
The Extracellular Matrix Proteins Tenascin-C and Tenascin-R Retard Oligodendrocyte Precursor Maturation and Myelin Regeneration in a Cuprizone-Induced Long-Term Demyelination Animal Model



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Genotyping
2.2. Immunological Reagents
2.3. Isolation and Cultivation of Primary Murine OPCs
2.4. Immunocytochemistry
2.5. Myelination of Electrospun Fibers
2.6. Cuprizone Model Induced Demyelination
2.7. Histochemistry of the Brain Slices
2.8. Molecular Biology
RNA Isolation, cDNA Synthesis, Polymerase Chain Reaction (PCR)
2.9. Electron Microscopy
2.10. Statistics
3. Results
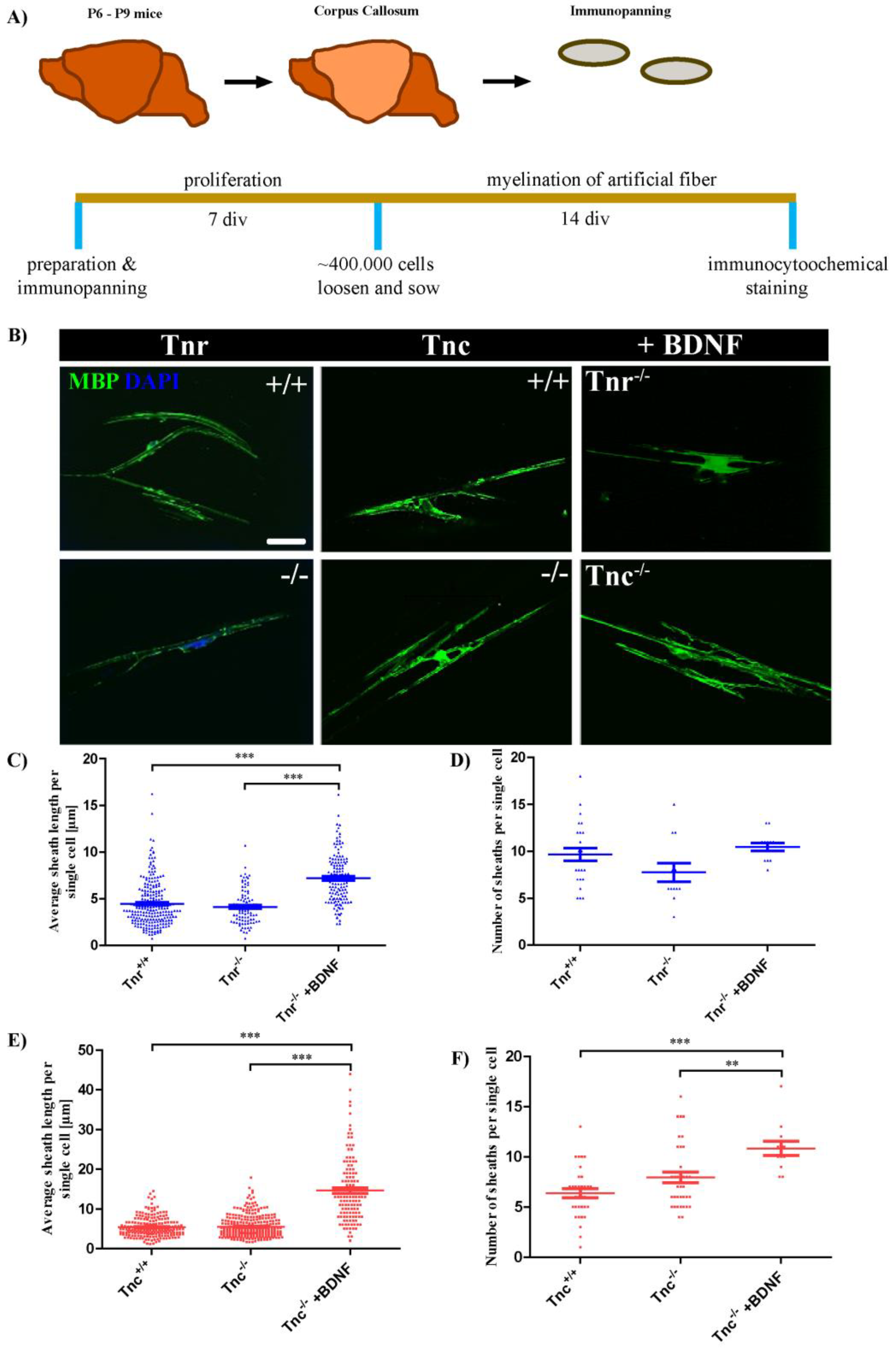
3.1. Tenascins Intervene in Myelination of Artificial Microfibers
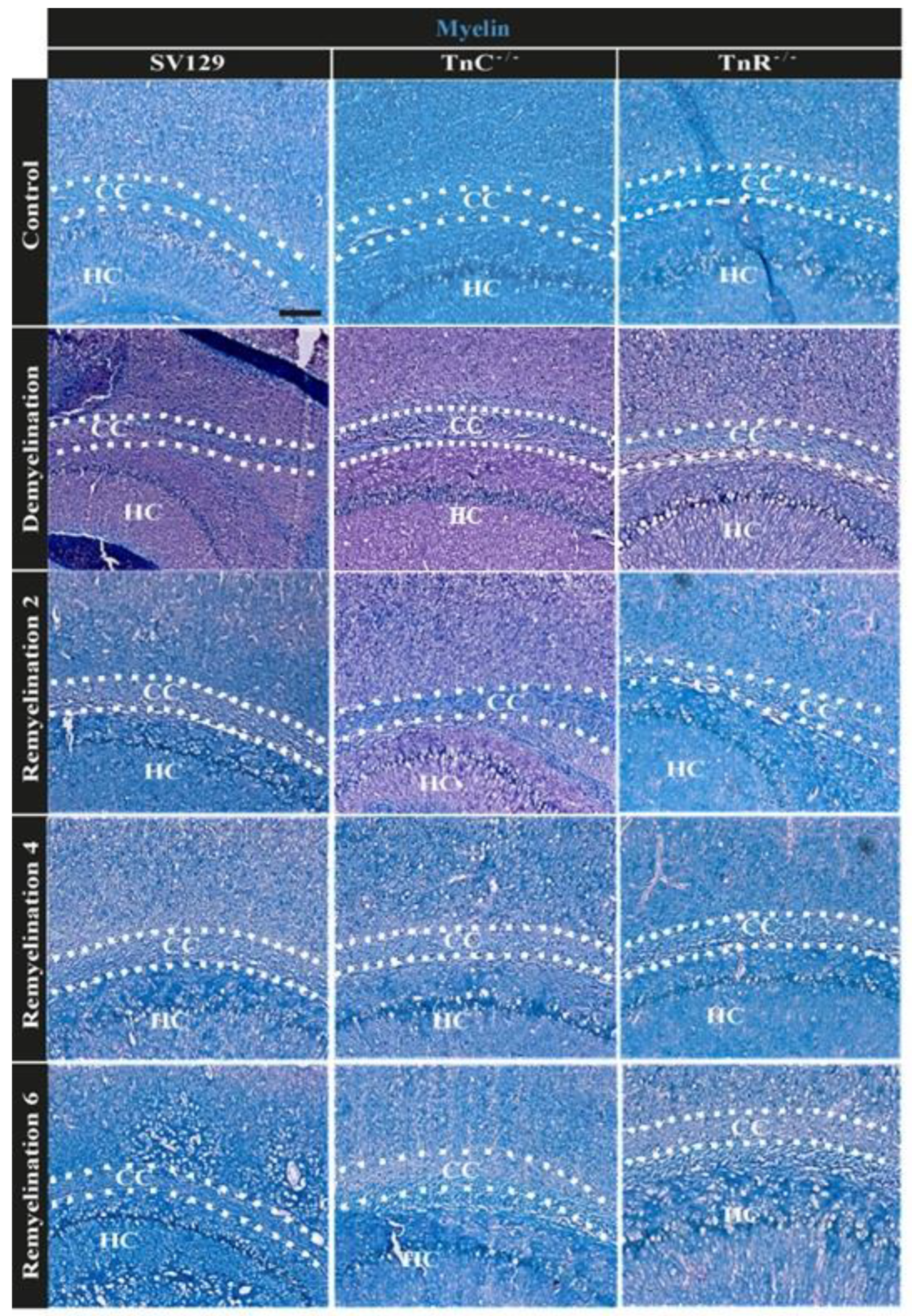
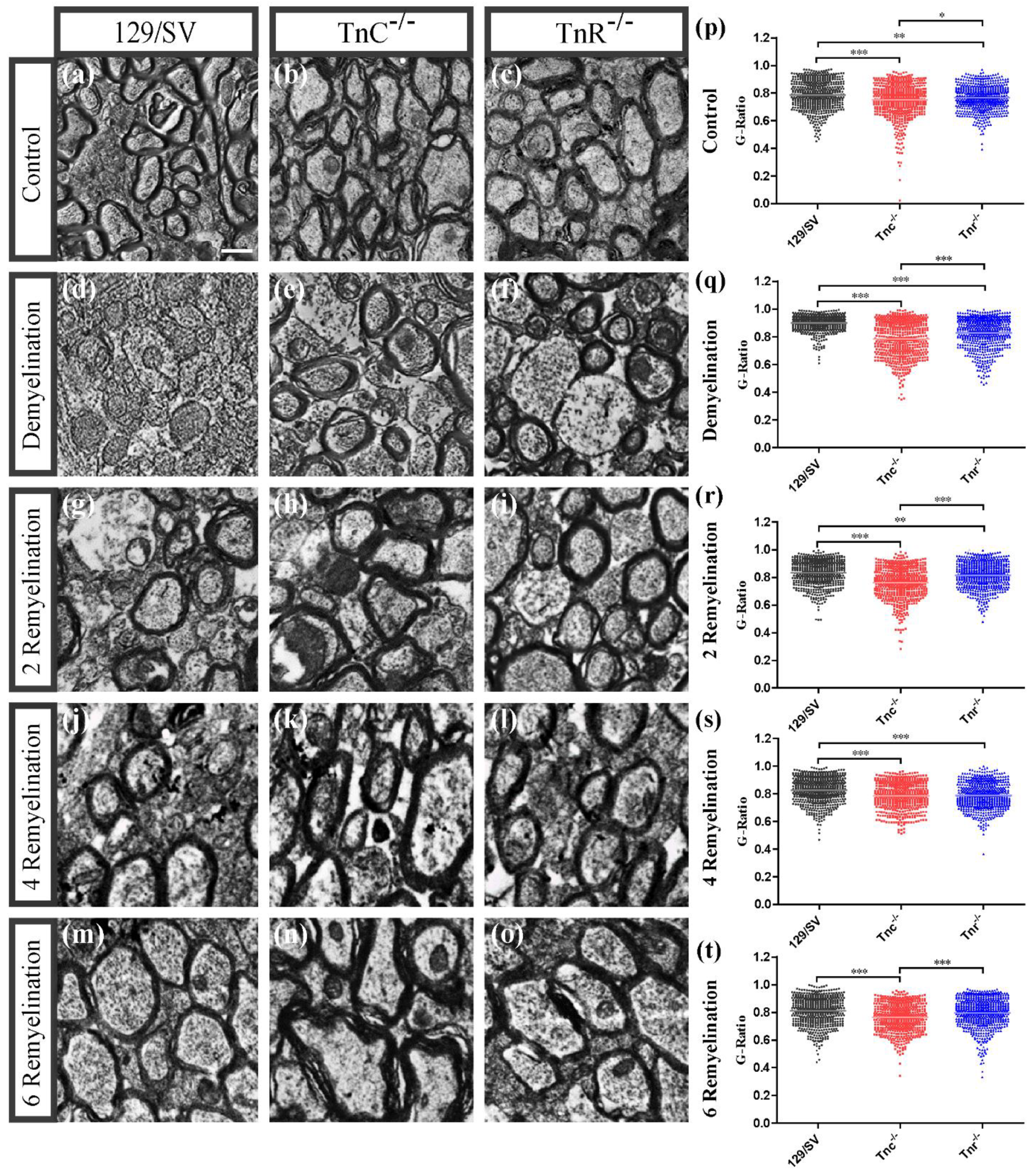
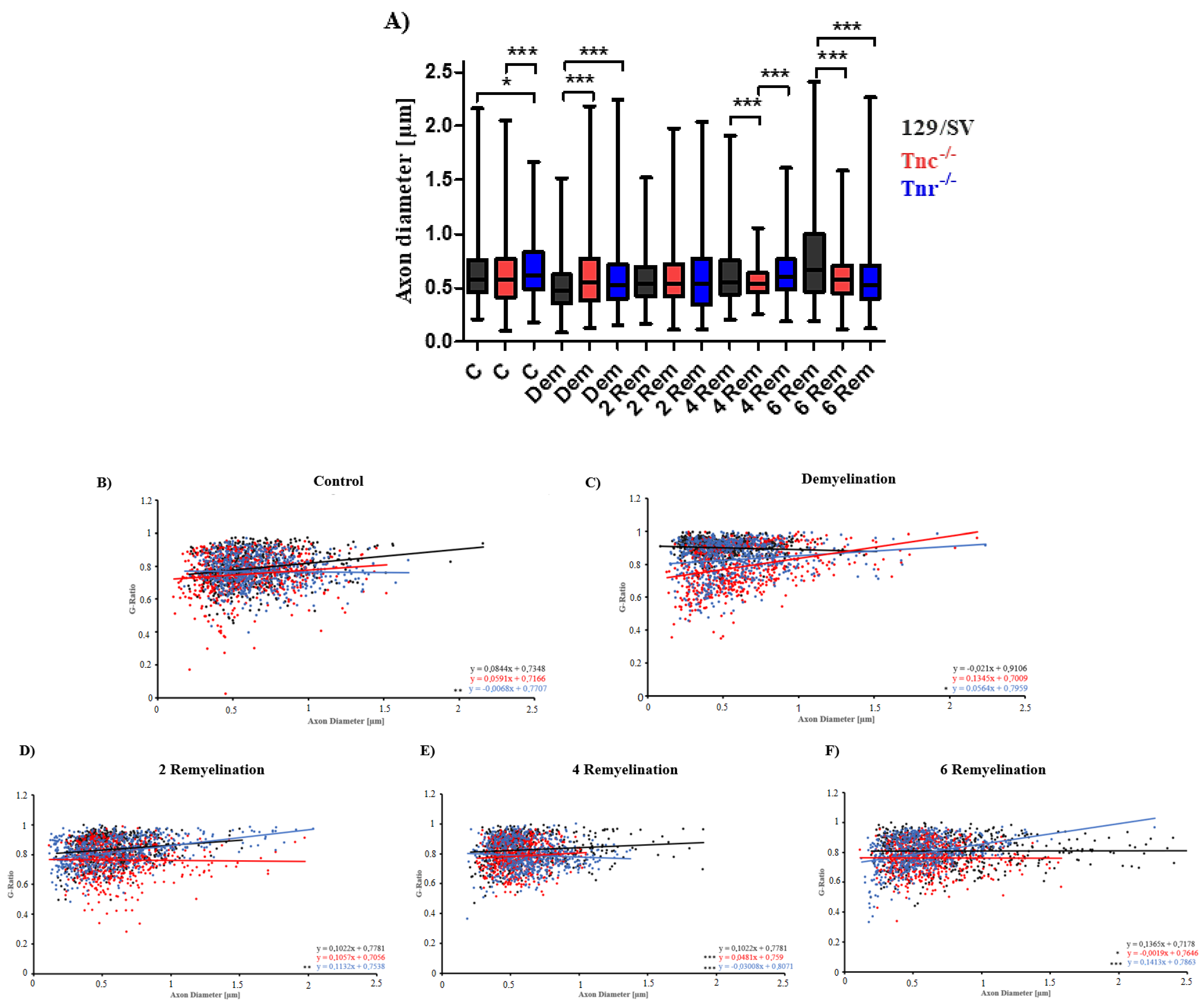
3.2. Electron Microscopy Analysis Revealed That Remyelination Is Accelerated in the Absence of Tenascins in the Cuprizone Animal Model
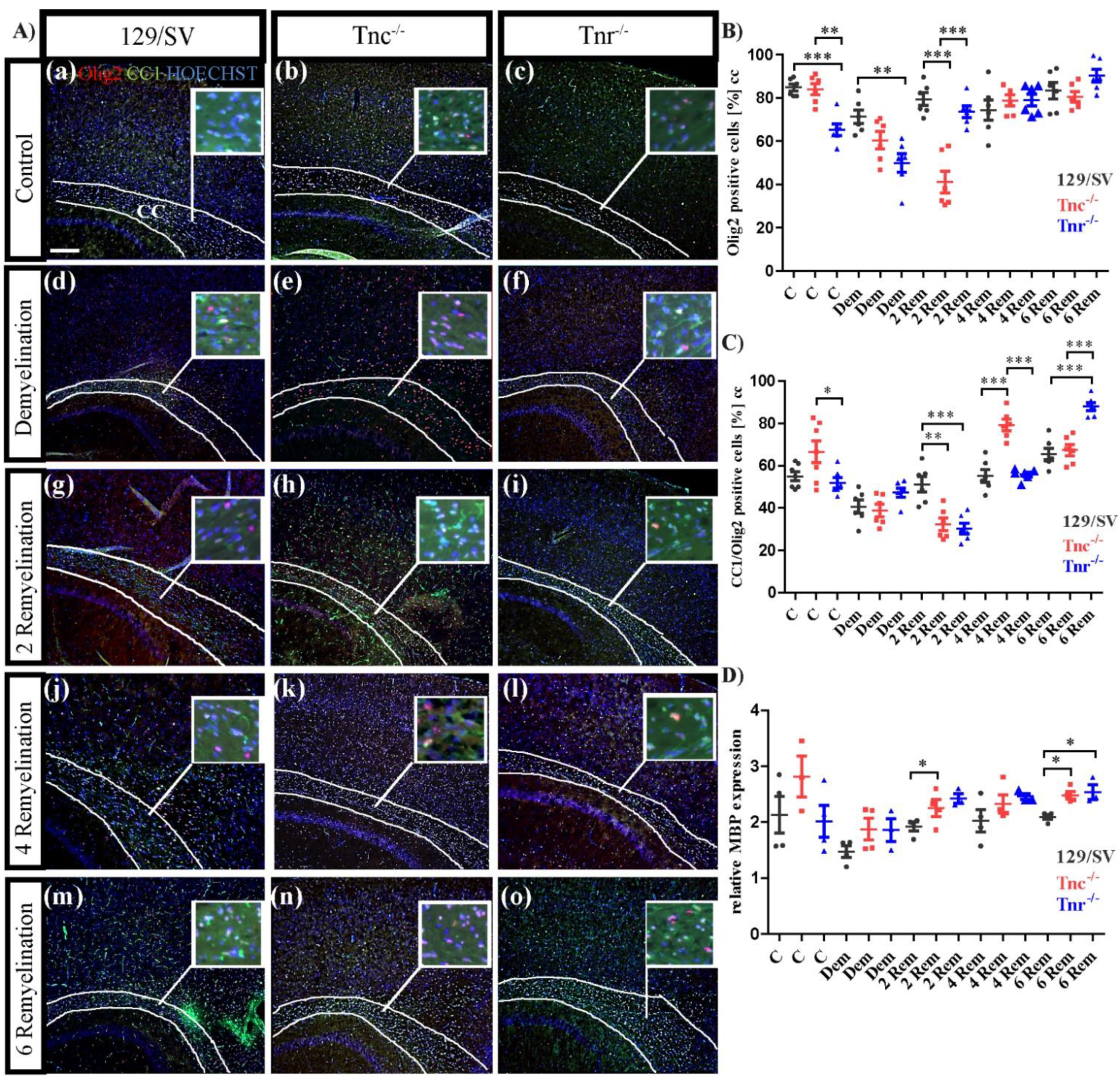
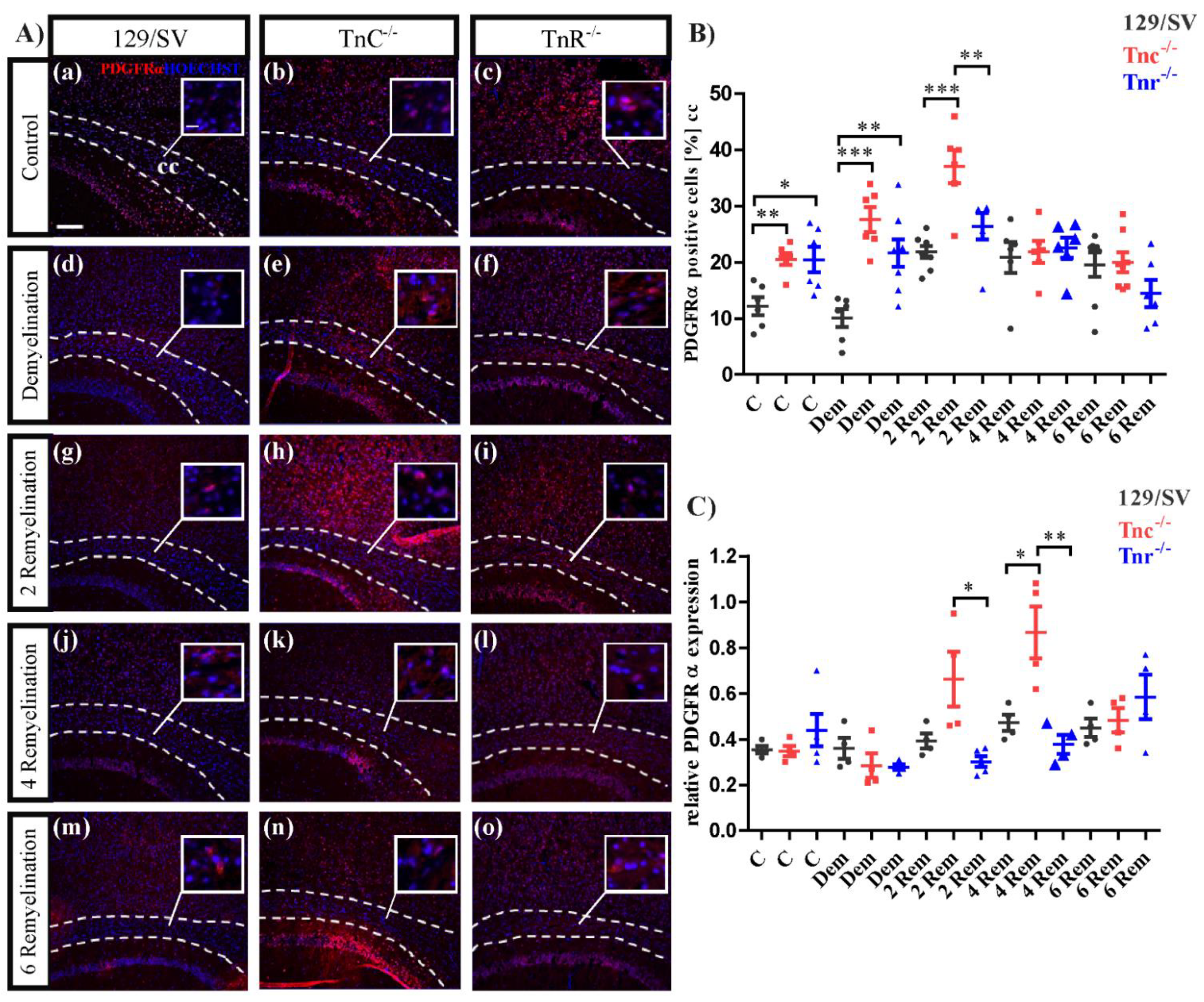
3.3. Tnc and Tnr Modulate Recruitment of OPCs to and Their Maturation in Myelin Lesions
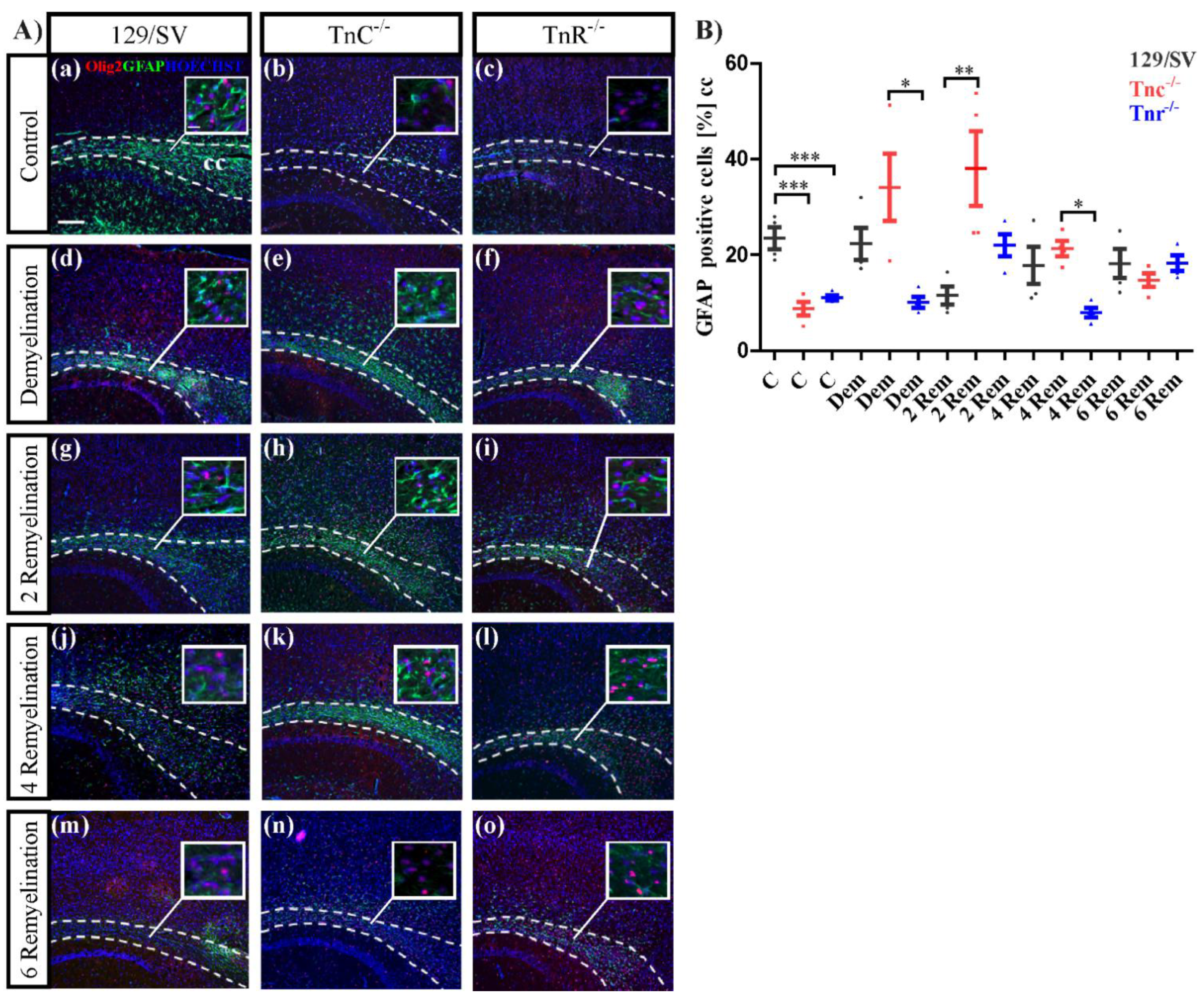
3.4. The Loss of Tnc Enhances Astrocyte Reactivity in Cuprizone-Induced CNS Lesions
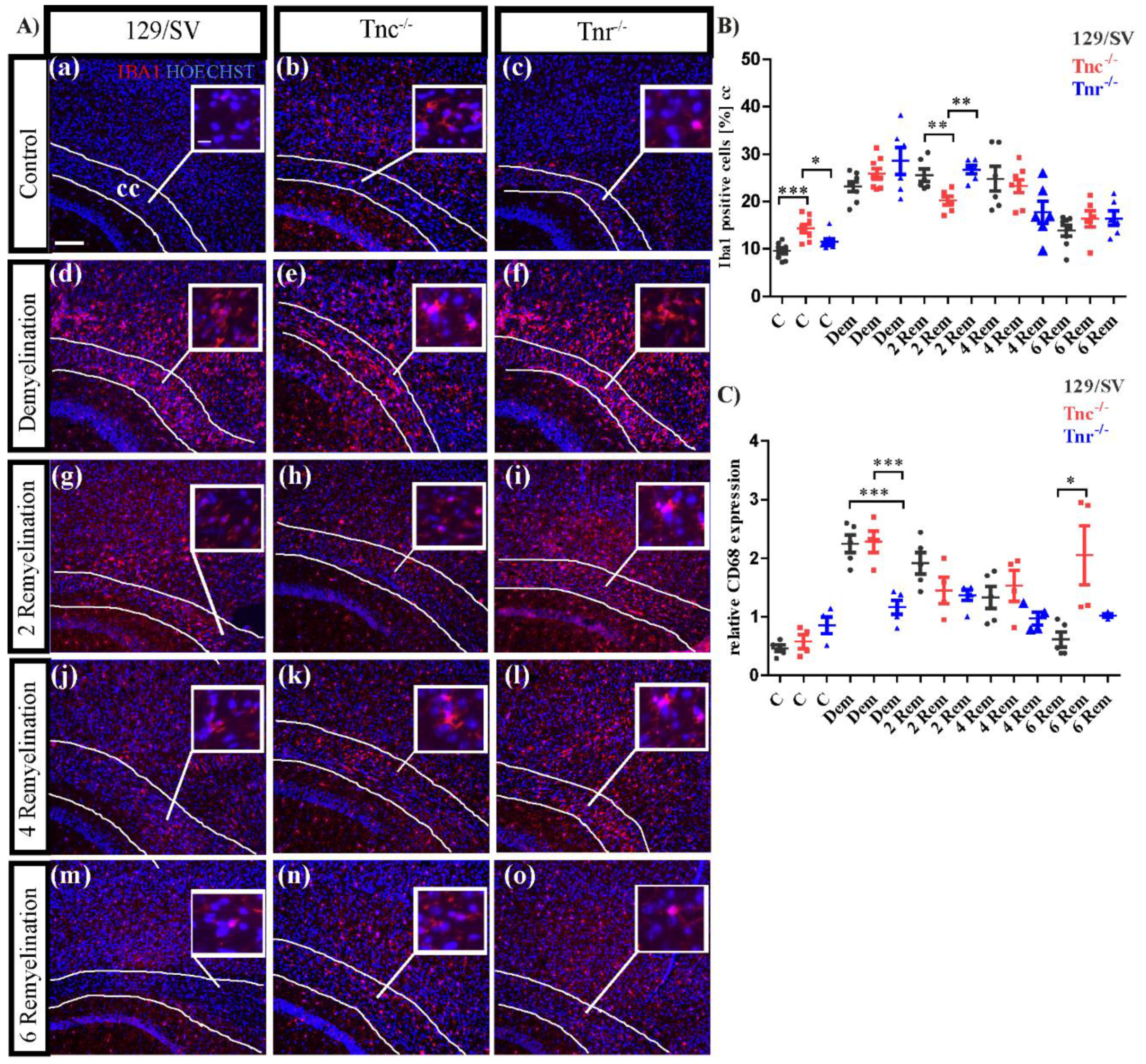
3.5. Tenascins Modulate Microglia and Leucocytes in Cuprizone-Induced Lesions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barateiro, A.; Fernandes, A. Temporal oligodendrocyte lineage progression: In vitro models of proliferation, differentiation and myelination. Biochim. Biophys. Acta 2014, 1843, 1917–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, S.A.; Kuypers, N.J. How to make an oligodendrocyte. Development 2015, 142, 3983–3995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miron, V.E.; Kuhlmann, T.; Antel, J.P. Cells of the oligodendroglial lineage, myelination, and remyelination. Biochim. Biophys. Acta 2011, 1812, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Nave, K.A. Myelination and support of axonal integrity by glia. Nature 2010, 468, 244–252. [Google Scholar] [CrossRef]
- Nave, K.A.; Werner, H.B. Ensheathment and Myelination of Axons: Evolution of Glial Functions. Annu. Rev. Neurosci. 2021, 44, 197–219. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.M.; Ffrench-Constant, C. Regenerating CNS myelin—From mechanisms to experimental medicines. Nat. Rev. Neurosci. 2017, 18, 753–769. [Google Scholar] [CrossRef]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef]
- Hagemeier, K.; Bruck, W.; Kuhlmann, T. Multiple sclerosis—Remyelination failure as a cause of disease progression. Histol. Histopathol. 2012, 27, 277–287. [Google Scholar] [CrossRef]
- Sospedra, M. B cells in multiple sclerosis. Curr. Opin. Neurol. 2018, 31, 256–262. [Google Scholar] [CrossRef]
- Pugliatti, M.; Sotgiu, S.; Rosati, G. The worldwide prevalence of multiple sclerosis. Clin. Neurol. Neurosurg. 2002, 104, 182–191. [Google Scholar] [CrossRef]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Patani, R.; Balaratnam, M.; Vora, A.; Reynolds, R. Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathol. Appl. Neurobiol. 2007, 33, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M.; Clarner, T.; Dang, J.; Copray, S.; Beyer, C. The cuprizone animal model: New insights into an old story. Acta Neuropathol. 2009, 118, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, G.K.; Morell, P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001, 11, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. Animal models of multiple sclerosis: The good, the bad and the bottom line. Nat. Neurosci. 2012, 15, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Jakovcevski, I.; Miljkovic, D.; Schachner, M.; Andjus, P.R. Tenascins and inflammation in disorders of the nervous system. Amino Acids 2013, 44, 1115–1127. [Google Scholar] [CrossRef]
- Wheeler, N.A.; Fuss, B. Extracellular cues influencing oligodendrocyte differentiation and (re)myelination. Exp. Neurol. 2016, 283, 512–530. [Google Scholar] [CrossRef] [Green Version]
- Dityatev, A.; Schachner, M.; Sonderegger, P. The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat. Rev. Neurosci. 2010, 11, 735–746. [Google Scholar] [CrossRef]
- Barros, C.S.; Franco, S.J.; Muller, U. Extracellular matrix: Functions in the nervous system. Cold Spring Harb. Perspect. Biol. 2011, 3, a005108. [Google Scholar] [CrossRef] [Green Version]
- Faissner, A.; Reinhard, J. The extracellular matrix compartment of neural stem and glial progenitor cells. Glia 2015, 63, 1330–1349. [Google Scholar] [CrossRef] [PubMed]
- Czopka, T.; Von Holst, A.; Schmidt, G.; Ffrench-Constant, C.; Faissner, A. Tenascin C and tenascin R similarly prevent the formation of myelin membranes in a RhoA-dependent manner, but antagonistically regulate the expression of myelin basic protein via a separate pathway. Glia 2009, 57, 1790–1801. [Google Scholar] [CrossRef] [PubMed]
- Tucker, R.P.; Drabikowski, K.; Hess, J.F.; Ferralli, J.; Chiquet-Ehrismann, R.; Adams, J.C. Phylogenetic analysis of the tenascin gene family: Evidence of origin early in the chordate lineage. BMC Evol. Biol. 2006, 6, 60. [Google Scholar] [CrossRef] [Green Version]
- Bartsch, S.; Bartsch, U.; Dorries, U.; Faissner, A.; Weller, A.; Ekblom, P.; Schachner, M. Expression of tenascin in the developing and adult cerebellar cortex. J. Neurosci. 1992, 12, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Faissner, A.; Roll, L.; Theocharidis, U. Tenascin-C in the matrisome of neural stem and progenitor cells. Mol. Cell. Neurosci. 2017, 81, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, B.W.; Gotz, B.; Faissner, A.; Ffrench-Constant, C. Tenascin-C inhibits oligodendrocyte precursor cell migration by both adhesion-dependent and adhesion-independent mechanisms. Mol. Cell. Neurosci. 1996, 7, 322–335. [Google Scholar] [CrossRef]
- Zhao, C.; Fancy, S.P.; Franklin, R.J.; Ffrench-Constant, C. Up-regulation of oligodendrocyte precursor cell alphaV integrin and its extracellular ligands during central nervous system remyelination. J. Neurosci. Res. 2009, 87, 3447–3455. [Google Scholar] [CrossRef]
- Fuss, B.; Wintergerst, E.S.; Bartsch, U.; Schachner, M. Molecular characterization and in situ mRNA localization of the neural recognition molecule J1-160/180: A modular structure similar to tenascin. J. Cell Biol. 1993, 120, 1237–1249. [Google Scholar] [CrossRef] [Green Version]
- Becker, T.; Anliker, B.; Becker, C.G.; Taylor, J.; Schachner, M.; Meyer, R.L.; Bartsch, U. Tenascin-R inhibits regrowth of optic fibers in vitro and persists in the optic nerve of mice after injury. Glia 2000, 29, 330–346. [Google Scholar] [CrossRef]
- Pesheva, P.; Gloor, S.; Schachner, M.; Probstmeier, R. Tenascin-R is an intrinsic autocrine factor for oligodendrocyte differentiation and promotes cell adhesion by a sulfatide-mediated mechanism. J. Neurosci. 1997, 17, 4642–4651. [Google Scholar] [CrossRef] [Green Version]
- Bauch, J.; Vom Ort, S.; Ulc, A.; Faissner, A. Tenascins interfere with remyelination in an ex vivo cerebellar explant model of demyelination. Front. Cell Dev. Biol. 2022; in press. [Google Scholar] [CrossRef] [PubMed]
- Watkins, T.A.; Emery, B.; Mulinyawe, S.; Barres, B.A. Distinct stages of myelination regulated by gamma-secretase and astrocytes in a rapidly myelinating CNS coculture system. Neuron 2008, 60, 555–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, S.; Hagemeier, K.; Ehrlich, M.; Kemming, C.; Trotter, J.; Kuhlmann, T. Recovery from Toxic-Induced Demyelination Does Not Require the NG2 Proteoglycan. PLoS ONE 2016, 11, e0163841. [Google Scholar] [CrossRef] [PubMed]
- Czopka, T.; von Holst, A.; Ffrench-Constant, C.; Faissner, A. Regulatory mechanisms that mediate tenascin C-dependent inhibition of oligodendrocyte precursor differentiation. J. Neurosci. 2010, 30, 12310–12322. [Google Scholar] [CrossRef] [Green Version]
- Sallouh, M.; Jarocki, M.; Sallouh, O.; Degen, P.; Faissner, A.; Weberskirch, R. The Synergistic Effect of Cationic Moieties and GRGDSF-Peptides in Hydrogels on Neural Stem Cell Behavior. Macromol. Biosci. 2017, 17I, 1600178. [Google Scholar] [CrossRef]
- Bechler, M.E.; Byrne, L.; Ffrench-Constant, C. CNS Myelin Sheath Lengths Are an Intrinsic Property of Oligodendrocytes. Curr. Biol. CB 2015, 25, 2411–2416. [Google Scholar] [CrossRef] [Green Version]
- Ghaiad, H.R.; Nooh, M.M.; El-Sawalhi, M.M.; Shaheen, A.A. Resveratrol Promotes Remyelination in Cuprizone Model of Multiple Sclerosis: Biochemical and Histological Study. Mol. Neurobiol. 2017, 54, 3219–3229. [Google Scholar] [CrossRef]
- Hillis, J.M.; Davies, J.; Mundim, M.V.; Al-Dalahmah, O.; Szele, F.G. Cuprizone demyelination induces a unique inflammatory response in the subventricular zone. J. Neuroinflamm. 2016, 13, 190. [Google Scholar] [CrossRef] [Green Version]
- Blakemore, W.F. Remyelination of the superior cerebellar peduncle in old mice following demyelination induced by cuprizone. J. Neurol. Sci. 1974, 22, 121–126. [Google Scholar] [CrossRef]
- Poliani, P.L.; Wang, Y.; Fontana, E.; Robinette, M.L.; Yamanishi, Y.; Gilfillan, S.; Colonna, M. TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Investig. 2015, 125, 2161–2170. [Google Scholar] [CrossRef] [Green Version]
- Zhan, J.; Mann, T.; Joost, S.; Behrangi, N.; Frank, M.; Kipp, M. The Cuprizone Model: Dos and Do Nots. Cells 2020, 9, 843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwin, S.K. Chronic demyelination inhibits remyelination in the central nervous system. An analysis of contributing factors. Lab. Investig. 1980, 43, 382–387. [Google Scholar] [PubMed]
- Armstrong, R.C.; Le, T.Q.; Flint, N.C.; Vana, A.C.; Zhou, Y.X. Endogenous cell repair of chronic demyelination. J. Neuropathol. Exp. Neurol. 2006, 65, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harsan, L.A.; Steibel, J.; Zaremba, A.; Agin, A.; Sapin, R.; Poulet, P.; Guignard, B.; Parizel, N.; Grucker, D.; Boehm, N.; et al. Recovery from chronic demyelination by thyroid hormone therapy: Myelinogenesis induction and assessment by diffusion tensor magnetic resonance imaging. J. Neurosci. 2008, 28, 14189–14201. [Google Scholar] [CrossRef] [PubMed]
- Safaiyan, S.; Besson-Girard, S.; Kaya, T.; Cantuti-Castelvetri, L.; Liu, L.; Ji, H.; Schifferer, M.; Gouna, G.; Usifo, F.; Kannaiyan, N.; et al. White matter aging drives microglial diversity. Neuron 2021, 109, 1100–1117. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Haro, D.; Mora-Loyola, E.; Soria-Ortiz, B.; Garcia-Colunga, J. Regional density of glial cells in the rat corpus callosum. Biol. Res. 2013, 46, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Mason, J.L.; Jones, J.J.; Taniike, M.; Morell, P.; Suzuki, K.; Matsushima, G.K. Mature oligodendrocyte apoptosis precedes IGF-1 production and oligodendrocyte progenitor accumulation and differentiation during demyelination/remyelination. J. Neurosci. Res. 2000, 61, 251–262. [Google Scholar] [CrossRef]
- Jarjour, A.A.; Boyd, A.; Dow, L.E.; Holloway, R.K.; Goebbels, S.; Humbert, P.O.; Williams, A.; Ffrench-Constant, C. The polarity protein Scribble regulates myelination and remyelination in the central nervous system. PLoS Biol. 2015, 13, e1002107. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, E.S. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 1963, 17, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.L.; Wood, R.J.; Nguyen, J.; Norman, E.M.L.; Jun, C.M.K.; Prawdiuk, A.R.; Biemond, M.; Nguyen, H.T.H.; Northfield, S.E.; Hughes, R.A.; et al. Targeting TrkB with a Brain-Derived Neurotrophic Factor Mimetic Promotes Myelin Repair in the Brain. J. Neurosci. 2018, 38, 7088–7099. [Google Scholar] [CrossRef] [Green Version]
- Montag-Sallaz, M.; Montag, D. Severe cognitive and motor coordination deficits in tenascin-R-deficient mice. Genes Brain Behav. 2003, 2, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, B.W.; Garcion, E.; Ferguson, J.; Frost, E.E.; Torres, E.M.; Dunnett, S.B.; Saga, Y.; Aizawa, S.; Faissner, A.; Kaur, R.; et al. Myelination and behaviour of tenascin-C null transgenic mice. Eur. J. Neurosci. 1999, 11, 3082–3092. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Trotter, J. Wrapping it up: The cell biology of myelination. Curr. Opin. Neurobiol. 2007, 17, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Kramer, E.M.; Thiele, C.; Stoffel, W.; Trotter, J. Assembly of myelin by association of proteolipid protein with cholesterol- and galactosylceramide-rich membrane domains. J. Cell Biol. 2000, 151, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Bauer, N.M.; Schafer, I.; White, R. Making myelin basic protein -from mRNA transport to localized translation. Front. Cell. Neurosci. 2013, 7, 169. [Google Scholar] [CrossRef] [Green Version]
- Boggs, J.M. Myelin basic protein: A multifunctional protein. Cell. Mol. Life Sci. CMLS 2006, 63, 1945–1961. [Google Scholar] [CrossRef]
- Krugmann, B.; Radulescu, A.; Appavou, M.S.; Koutsioubas, A.; Stingaciu, L.R.; Dulle, M.; Forster, S.; Stadler, A.M. Membrane stiffness and myelin basic protein binding strength as molecular origin of multiple sclerosis. Sci. Rep. 2020, 10, 16691. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Dzyubenko, E.; Manrique-Castano, D.; Kleinschnitz, C.; Faissner, A.; Hermann, D.M. Role of immune responses for extracellular matrix remodeling in the ischemic brain. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418818092. [Google Scholar] [CrossRef]
- Wiemann, S.; Reinhard, J.; Faissner, A. Immunomodulatory role of the extracellular matrix protein tenascin-C in neuroinflammation. Biochem. Soc. Trans. 2019, 47, 1651–1660. [Google Scholar] [CrossRef]
- Angelov, D.N.; Walther, M.; Streppel, M.; Guntinas-Lichius, O.; Neiss, W.F.; Probstmeier, R.; Pesheva, P. Tenascin-R is antiadhesive for activated microglia that induce downregulation of the protein after peripheral nerve injury: A new role in neuronal protection. J. Neurosci. 1998, 18, 6218–6229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijesundera, K.K.; Juniantito, V.; Golbar, H.M.; Fujisawa, K.; Tanaka, M.; Ichikawa, C.; Izawa, T.; Kuwamura, M.; Yamate, J. Expressions of Iba1 and galectin-3 (Gal-3) in thioacetamide (TAA)-induced acute rat liver lesions. Exp. Toxicol. Pathol. 2013, 65, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Garwood, J.; Garcion, E.; Dobbertin, A.; Heck, N.; Calco, V.; Ffrench-Constant, C.; Faissner, A. The extracellular matrix glycoprotein Tenascin-C is expressed by oligodendrocyte precursor cells and required for the regulation of maturation rate, survival and responsiveness to platelet-derived growth factor. Eur. J. Neurosci. 2004, 20, 2524–2540. [Google Scholar] [CrossRef] [PubMed]
- Rathjen, F.G.; Hodge, R. Early Days of Tenascin-R Research: Two Approaches Discovered and Shed Light on Tenascin-R. Front. Immunol. 2020, 11, 612482. [Google Scholar] [CrossRef] [PubMed]
- VonDran, M.W.; Singh, H.; Honeywell, J.Z.; Dreyfus, C.F. Levels of BDNF impact oligodendrocyte lineage cells following a cuprizone lesion. J. Neurosci. 2011, 31, 14182–14190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulmer, C.G.; VonDran, M.W.; Stillman, A.A.; Huang, Y.; Hempstead, B.L.; Dreyfus, C.F. Astrocyte-derived BDNF supports myelin protein synthesis after cuprizone-induced demyelination. J. Neurosci. 2014, 34, 8186–8196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitta, K.S.; Lercher, L.D.; Sainato, D.M.; Patel, A.; Huang, Y.; McAuliffe, G.; Dreyfus, C.F. CHPG enhances BDNF and myelination in cuprizone-treated mice through astrocytic metabotropic glutamate receptor 5. Glia 2021, 69, 1950–1965. [Google Scholar] [CrossRef]
- Wenk, M.B.; Midwood, K.S.; Schwarzbauer, J.E. Tenascin-C suppresses Rho activation. J. Cell Biol. 2000, 150, 913–920. [Google Scholar] [CrossRef]
- Czopka, T.; Hennen, E.; von Holst, A.; Faissner, A. Novel conserved oligodendrocyte surface epitope identified by monoclonal antibody 4860. Cell Tissue Res. 2009, 338, 161–170. [Google Scholar] [CrossRef]
- Ulc, A.; Gottschling, C.; Schäfer, I.; Wegrzyn, D.; van Leeuwen, S.; Luft, V.; Reinhard, J.; Faissner, A. Involvement of the guanine nucleotide exchange factor Vav3 in central nervous system development and plasticity. Biol. Chem. 2017, 398, 663–675. [Google Scholar] [CrossRef]
- Ulc, A.; Zeug, A.; Bauch, J.; van Leeuwen, S.; Kuhlmann, T.; Ffrench-Constant, C.; Ponimaskin, E.; Faissner, A. The guanine nucleotide exchange factor Vav3 modulates oligodendrocyte precursor differentiation and supports remyelination in white matter lesions. Glia 2019, 67, 376–392. [Google Scholar] [CrossRef] [PubMed]
- Chiquet-Ehrismann, R.; Tucker, R.P. Tenascins and the importance of adhesion modulation. Cold Spring Harb. Perspect. Biol. 2011, 3, a004960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackie, E.J.; Tucker, R.P. The tenascin-C knockout revisited. J. Cell Sci. 1999, 112 Pt 22, 3847–3853. [Google Scholar] [CrossRef]
- Garcion, E.; Faissner, A.; Ffrench-Constant, C. Knockout mice reveal a contribution of the extracellular matrix molecule tenascin-C to neural precursor proliferation and migration. Development 2001, 128, 2485–2496. [Google Scholar] [CrossRef] [PubMed]
- Garcion, E.; Halilagic, A.; Faissner, A.; Ffrench-Constant, C. Generation of an environmental niche for neural stem cell development by the extracellular matrix molecule tenascin C. Development 2004, 131, 3423–3432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momcilovic, M.; Stamenkovic, V.; Jovanovic, M.; Andjus, P.R.; Jakovcevski, I.; Schachner, M.; Miljkovic, D. Tenascin-C deficiency protects mice from experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2017, 302, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Nakano, F.; Kawakita, F.; Liu, L.; Nakatsuka, Y.; Nishikawa, H.; Okada, T.; Shiba, M.; Suzuki, H. Link Between Receptors That Engage in Developing Vasospasm After Subarachnoid Hemorrhage in Mice. Acta Neurochir. Suppl. 2020, 127, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, S.; Reinhard, J.; Reinehr, S.; Cibir, Z.; Joachim, S.C.; Faissner, A. Loss of the Extracellular Matrix Molecule Tenascin-C Leads to Absence of Reactive Gliosis and Promotes Anti-inflammatory Cytokine Expression in an Autoimmune Glaucoma Mouse Model. Front. Immunol. 2020, 11, 566279. [Google Scholar] [CrossRef]
- Wiemann, S.; Yousf, A.; Joachim, S.C.; Peters, C.; Mueller-Buehl, A.M.; Wagner, N.; Reinhard, J. Knock-Out of Tenascin-C Ameliorates Ischemia-Induced Rod-Photoreceptor Degeneration and Retinal Dysfunction. Front. Neurosci. 2021, 15, 642176. [Google Scholar] [CrossRef]
- Roll, L.; Faissner, A. Tenascins in CNS lesions. Semin. Cell Dev. Biol. 2019, 89, 118–124. [Google Scholar] [CrossRef]
- Skripuletz, T.; Lindner, M.; Kotsiari, A.; Garde, N.; Fokuhl, J.; Linsmeier, F.; Trebst, C.; Stangel, M. Cortical demyelination is prominent in the murine cuprizone model and is strain-dependent. Am. J. Pathol. 2008, 172, 1053–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torkildsen, O.; Brunborg, L.A.; Myhr, K.M.; Bo, L. The cuprizone model for demyelination. Acta Neurol. Scand. Suppl. 2008, 188, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Dalcq, M.; Williams, A.; Stadelmann, C.; Stankoff, B.; Zalc, B.; Lubetzki, C. From fish to man: Understanding endogenous remyelination in central nervous system demyelinating diseases. Brain A J. Neurol. 2008, 131, 1686–1700. [Google Scholar] [CrossRef] [Green Version]
- Brown, T.L.; Macklin, W.B. The Actin Cytoskeleton in Myelinating Cells. Neurochem. Res. 2020, 45, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.; Heine, S.; Haastert, K.; Garde, N.; Fokuhl, J.; Linsmeier, F.; Grothe, C.; Baumgartner, W.; Stangel, M. Sequential myelin protein expression during remyelination reveals fast and efficient repair after central nervous system demyelination. Neuropathol. Appl. Neurobiol. 2008, 34, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Sachs, H.H.; Bercury, K.K.; Popescu, D.C.; Narayanan, S.P.; Macklin, W.B. A new model of cuprizone-mediated demyelination/remyelination. ASN Neuro 2014, 6, 1759091414551955. [Google Scholar] [CrossRef] [PubMed]
- Sen, M.K.; Mahns, D.A.; Coorssen, J.R.; Shortland, P.J. The roles of microglia and astrocytes in phagocytosis and myelination: Insights from the cuprizone model of multiple sclerosis. Glia 2022, 70, 1215–1250. [Google Scholar] [CrossRef]
- Steindler, D.A.; Settles, D.; Erickson, H.P.; Laywell, E.D.; Yoshiki, A.; Faissner, A.; Kusakabe, M. Tenascin knockout mice: Barrels, boundary molecules, and glial scars. J. Neurosci. 1995, 15, 1971–1983. [Google Scholar] [CrossRef]
- Karus, M.; Denecke, B.; Ffrench-Constant, C.; Wiese, S.; Faissner, A. The extracellular matrix molecule tenascin C modulates expression levels and territories of key patterning genes during spinal cord astrocyte specification. Development 2011, 138, 5321–5331. [Google Scholar] [CrossRef] [Green Version]
- Roll, L.; Eysel, U.T.; Faissner, A. Laser Lesion in the Mouse Visual Cortex Induces a Stem Cell Niche-Like Extracellular Matrix, Produced by Immature Astrocytes. Front. Cell. Neurosci. 2020, 14, 102. [Google Scholar] [CrossRef]
- Gutowski, N.J.; Newcombe, J.; Cuzner, M.L. Tenascin-R and C in multiple sclerosis lesions: Relevance to extracellular matrix remodelling. Neuropathol. Appl. Neurobiol. 1999, 25, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Nash, B.; Thomson, C.E.; Linington, C.; Arthur, A.T.; McClure, J.D.; McBride, M.W.; Barnett, S.C. Functional duality of astrocytes in myelination. J. Neurosci. 2011, 31, 13028–13038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, H.; Huang, W.; Schachner, M.; Guan, Y.; Guo, J.; Yan, J.; Qin, J.; Bai, X.; Zhang, L. Beta 1 integrin-mediated effects of tenascin-R domains EGFL and FN6-8 on neural stem/progenitor cell proliferation and differentiation in vitro. J. Biol. Chem. 2008, 283, 27927–27936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, R.P.; Chiquet-Ehrismann, R. Tenascin-C: Its functions as an integrin ligand. Int. J. Biochem. Cell B 2015, 65, 165–168. [Google Scholar] [CrossRef]
- Dzyubenko, E.; Gottschling, C.; Faissner, A. Neuron-Glia Interactions in Neural Plasticity: Contributions of Neural Extracellular Matrix and Perineuronal Nets. Neural Plast. 2016, 2016, 5214961. [Google Scholar] [CrossRef] [Green Version]
- Apostolova, I.; Irintchev, A.; Schachner, M. Tenascin-R restricts posttraumatic remodeling of motoneuron innervation and functional recovery after spinal cord injury in adult mice. J. Neurosci. 2006, 26, 7849–7859. [Google Scholar] [CrossRef] [Green Version]
- Lau, L.W.; Keough, M.B.; Haylock-Jacobs, S.; Cua, R.; Doring, A.; Sloka, S.; Stirling, D.P.; Rivest, S.; Yong, V.W. Chondroitin sulfate proteoglycans in demyelinated lesions impair remyelination. Ann. Neurol. 2012, 72, 419–432. [Google Scholar] [CrossRef]
- Keough, M.B.; Rogers, J.A.; Zhang, P.; Jensen, S.K.; Stephenson, E.L.; Chen, T.; Hurlbert, M.G.; Lau, L.W.; Rawji, K.S.; Plemel, J.R.; et al. An inhibitor of chondroitin sulfate proteoglycan synthesis promotes central nervous system remyelination. Nat. Commun. 2016, 7, 11312. [Google Scholar] [CrossRef]
- Ghorbani, S.; Yong, V.W. The extracellular matrix as modifier of neuroinflammation and remyelination in multiple sclerosis. Brain A J. Neurol. 2021, 144, 1958–1973. [Google Scholar] [CrossRef]
- Kakinuma, Y.; Saito, F.; Osawa, S.; Miura, M. A mechanism of impaired mobility of oligodendrocyte progenitor cells by tenascin C through modification of wnt signaling. FEBS Lett. 2004, 568, 60–64. [Google Scholar] [CrossRef] [Green Version]
- Pringle, N.P.; Mudhar, H.S.; Collarini, E.J.; Richardson, W.D. PDGF receptors in the rat CNS: During late neurogenesis, PDGF alpha-receptor expression appears to be restricted to glial cells of the oligodendrocyte lineage. Development 1992, 115, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Lin, X.H.; Giese, N.; Heldin, C.H.; Stallcup, W.B. Co-localization of NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells in the developing rat brain. J. Neurosci. Res. 1996, 43, 299–314. [Google Scholar] [CrossRef]
- Pesheva, P.; Spiess, E.; Schachner, M. J1-160 and J1-180 are oligodendrocyte-secreted nonpermissive substrates for cell adhesion. J. Cell Biol. 1989, 109, 1765–1778. [Google Scholar] [CrossRef] [PubMed]
- Haage, V.; Elmadany, N.; Roll, L.; Faissner, A.; Gutmann, D.H.; Semtner, M.; Kettenmann, H. Tenascin C regulates multiple microglial functions involving TLR4 signaling and HDAC1. Brain Behav. Immun. 2019, 81, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, R.; Sarkar, S.; Dzikowski, L.; Rawji, K.S.; Khan, L.; Faissner, A.; Bose, P.; Yong, V.W. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. Oncoimmunology 2018, 7, e1478647. [Google Scholar] [CrossRef] [Green Version]
- Manrique-Castano, D.; Dzyubenko, E.; Borbor, M.; Vasileiadou, P.; Kleinschnitz, C.; Roll, L.; Faissner, A.; Hermann, D.M. Tenascin-C preserves microglia surveillance and restricts leukocyte and, more specifically, T cell infiltration of the ischemic brain. Brain Behav. Immun. 2020, 91, 639–648. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauch, J.; Faissner, A. The Extracellular Matrix Proteins Tenascin-C and Tenascin-R Retard Oligodendrocyte Precursor Maturation and Myelin Regeneration in a Cuprizone-Induced Long-Term Demyelination Animal Model. Cells 2022, 11, 1773. https://doi.org/10.3390/cells11111773
Bauch J, Faissner A. The Extracellular Matrix Proteins Tenascin-C and Tenascin-R Retard Oligodendrocyte Precursor Maturation and Myelin Regeneration in a Cuprizone-Induced Long-Term Demyelination Animal Model. Cells. 2022; 11(11):1773. https://doi.org/10.3390/cells11111773
Chicago/Turabian StyleBauch, Juliane, and Andreas Faissner. 2022. "The Extracellular Matrix Proteins Tenascin-C and Tenascin-R Retard Oligodendrocyte Precursor Maturation and Myelin Regeneration in a Cuprizone-Induced Long-Term Demyelination Animal Model" Cells 11, no. 11: 1773. https://doi.org/10.3390/cells11111773
APA StyleBauch, J., & Faissner, A. (2022). The Extracellular Matrix Proteins Tenascin-C and Tenascin-R Retard Oligodendrocyte Precursor Maturation and Myelin Regeneration in a Cuprizone-Induced Long-Term Demyelination Animal Model. Cells, 11(11), 1773. https://doi.org/10.3390/cells11111773