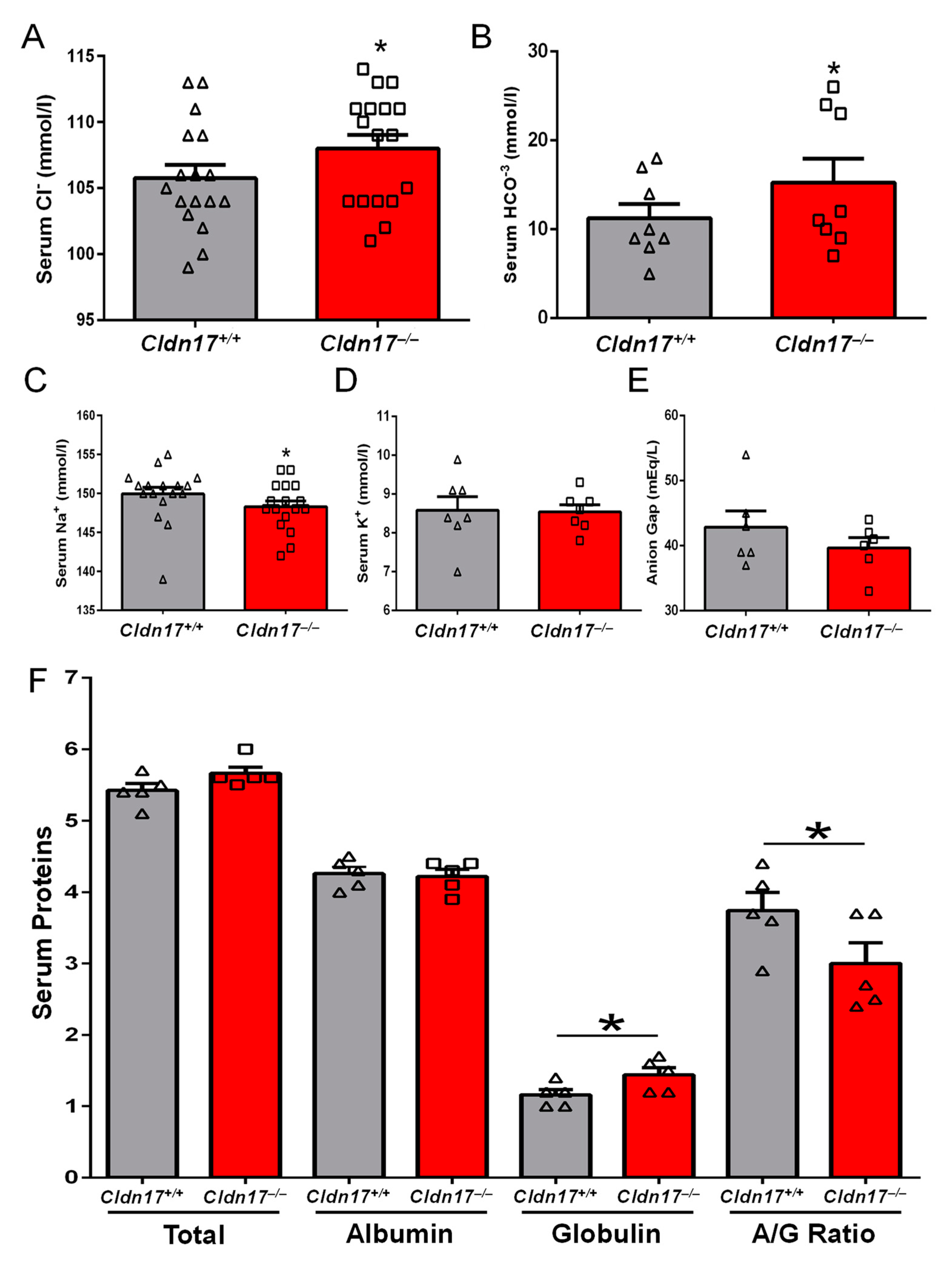
3.2. Impact of Cldn17 Loss on Blood and Urine Parameters
3.3. Cldn17−/− Mice Exhibit Hypophagia and Polydipsia
3.4. Cldn17−/− Mice Display Kidney Injury
3.5. Cldn17 Loss Affects the Expression of other Cldns in Mouse Kidneys
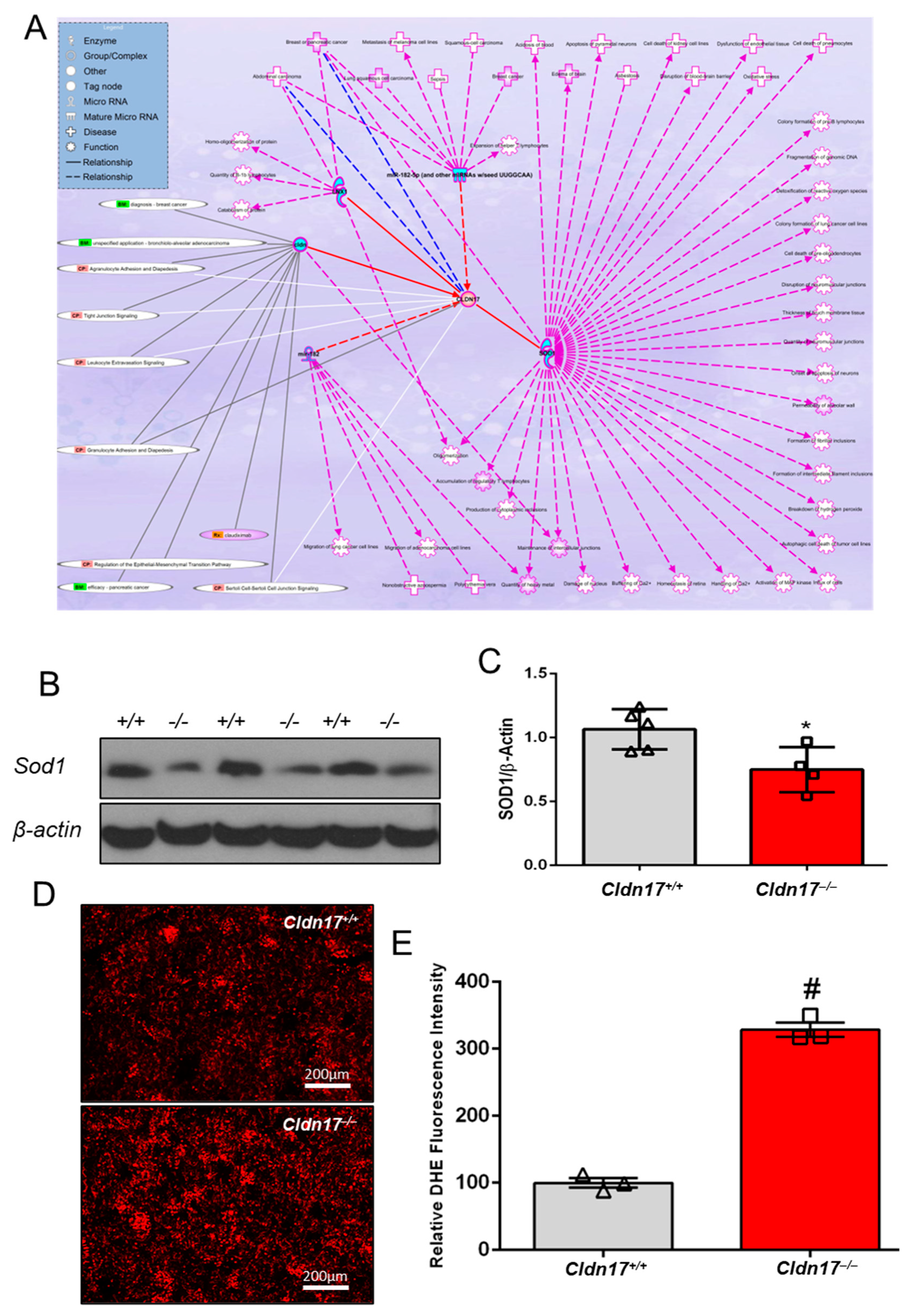
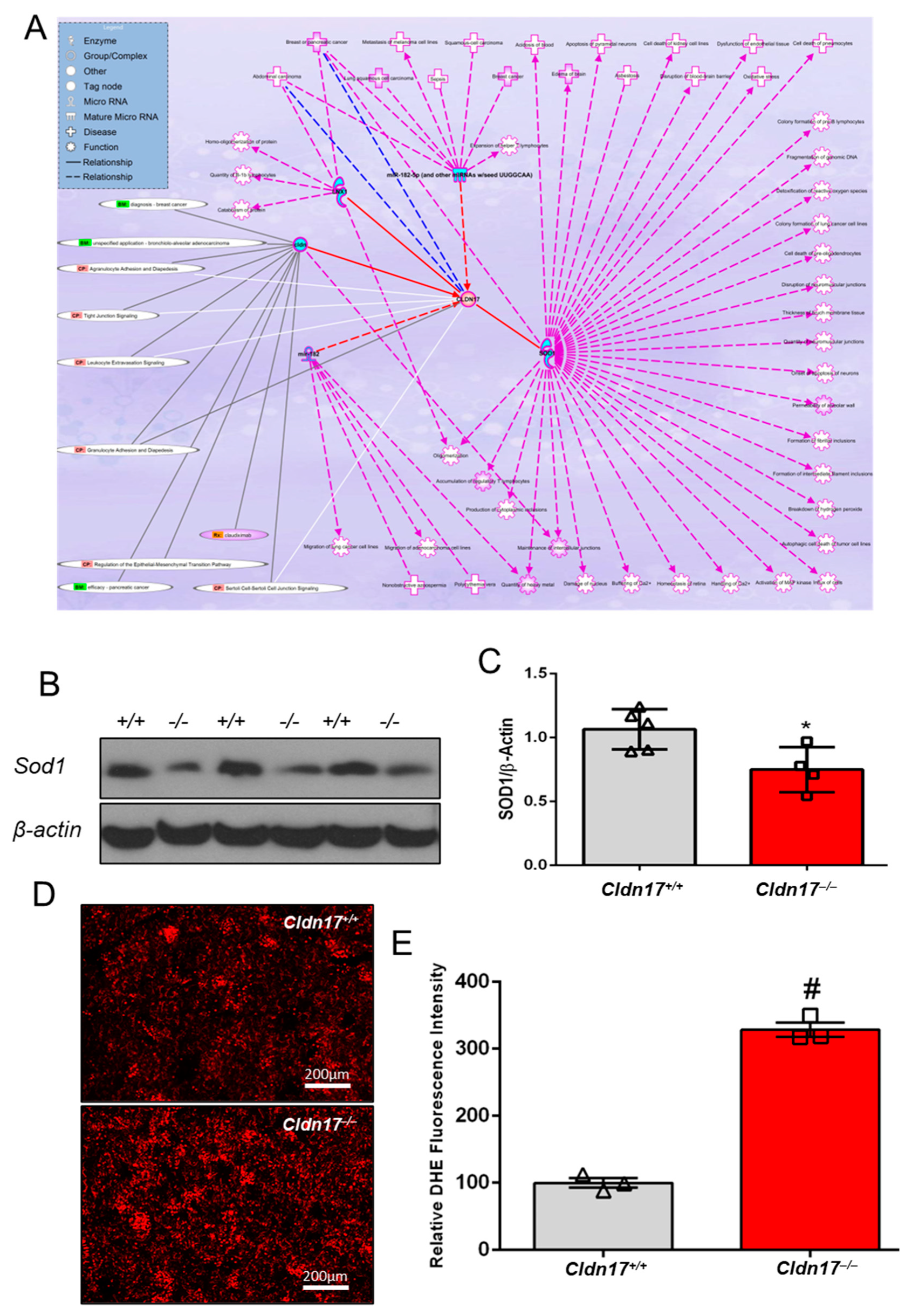
3.6. CLDN17 Co-Expression with Other Genes in Physiology and Pathology
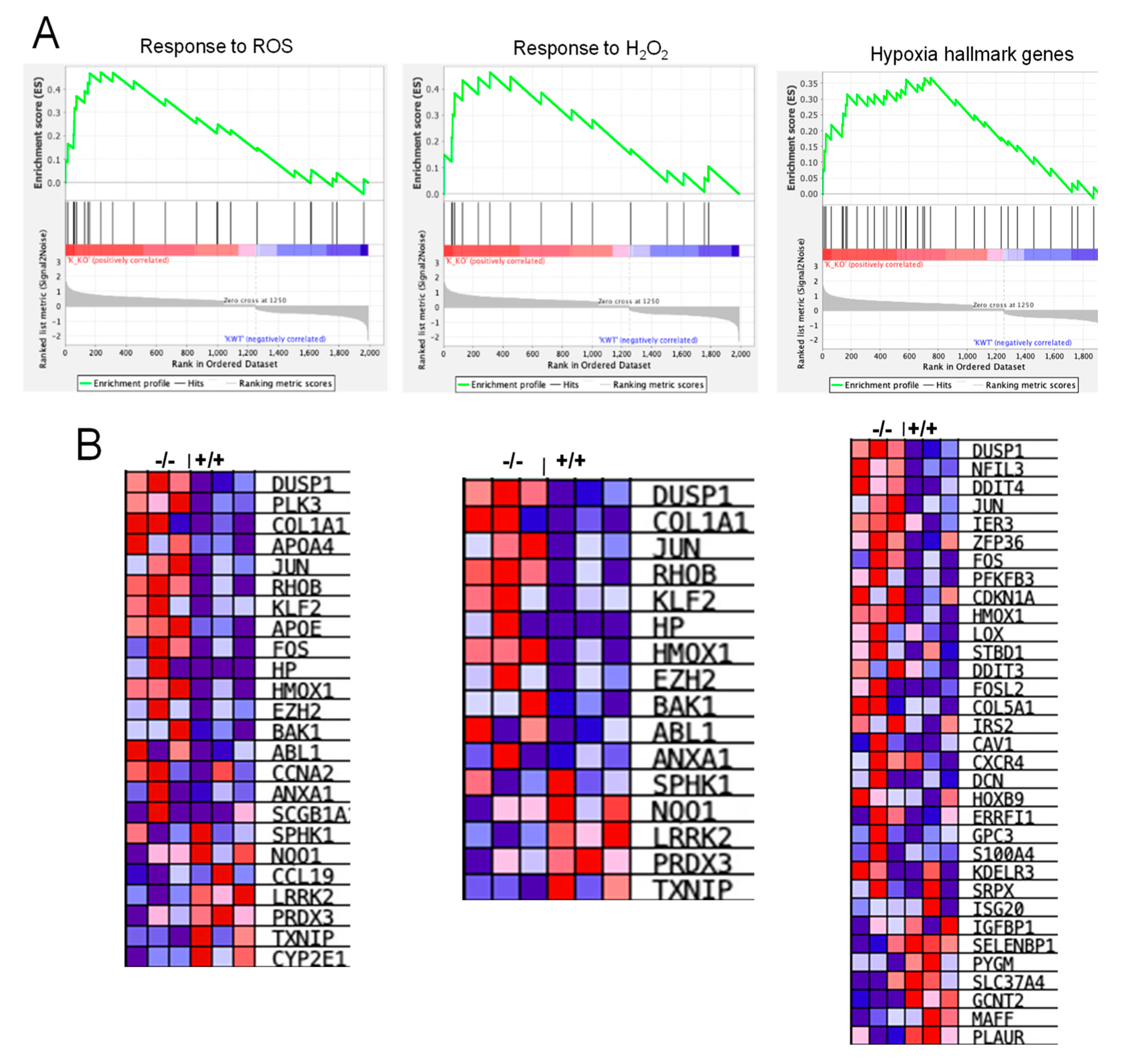
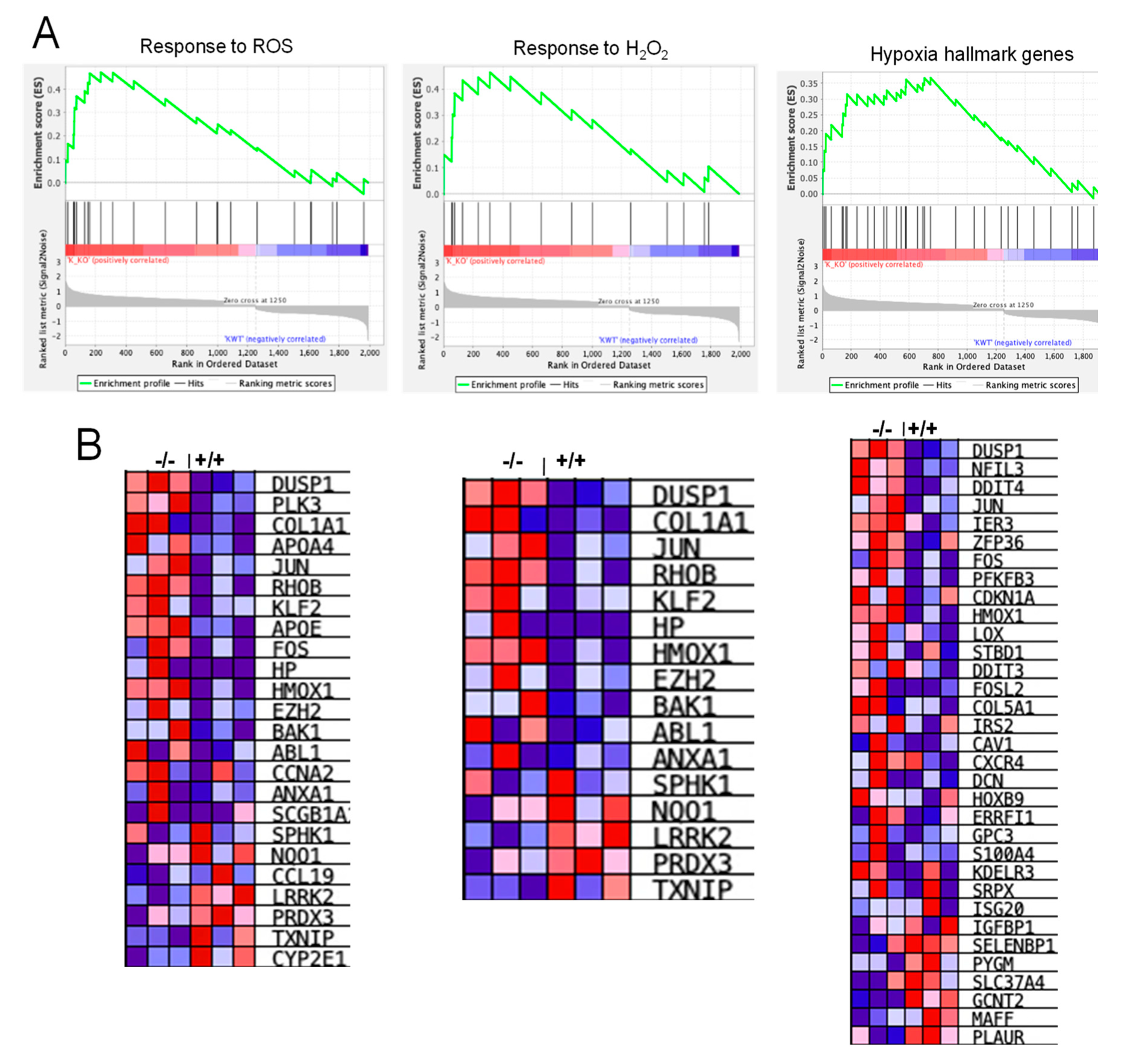
3.7. RNA-Seq Analysis Revealed Alterations in Genes Involved in Inflammation, Hypoxia, and ROS Generation
3.8. Cldn17 Loss Did Not Affect the Expression of Vasopressin and Angiotensin Pathway Genes in Mice
3.9. Cldn17 Deficiency Promotes ROS Generation in the Kidneys
4. Discussion
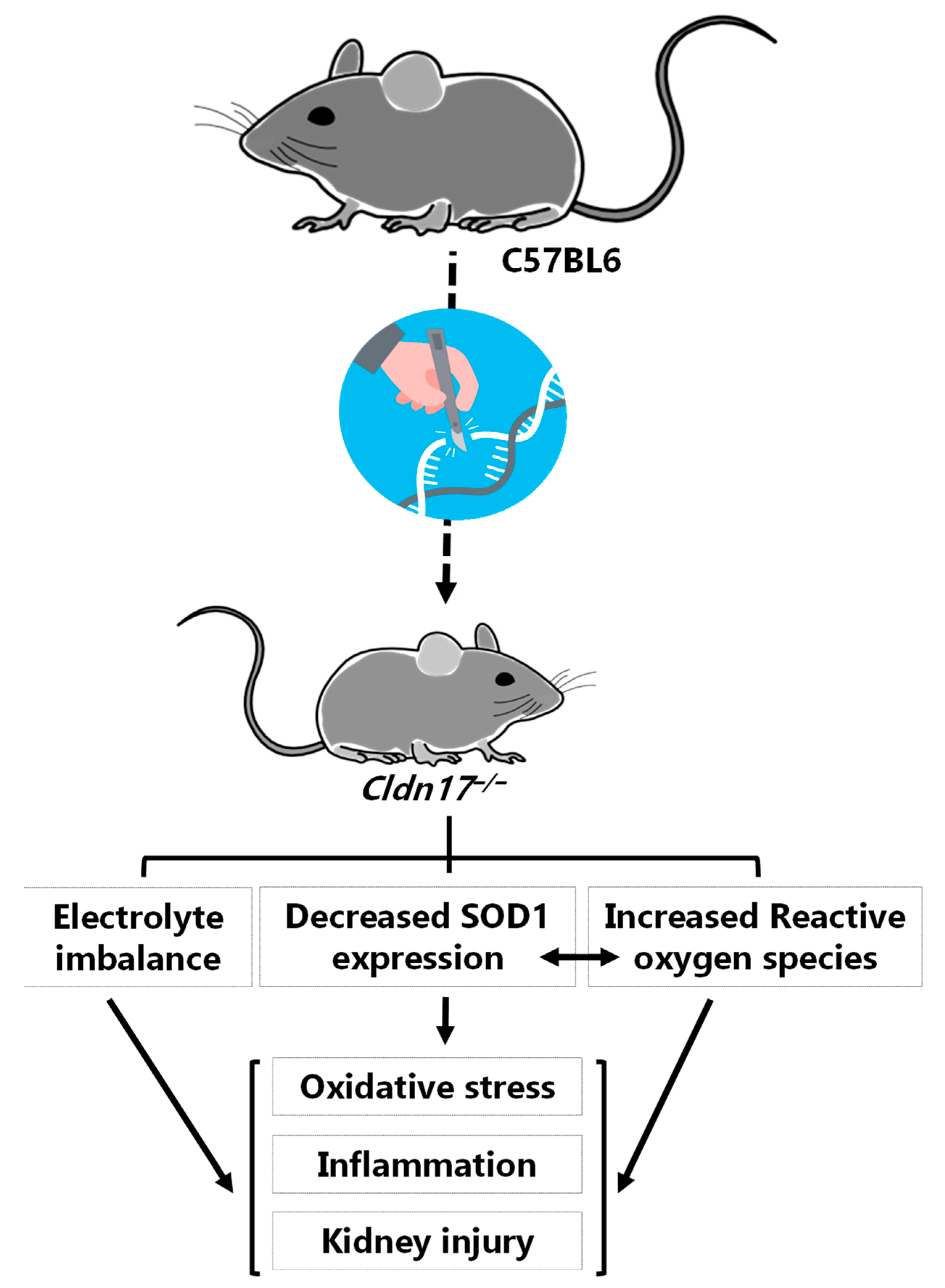
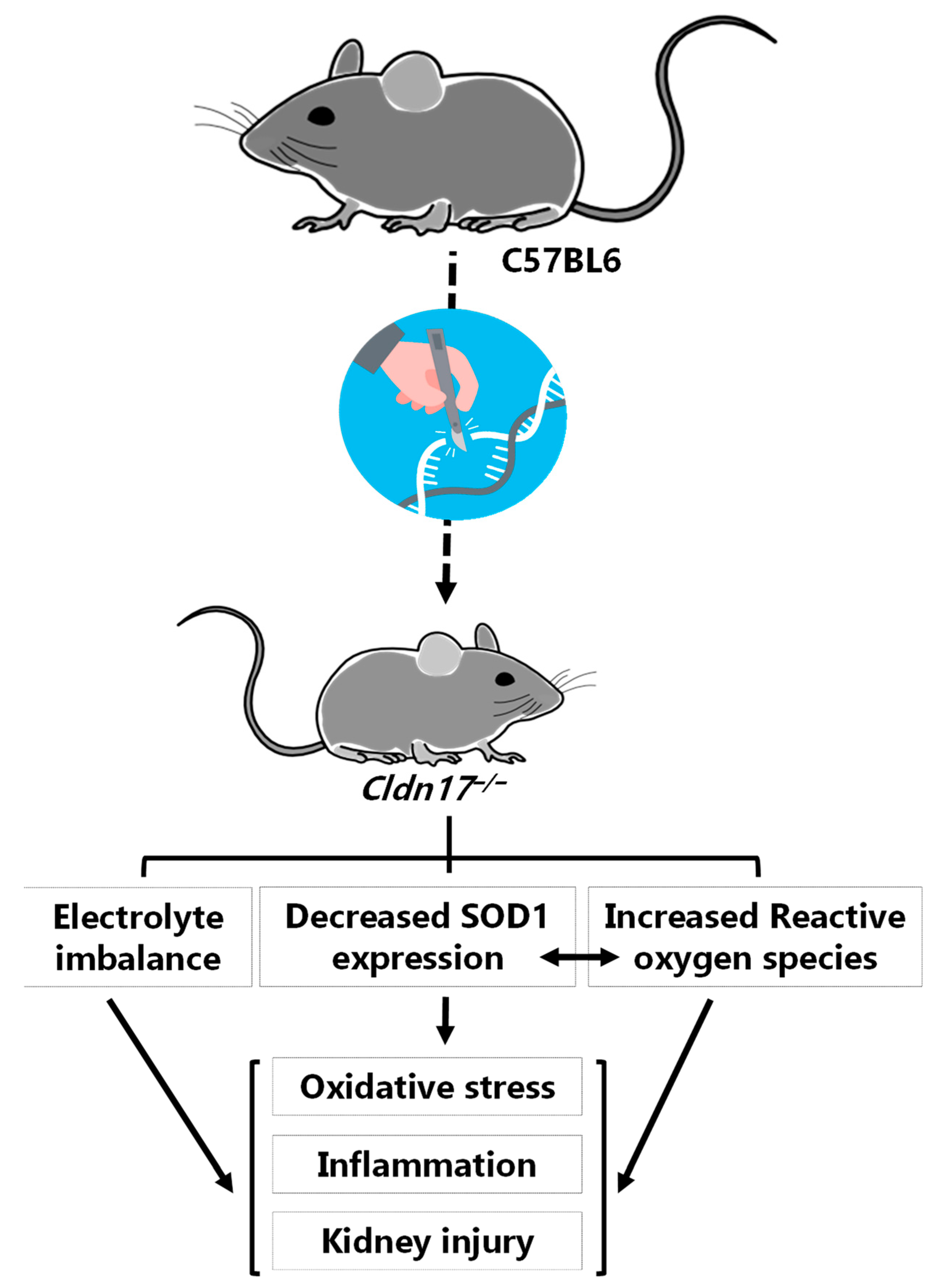
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Günzel, D. Claudins: Vital partners in transcellular and paracellular transport coupling. Pflug. Arch. 2017, 469, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Elkouby-Naor, L.; Ben-Yosef, T. Functions of claudin tight junction proteins and their complex interactions in various physiological systems. Int. Rev. Cell Mol. Biol 2010, 279, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Shekarabi, M.; Zhang, J.; Khanna, A.R.; Ellison, D.H.; Delpire, E.; Kahle, K.T. WNK Kinase Signaling in Ion Homeostasis and Human Disease. Cell Metab 2017, 25, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawedia, J.D.; Nieman, M.L.; Boivin, G.P.; Melvin, J.E.; Kikuchi, K.; Hand, A.R.; Lorenz, J.N.; Menon, A.G. Interaction between transcellular and paracellular water transport pathways through Aquaporin 5 and the tight junction complex. Proc. Natl. Acad. Sci. USA 2007, 104, 3621–3626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park-Windhol, C.; D’Amore, P.A. Disorders of Vascular Permeability. Annu. Rev. Pathol. 2016, 11, 251–281. [Google Scholar] [CrossRef] [PubMed]
- Wen, Q.; Tang, E.I.; Gao, Y.; Jesus, T.T.; Chu, D.S.; Lee, W.M.; Wong, C.K.C.; Liu, Y.X.; Xiao, X.; Silvestrini, B.; et al. Signaling pathways regulating blood-tissue barriers-Lesson from the testis. Biochim. Biophys Acta Biomembr. 2018, 1860, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.A. Claudins and alveolar epithelial barrier function in the lung. Ann. N. Y. Acad. Sci. 2012, 1257, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Bhattad, P.B.; Jain, V. Renal Artery Stenosis As Etiology of Recurrent Flash Pulmonary Edema and Role of Imaging in Timely Diagnosis and Management. Cureus 2020, 12, e7609. [Google Scholar] [CrossRef] [Green Version]
- Owen-Woods, C.; Joulia, R.; Barkaway, A.; Rolas, L.; Ma, B.; Nottebaum, A.F.; Arkill, K.P.; Stein, M.; Girbl, T.; Golding, M.; et al. Local microvascular leakage promotes trafficking of activated neutrophils to remote organs. J. Clin. Investig. 2020, 130, 2301–2318. [Google Scholar] [CrossRef]
- Lee, K.S.; Kim, S.R.; Park, S.J.; Park, H.S.; Min, K.H.; Lee, M.H.; Jin, S.M.; Jin, G.Y.; Yoo, W.H.; Lee, Y.C. Hydrogen peroxide induces vascular permeability via regulation of vascular endothelial growth factor. Am. J. Respir Cell Mol. Biol. 2006, 35, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharm. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ikari, A.; Atomi, K.; Yamazaki, Y.; Sakai, H.; Hayashi, H.; Yamaguchi, M.; Sugatani, J. Hyperosmolarity-induced up-regulation of claudin-4 mediated by NADPH oxidase-dependent H2O2 production and Sp1/c-Jun cooperation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 2617–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddei, A.; Giampietro, C.; Conti, A.; Orsenigo, F.; Breviario, F.; Pirazzoli, V.; Potente, M.; Daly, C.; Dimmeler, S.; Dejana, E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 2008, 10, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Artham, S.; Sabbineni, H.; Al-Azayzih, A.; Peng, X.D.; Hay, N.; Adams, R.H.; Byzova, T.V.; Somanath, P.R. Akt1 promotes stimuli-induced endothelial-barrier protection through FoxO-mediated tight-junction protein turnover. Cell Mol. Life Sci. 2016, 73, 3917–3933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Sabbineni, H.; Artham, S.; Somanath, P.R. Modulation of long-term endothelial-barrier integrity is conditional to the cross-talk between Akt and Src signaling. J. Cell Physiol. 2017, 232, 2599–2609. [Google Scholar] [CrossRef] [PubMed]
- Alwhaibi, A.; Verma, A.; Adil, M.S.; Somanath, P.R. The unconventional role of Akt1 in the advanced cancers and in diabetes-promoted carcinogenesis. Pharm. Res. 2019, 145, 104270. [Google Scholar] [CrossRef]
- Adil, M.S.; Narayanan, S.P.; Somanath, P.R. Cell-cell junctions: Structure and regulation in physiology and pathology. Tissue Barriers 2020, 9, 1848212. [Google Scholar] [CrossRef]
- Rosenthal, R.; Günzel, D.; Theune, D.; Czichos, C.; Schulzke, J.D.; Fromm, M. Water channels and barriers formed by claudins. Ann. N. Y. Acad. Sci. 2017, 1397, 100–109. [Google Scholar] [CrossRef]
- Conrad, M.P.; Piontek, J.; Günzel, D.; Fromm, M.; Krug, S.M. Molecular basis of claudin-17 anion selectivity. Cell Mol. Life Sci. 2016, 73, 185–200. [Google Scholar] [CrossRef]
- Marcelino Cereijido, J.M.A. Tight Junctions, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2001. [Google Scholar]
- Shabalina, S.A.; Ogurtsov, A.Y.; Spiridonov, A.N.; Novichkov, P.S.; Spiridonov, N.A.; Koonin, E.V. Distinct patterns of expression and evolution of intronless and intron-containing mammalian genes. Mol. Biol. Evol. 2010, 27, 1745–1749. [Google Scholar] [CrossRef] [Green Version]
- Günzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [Green Version]
- Kielgast, F.; Schmidt, H.; Braubach, P.; Winkelmann, V.E.; Thompson, K.E.; Frick, M.; Dietl, P.; Wittekindt, O.H. Glucocorticoids Regulate Tight Junction Permeability of Lung Epithelia by Modulating Claudin 8. Am. J. Respir Cell Mol. Biol. 2016, 54, 707–717. [Google Scholar] [CrossRef]
- Li, Z.H.; Xia, T.H.; Kang, Z.J.; Deng, X.; Wang, Y. Expression and significance of tight junction proteins in the kidney in a mouse model of renal ischemia-reperfusion injury. Zhongguo Dang Dai Er Ke Za Zhi 2018, 20, 1055–1062. [Google Scholar]
- Xu, Y.N.; Deng, M.S.; Liu, Y.F.; Yao, J.; Xiao, Z.Y. Tight junction protein CLDN17 serves as a tumor suppressor to reduce the invasion and migration of oral cancer cells by inhibiting epithelial-mesenchymal transition. Arch Oral Biol. 2022, 133, 105301. [Google Scholar] [CrossRef]
- Sun, L.; Feng, L.; Cui, J. Increased expression of claudin-17 promotes a malignant phenotype in hepatocyte via Tyk2/Stat3 signaling and is associated with poor prognosis in patients with hepatocellular carcinoma. Diagn Pathol. 2018, 13, 72. [Google Scholar] [CrossRef]
- Gao, M.; Li, W.; Wang, H.; Wang, G. The distinct expression patterns of claudin-10, -14, -17 and E-cadherin between adjacent non-neoplastic tissues and gastric cancer tissues. Diagn Pathol. 2013, 8, 205. [Google Scholar] [CrossRef] [Green Version]
- Berndt, P.; Winkler, L.; Cording, J.; Breitkreuz-Korff, O.; Rex, A.; Dithmer, S.; Rausch, V.; Blasig, R.; Richter, M.; Sporbert, A.; et al. Tight junction proteins at the blood-brain barrier: Far more than claudin-5. Cell Mol. Life Sci. 2019, 76, 1987–2002. [Google Scholar] [CrossRef]
- Alharthi, A.; Verma, A.; Sabbineni, H.; Adil, M.S.; Somanath, P.R. Distinct effects of pharmacological inhibition of stromelysin1 on endothelial-to-mesenchymal transition and myofibroblast differentiation. J. Cell Physiol. 2021, 236, 5147–5161. [Google Scholar] [CrossRef]
- Gah, A.; Adil, M.S.; Sabbineni, H.; Verma, A.; Somanath, P.R. Differential regulation of TGFβ type-I receptor expressions in TGFβ1-induced myofibroblast differentiation. Can J. Physiol. Pharm. 2020, 98, 841–848. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Wang, Q.; Zou, M.H. Measurement of Reactive Oxygen Species (ROS) and Mitochondrial ROS in AMPK Knockout Mice Blood Vessels. Methods Mol. Biol. 2018, 1732, 507–517. [Google Scholar] [CrossRef]
- Brandner, J.M.; Kief, S.; Grund, C.; Rendl, M.; Houdek, P.; Kuhn, C.; Tschachler, E.; Franke, W.W.; Moll, I. Organization and formation of the tight junction system in human epidermis and cultured keratinocytes. Eur. J. Cell Biol. 2002, 81, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Cong, X.; Kong, W. Endothelial tight junctions and their regulatory signaling pathways in vascular homeostasis and disease. Cell. Signal. 2020, 66, 109485. [Google Scholar] [CrossRef]
- Rudraraju, M.; Narayanan, S.P.; Somanath, P.R. Regulation of blood-retinal barrier cell-junctions in diabetic retinopathy. Pharm. Res. 2020, 161, 105115. [Google Scholar] [CrossRef]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef]
- Chen, J.; Somanath, P.R.; Razorenova, O.; Chen, W.S.; Hay, N.; Bornstein, P.; Byzova, T.V. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat. Med. 2005, 11, 1188–1196. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Alwhaibi, A.; Artham, S.; Verma, A.; Somanath, P.R. Endothelial Akt1 loss promotes prostate cancer metastasis via beta-catenin-regulated tight-junction protein turnover. Br. J. Cancer 2018, 118, 1464–1475. [Google Scholar] [CrossRef]
- Krug, S.M.; Günzel, D.; Conrad, M.P.; Rosenthal, R.; Fromm, A.; Amasheh, S.; Schulzke, J.D.; Fromm, M. Claudin-17 forms tight junction channels with distinct anion selectivity. Cell Mol. Life Sci. 2012, 69, 2765–2778. [Google Scholar] [CrossRef]
- Runkle, E.A.; Mu, D. Tight junction proteins: From barrier to tumorigenesis. Cancer Lett. 2013, 337, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Portalatin, M.; Winstead, N. Medical management of constipation. Clin. Colon. Rectal Surg. 2012, 25, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Torgerson, D.G.; Ballard, P.L.; Keller, R.L.; Oh, S.S.; Huntsman, S.; Hu, D.; Eng, C.; Burchard, E.G.; Ballard, R.A. Ancestry and genetic associations with bronchopulmonary dysplasia in preterm infants. Am. J. Physiol Lung Cell Mol. Physiol. 2018, 315, L858–L869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Du, G.; Wang, Y.; Shi, L.; Mi, J.; Tang, G. Integrative analysis of mRNA and miRNA expression profiles in oral lichen planus: Preliminary results. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2017, 124, 390–402.e317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunton-Stasyshyn, R.K.; Saccon, R.A.; Fratta, P.; Fisher, E.M. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist 2015, 21, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veith, A.; Moorthy, B. Role of cytochrome p450s in the generation and metabolism of reactive oxygen species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adil, M.S.; Parvathagiri, V.; Verma, A.; Liu, F.; Rudraraju, M.; Narayanan, S.P.; Somanath, P.R. Claudin-17 Deficiency in Mice Results in Kidney Injury Due to Electrolyte Imbalance and Oxidative Stress. Cells 2022, 11, 1782. https://doi.org/10.3390/cells11111782
Adil MS, Parvathagiri V, Verma A, Liu F, Rudraraju M, Narayanan SP, Somanath PR. Claudin-17 Deficiency in Mice Results in Kidney Injury Due to Electrolyte Imbalance and Oxidative Stress. Cells. 2022; 11(11):1782. https://doi.org/10.3390/cells11111782
Chicago/Turabian StyleAdil, Mir S., Varun Parvathagiri, Arti Verma, Fang Liu, Madhuri Rudraraju, S. Priya Narayanan, and Payaningal R. Somanath. 2022. "Claudin-17 Deficiency in Mice Results in Kidney Injury Due to Electrolyte Imbalance and Oxidative Stress" Cells 11, no. 11: 1782. https://doi.org/10.3390/cells11111782