
Hedgehog Signaling: Linking Embryonic Lung Development and Asthmatic Airway Remodeling

 , , , , and
, , , , and
Abstract
:1. Introduction
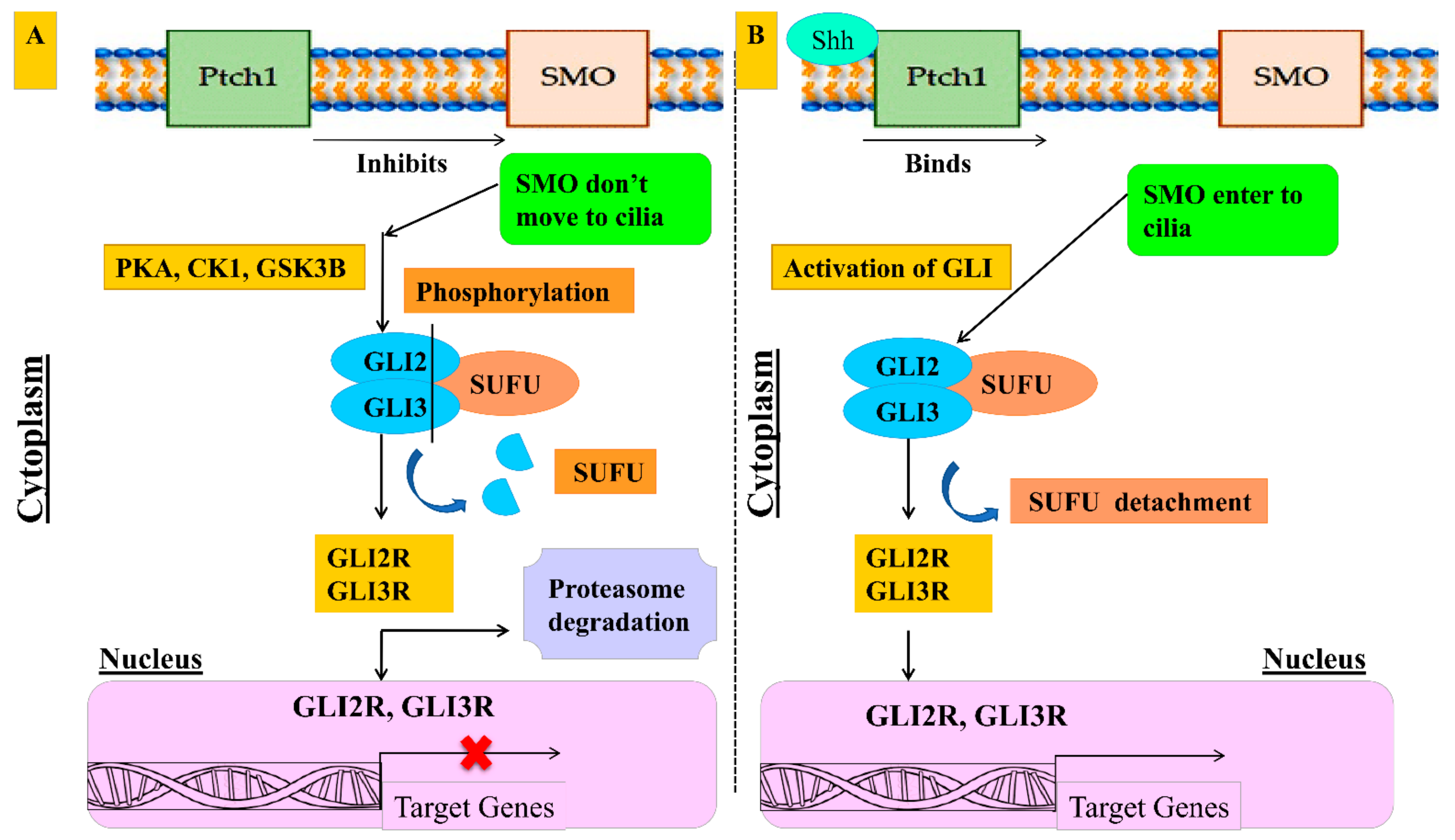
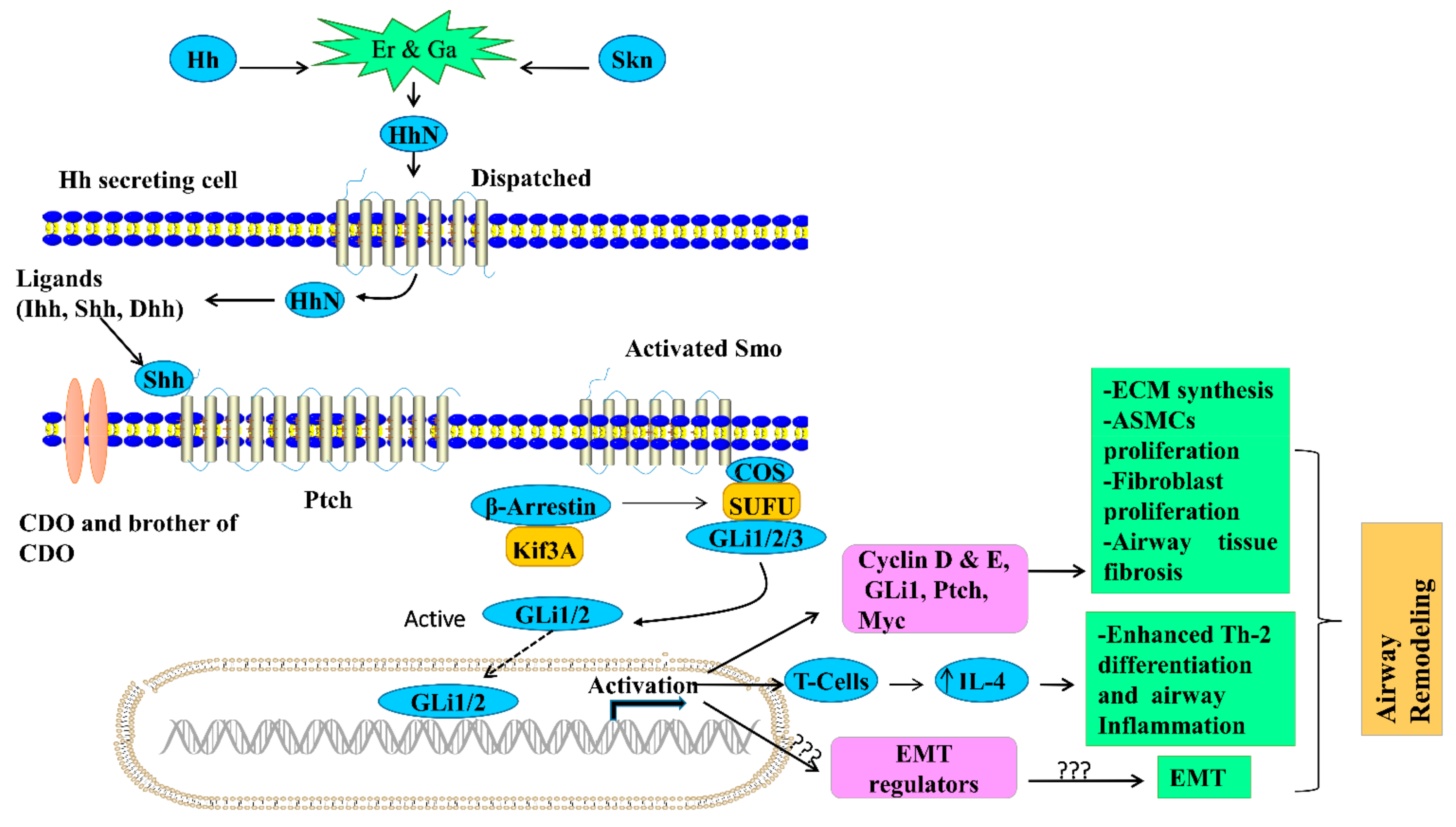
1.1. Regulation of Hedgehog Signaling
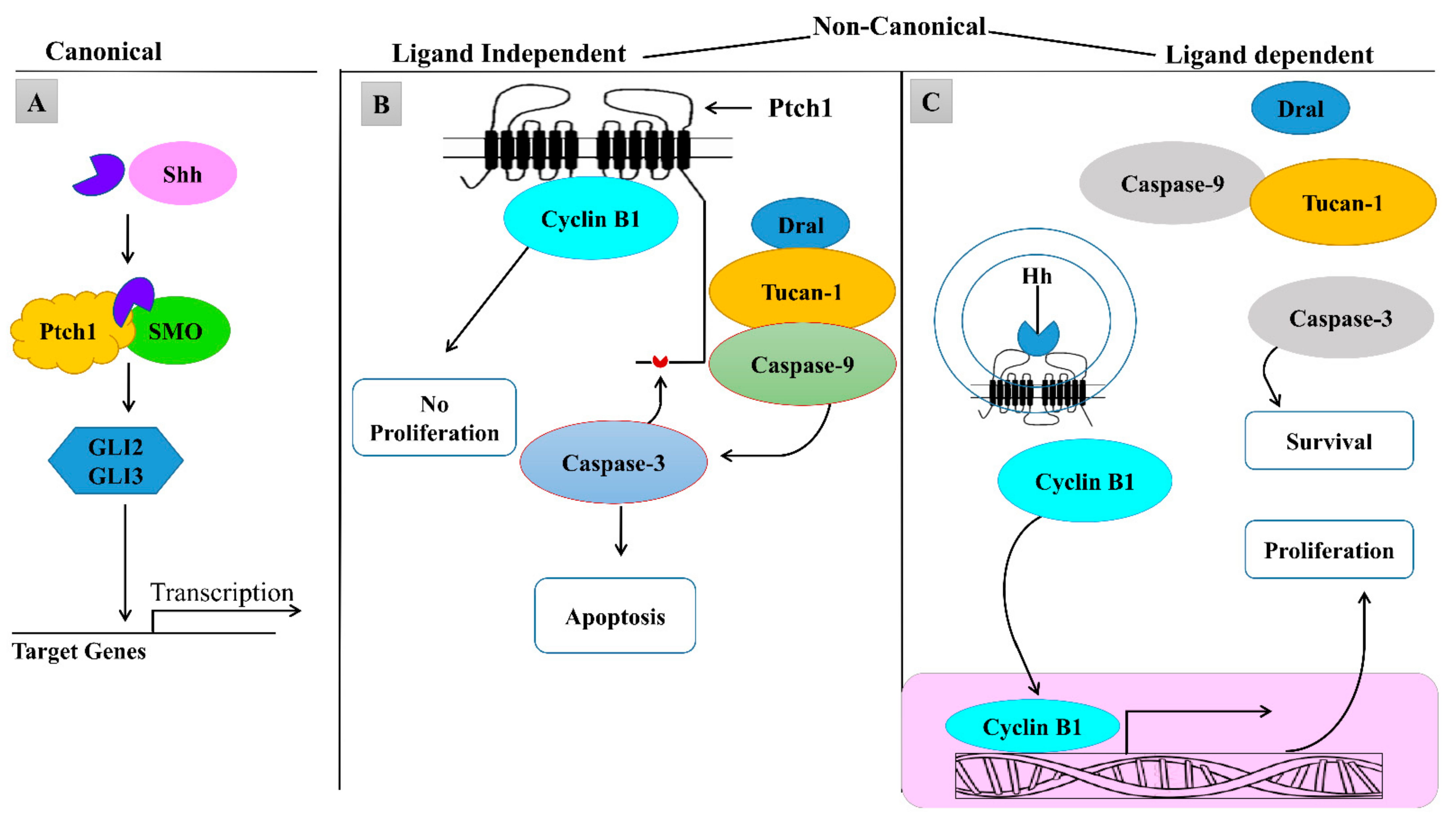
1.2. Canonical and Non-Canonical Hedgehog Signaling
2. Role of Sonic Hedgehog in Lung Development
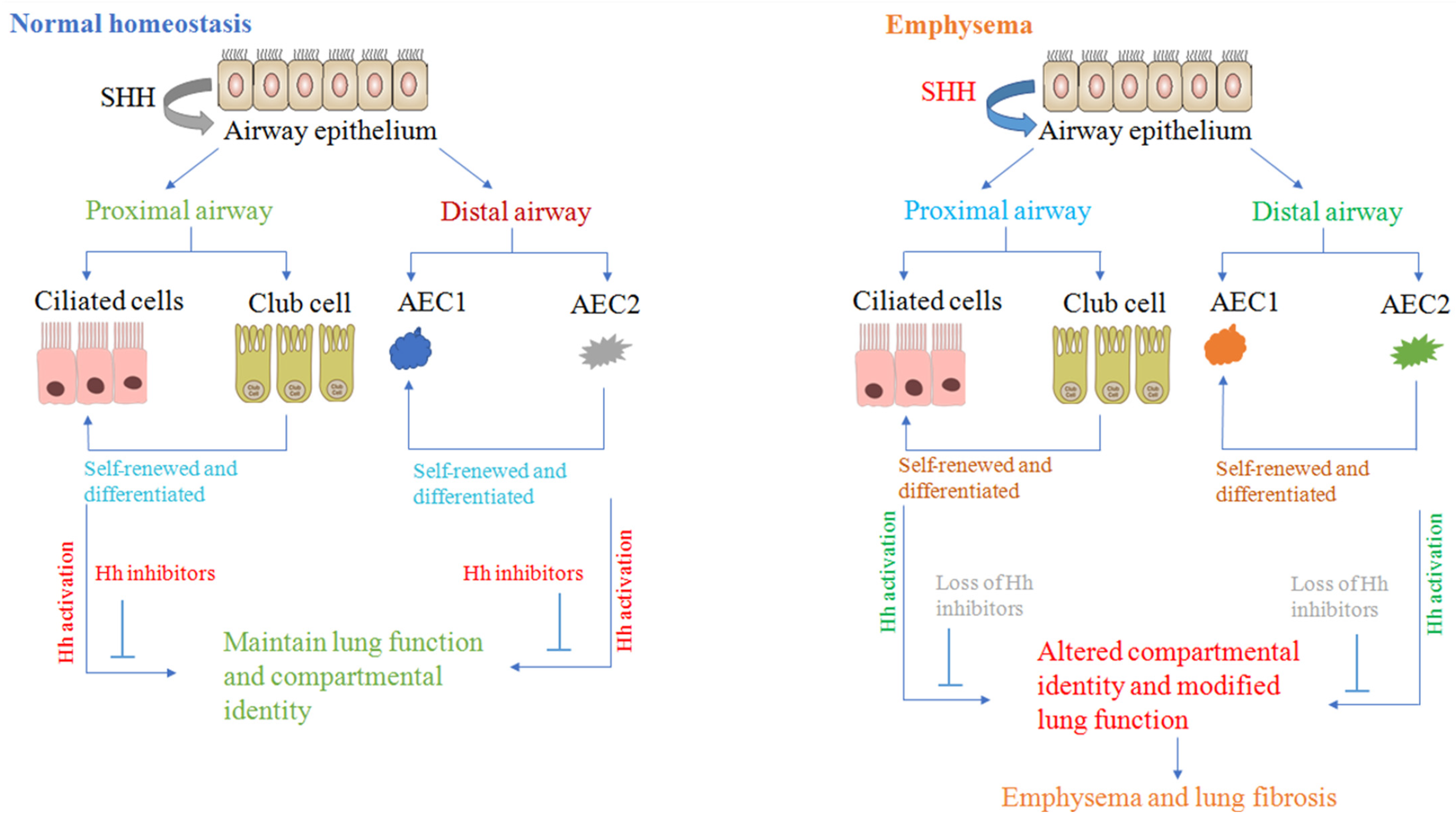
2.1. Sonic Hedgehog Regulates Fetal Lung Development and Airway Repair
2.2. Sonic Hedgehog Regulates Mesenchymal Proliferation and Branching Morphogenesis
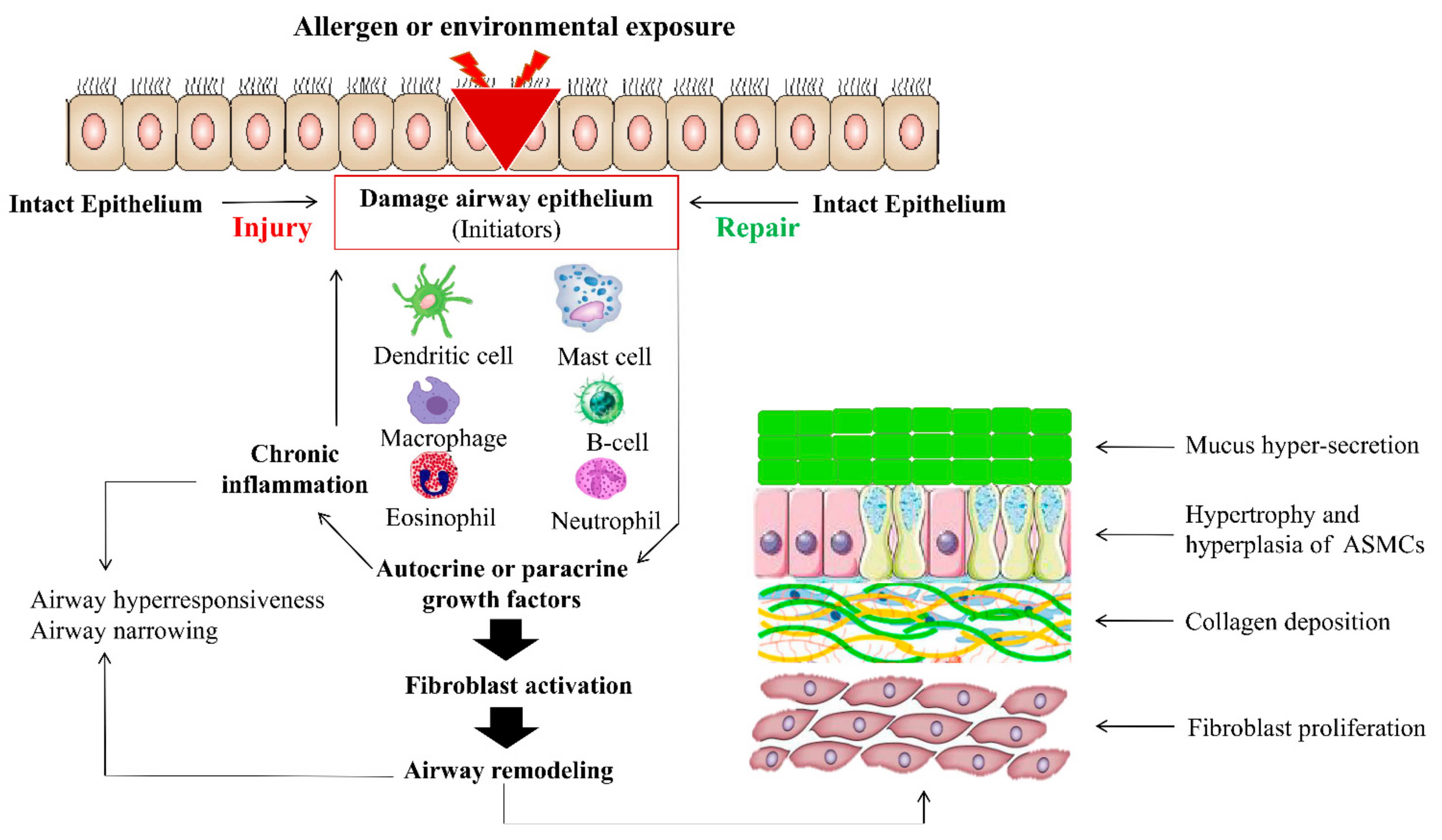
3. Role of Sonic Hedgehog in Asthmatic Airway Remodeling
3.1. Hedgehog Signaling in Tissue or Pulmonary Fibrosis
3.2. Hh Signaling in Inflammation, COPD, and Asthma
3.3. Sonic Hedgehog in ECM Production and EMT
4. Role of Hedgehog Signaling as a Pathogen Target and Used in Wound Repair, Immune System Pathway, and Immune Diseases
4.1. Hedgehog Signaling Used as a Pathogen Target
4.2. Hedgehog Signaling Regulates Wound Repair
4.3. Role of Hh Signaling in Immune System Diseases
5. Potentially Therapeutic Significance
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hogan, B.L.; Barkauskas, C.E.; Chapman, H.A.; Epstein, J.A.; Jain, R.; Hsia, C.C.; Niklason, L.; Calle, E.; Le, A.; Randell, S.H.; et al. Repair and Regeneration of the Respiratory System: Complexity, Plasticity, and Mechanisms of Lung Stem Cell Function. Cell Stem Cell 2014, 15, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Ward, H.; Nicholas, T. Alveolar type I and type II cells, Australian and New Zealand. J. Med. 1984, 14, 731–734. [Google Scholar]
- Fehrenbach, H. Alveolar epithelial type II cell: Defender of the alveolus revisited. Respir. Res. 2001, 2, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Cassandras, M.; Peng, T. The Role of Hedgehog Signaling in Adult Lung Regeneration and Maintenance. J. Dev. Biol. 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; De Mochel, N.S.R.; Christenson, S.A.; Cassandras, M.; Moon, R.; Brumwell, A.N.; Byrnes, L.E.; Li, A.; Yokosaki, Y.; Shan, P.; et al. Expansion of hedgehog disrupts mesenchymal identity and induces emphysema phenotype. J. Clin. Investig. 2018, 128, 4343–4358. [Google Scholar] [CrossRef]
- Peng, T.; Frank, D.B.; Kadzik, R.S.; Morley, M.P.; Rathi, K.; Wang, T.; Zhou, S.; Cheng, L.; Lu, M.M.; Morrisey, E.E. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature 2015, 526, 578–582. [Google Scholar] [CrossRef]
- Hussain, M.; Xu, C.; Ahmad, M.; Yang, Y.; Lu, M.; Wu, X.; Tang, L.; Wu, X. Notch Signaling: Linking Embryonic Lung Development and Asthmatic Airway Remodeling. Mol. Pharmacol. 2017, 92, 676–693. [Google Scholar] [CrossRef]
- Balzar, S.; Fajt, M.L.; Comhair, S.A.; Erzurum, S.C.; Bleecker, E.; Busse, W.W.; Castro, M.; Gaston, B.; Israel, E.; Schwartz, L.B. Mast cell phenotype, location, and activation in severe asthma: Data from the severe asthma research program. Am. J. Respir. Crit. Care Med. 2011, 183, 299–309. [Google Scholar] [CrossRef]
- Berair, R.; Saunders, R.; Brightling, C.E. Origins of increased airway smooth muscle mass in asthma. BMC Med. 2013, 11, 145. [Google Scholar] [CrossRef]
- Kim, V.; Desai, P.; Newell, J.D.; Make, B.J.; Washko, G.R.; Silverman, E.K.; Crapo, J.D.; Bhatt, S.P.; Criner, G.J. Airway wall thickness is increased in COPD patients with bronchodilator responsiveness. Respir. Res. 2014, 15, 84. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Patterson, E.B. The Role of Mitogen-Activated Protein Kinases in Asthma. Curr. Immunol. Rev. 2015, 11, 132–146. [Google Scholar] [CrossRef]
- Wang, X.; Xu, C.; Ji, J.; Cai, Y.; Shu, Y.; Chao, Y.; Wu, X.; Zou, C.; Wu, X.; Tang, L. IL-4/IL-13 upregulates Sonic hedgehog expression to induce allergic airway epithelial remodeling. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2020, 318, L888–L899. [Google Scholar] [CrossRef] [PubMed]
- James, A.L.; Elliot, J.G.; Jones, R.L.; Carroll, M.L.; Mauad, T.; Bai, T.R.; Abramson, M.; McKay, K.O.; Green, F.H. Airway Smooth Muscle Hypertrophy and Hyperplasia in Asthma. Am. J. Respir. Crit. Care Med. 2012, 185, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Brannan, J.D.P.; Lougheed, M.D.M. Airway Hyperresponsiveness in Asthma: Mechanisms, Clinical Significance, and Treatment. Front. Physiol. 2012, 3, 460. [Google Scholar] [CrossRef] [PubMed]
- Pain, M.; Bermudez, O.; Lacoste, P.; Royer, P.-J.; Botturi, K.; Tissot, A.; Brouard, S.; Eickelberg, O.; Magnan, A. Tissue remodelling in chronic bronchial diseases: From the epithelial to mesenchymal phenotype. Eur. Respir. Rev. 2014, 23, 118–130. [Google Scholar] [CrossRef]
- Harris, L.G.; Samant, R.S.; Shevde, L.A. Hedgehog Signaling: Networking to Nurture a Promalignant Tumor Microenvironment. Mol. Cancer Res. 2011, 9, 1165–1174. [Google Scholar] [CrossRef]
- Ancel, J.; Belgacemi, R.; Perotin, J.-M.; Diabasana, Z.; Dury, S.; Dewolf, M.; Bonnomet, A.; Lalun, N.; Birembaut, P.; Polette, M.; et al. Sonic hedgehog signalling as a potential endobronchial biomarker in COPD. Respir. Res. 2020, 21, 1–11. [Google Scholar] [CrossRef]
- Tong, X.; Lindemann, A.; Monteiro, A. Differential Involvement of Hedgehog Signaling in Butterfly Wing and Eyespot Development. PLoS ONE 2012, 7, e51087. [Google Scholar] [CrossRef]
- Heretsch, P.; Tzagkaroulaki, L.; Giannis, A. Modulators of the hedgehog signaling pathway. Bioorgan. Med. Chem. 2010, 18, 6613–6624. [Google Scholar] [CrossRef]
- Shin, K.; Lee, J.; Guo, N.; Kim, J.; Lim, A.; Qu, L.; Mysorekar, I.U.; Beachy, P.A. Hedgehog/Wnt feedback supports regenerative proliferation of epithelial stem cells in bladder. Nature 2011, 472, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-C.; Levine, C.M.; Zahid, S.; Wilson, E.L.; Joyner, A.L. Sonic hedgehog signals to multiple prostate stromal stem cells that replenish distinct stromal subtypes during regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 20611–20616. [Google Scholar] [CrossRef] [PubMed]
- Neureiter, D. New in Hedgehog signaling: A possible role in aging, and chronic degenerative and inflammatory diseases? BioEssays 2012, 34, 828–829. [Google Scholar] [CrossRef]
- Thayer, S.P.; Iano, M.P.d.L.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Castillo, C.F.; Yajnik, V. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Park, K.-S.; Martelotto, L.G.; Peifer, M.; Sos, M.L.; Karnezis, A.N.; Mahjoub, M.R.; Bernard, K.; Conklin, J.F.; Szczepny, A.; Yuan, J.; et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat. Med. 2011, 17, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Bolaños, A.L.; Milla, C.M.; Lira, J.C.; Ramírez, R.; Checa, M.; Barrera, L.; García-Alvarez, J.; Carbajal, V.; Becerril, C.; Gaxiola, M. Role of Sonic Hedgehog in idiopathic pulmonary fibrosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2012, 303, L978–L990. [Google Scholar] [CrossRef]
- Kugler, M.C.; Joyner, A.L.; Loomis, C.A.; Munger, J.S. Sonic hedgehog signaling in the lung. From development to disease. Am. J. Respir. Cell Mol. Biol. 2015, 52, 1–13. [Google Scholar] [CrossRef]
- Dimou, A.; Bamias, A.; Gogas, H.; Syrigos, K. Inhibition of the Hedgehog pathway in lung cancer. Lung Cancer 2019, 133, 56–61. [Google Scholar] [CrossRef]
- Kugler, M.C.; Yie, T.-A.; Cai, Y.; Berger, J.Z.; Loomis, C.A.; Munger, J. The Hedgehog target Gli1 is not required for bleomycin-induced lung fibrosis. Exp. Lung Res. 2019, 45, 22–29. [Google Scholar] [CrossRef]
- McMahon, A.P.; Ingham, P.W.; Tabin, C.J. 1 Developmental roles and clinical significance of Hedgehog signaling. Curr. Top. Dev. Biol. 2003, 53, 1–114. [Google Scholar] [CrossRef]
- Lai, L.P.; Mitchell, J. Indian hedgehog: Its roles and regulation in endochondral bone development. J. Cell. Biochem. 2005, 96, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S. Hedgehog Signaling in Skeletal Development: Roles of Indian Hedgehog and the Mode of Its Action. Int. J. Mol. Sci. 2020, 21, 6665. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, S.; Stecca, B. Cooperative integration between HEDGEHOG-GLI signalling and other oncogenic pathways: Implications for cancer therapy. Expert Rev. Mol. Med. 2015, 17, e5. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.B.; Auerbach, W.; Lee, J.S.; Stephen, D.; Joyner, A.L. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 2002, 129, 4753–4761. [Google Scholar] [CrossRef]
- Wang, B.; Li, Y. Evidence for the direct involvement of βTrCP in GLI3 protein processing. Proc. Natl. Acad. Sci. USA 2006, 103, 33–38. [Google Scholar] [CrossRef]
- Svärd, J.; Henricson, K.H.; Persson-Lek, M.; Rozell, B.; Lauth, M.; Bergström, Å.; Ericson, J.; Toftgård, R.; Teglund, S. Genetic Elimination of Suppressor of Fused Reveals an Essential Repressor Function in the Mammalian Hedgehog Signaling Pathway. Dev. Cell 2006, 10, 187–197. [Google Scholar] [CrossRef]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef]
- Hu, L.; Lin, X.; Lu, H.; Chen, B.; Bai, Y. An Overview of Hedgehog Signaling in Fibrosis. Mol. Pharmacol. 2014, 87, 174–182. [Google Scholar] [CrossRef]
- Zou, Y.; Song, W.; Zhou, L.; Mao, Y.; Hong, W. House dust mite induces Sonic hedgehog signaling that mediates epithelial-mesenchymal transition in human bronchial epithelial cells. Mol. Med. Rep. 2019, 20, 4674–4682. [Google Scholar] [CrossRef]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog Signal Transduction Network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef]
- Miller, L.-A.D.; Wert, S.E.; Whitsett, J.A. Immunolocalization of Sonic Hedgehog (Shh) in Developing Mouse Lung. J. Histochem. Cytochem. 2001, 49, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Morrisey, E.E.; Hogan, B.L. Preparing for the First Breath: Genetic and Cellular Mechanisms in Lung Development. Dev. Cell 2010, 18, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, H.; Teng, H.; Shi, J.; Zhang, Y. Expression of SHH signaling pathway components in the developing human lung. Histochem. Cell Biol. 2010, 134, 327–335. [Google Scholar] [CrossRef]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.; Palumbo, K.; Cordazzo, C.; Dees, C.; Akhmetshina, A.; Tomcik, M.; Zerr, P.; Avouac, J.; Gusinde, J.; Zwerina, J.; et al. Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis. Arthritis Care Res. 2012, 64, 2724–2733. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A.; Kaminski, N. Idiopathic Pulmonary Fibrosis: Aberrant Recapitulation of Developmental Programs? PLoS Med. 2008, 5, e62. [Google Scholar] [CrossRef] [PubMed]
- Fitch, P.M.; Howie, S.E.M.; Wallace, W.A.H. Oxidative damage and TGF-β differentially induce lung epithelial cell sonic hedgehog and tenascin-C expression: Implications for the regulation of lung remodelling in idiopathic interstitial lung disease. Int. J. Exp. Pathol. 2010, 92, 8–17. [Google Scholar] [CrossRef]
- Cigna, N.; Moshai, E.F.; Brayer, S.; Marchal-Somme, J.; Wémeau-Stervinou, L.; Fabre, A.; Mal, H.; Lesèche, G.; Dehoux, M.; Soler, P. The hedgehog system machinery controls transforming growth factor-β–dependent myofibroblastic differentiation in humans: Involvement in idiopathic pulmonary fibrosis. Am. J. Pathol. 2012, 181, 2126–2137. [Google Scholar] [CrossRef]
- Liu, J.A.-J.; Ngan, E.S.-W. Hedgehog and Notch Signaling in Enteric Nervous System Development. Neurosignals 2014, 22, 1–13. [Google Scholar] [CrossRef]
- Fisher, C.E.; Ahmad, S.A.; Fitch, P.M.; Lamb, J.R.; Howie, S.E. FITC-induced murine pulmonary inflammation: CC10 up-regulation and concurrent Shh expression. Cell Biol. Int. 2005, 29, 868–876. [Google Scholar] [CrossRef]
- Krause, A.; Xu, Y.; Joh, J.; Hubner, R.; Gess, A.; Ilic, T.; Worgall, S. Overexpression of Sonic Hedgehog in the Lung Mimics the Effect of Lung Injury and Compensatory Lung Growth on Pulmonary Sca-1 and CD34 Positive Cells. Mol. Ther. 2010, 18, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kugler, M.C.; Loomis, C.A.; Samdani, R.; Zhao, Z.; Chen, G.J.; Brandt, J.P.; Brownell, I.; Joyner, A.L.; Rom, W.N. Hedgehog signaling in neonatal and adult lung. Am. J. Respir. Cell Mol. Biol. 2013, 48, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Litingtung, Y.; Lei, L.; Westphal, H.; Chiang, C. Sonic hedgehog is essential to foregut development. Nat. Genet. 1998, 20, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.-A.D.; Wert, S.E.; Clark, J.C.; Xu, Y.; Perl, A.-K.T.; Whitsett, J.A. Role ofSonic hedgehog in patterning of tracheal-bronchial cartilage and the peripheral lung. Dev. Dyn. 2004, 231, 57–71. [Google Scholar] [CrossRef]
- Weaver, M.; Batts, L.; Hogan, B.L. Tissue interactions pattern the mesenchyme of the embryonic mouse lung. Dev. Biol. 2003, 258, 169–184. [Google Scholar] [CrossRef]
- Dixit, R.; Ai, X.; Fine, A. Derivation of lung mesenchymal lineages from the fetal mesothelium requires hedgehog signaling for mesothelial cell entry. Development 2013, 140, 4398–4406. [Google Scholar] [CrossRef]
- Wang, K.; Pan, L.; Che, X.; Cui, D.; Li, C. Sonic Hedgehog/GLI1 signaling pathway inhibition restricts cell migration and invasion in human gliomas. Neurol. Res. 2010, 32, 975–980. [Google Scholar] [CrossRef]
- Park, H.; Bai, C.; Platt, K.; Matise, M.; Beeghly, A.; Hui, C.; Nakashima, M.; Joyner, A. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development 2000, 127, 1593–1605. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, H.; Choi, S.C.; Litingtung, Y.; Chiang, C. Sonic hedgehog signaling regulates GLI3 processing, mesenchymal proliferation, and differentiation during mouse lung organogenesis. Dev. Biol. 2004, 270, 214–231. [Google Scholar] [CrossRef]
- Lin, C.; Chen, M.-H.; Yao, E.; Song, H.; Gacayan, R.; Hui, C.-C.; Chuang, P.-T. Differential regulation of Gli proteins by Sufu in the lung affects PDGF signaling and myofibroblast development. Dev. Biol. 2014, 392, 324–333. [Google Scholar] [CrossRef]
- Chuang, P.-T.; Kawcak, T.; McMahon, A.P. Feedback control of mammalian Hedgehog signaling by the Hedgehog-binding protein, Hip1, modulates Fgf signaling during branching morphogenesis of the lung. Genes Dev. 2003, 17, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Pepicelli, C.V.; Lewis, P.M.; McMahon, A.P. Sonic hedgehog regulates branching morphogenesis in the mammalian lung. Curr. Biol. 1998, 8, 1083–1086. [Google Scholar] [CrossRef]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, L.V.; Milenkovicć, L.; Higgins, K.M.; Scott, M.P. Altered Neural Cell Fates and Medulloblastoma in Mouse patched Mutants. Science 1997, 277, 1109–1113. [Google Scholar] [CrossRef]
- Motoyama, J.; Liu, J.; Mo, R.; Ding, Q.; Post, M.; Hui, C.-C. Essential function of Gli2 and Gli3 in the formation of lung, trachea and oesophagus. Nat. Genet. 1998, 20, 54–57. [Google Scholar] [CrossRef]
- Grindley, J.C.; Bellusci, S.; Perkins, D.; Hogan, B.L. Evidence for the Involvement of the Gli Gene Family in Embryonic Mouse Lung Development. Dev. Biol. 1997, 188, 337–348. [Google Scholar] [CrossRef]
- Bellusci, S.; Grindley, J.; Emoto, H.; Itoh, N.; Hogan, B. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development 1997, 124, 4867–4878. [Google Scholar] [CrossRef]
- Stewart, G.A.; Hoyne, G.; Ahmad, S.A.; Jarman, E.; Wallace, W.A.H.; Harrison, D.; Haslett, C.; Lamb, J.R.; Howie, S.E.M. Expression of the developmental Sonic hedgehog (Shh) signalling pathway is up-regulated in chronic lung fibrosis and the Shh receptor patched 1 is present in circulating T lymphocytes. J. Pathol. 2003, 199, 488–495. [Google Scholar] [CrossRef]
- Omenetti, A.; Porrello, A.; Jung, Y.; Yang, L.; Popov, Y.; Choi, S.S.; Witek, R.P.; Alpini, G.; Venter, J.; Vandongen, H.M.; et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J. Clin. Investig. 2008, 118, 3331–3342. [Google Scholar] [CrossRef]
- Fabian, S.L.; Penchev, R.R.; St-Jacques, B.; Rao, A.N.; Sipilä, P.; West, K.A.; McMahon, A.P.; Humphreys, B.D. Hedgehog-Gli Pathway Activation during Kidney Fibrosis. Am. J. Pathol. 2012, 180, 1441–1453. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.E.; Eickelberg, O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012, 380, 680–688. [Google Scholar] [CrossRef]
- Coon, D.R.; Roberts, D.J.; Loscertales, M.; Kradin, R. Differential epithelial expression of SHH and FOXF1 in usual and nonspecific interstitial pneumonia. Exp. Mol. Pathol. 2006, 80, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Howard, T.D.; Moore, W.C.; Ampleford, E.J.; Li, H.; Busse, W.W.; Calhoun, W.J.; Castro, M.; Chung, K.F.; Erzurum, S.C.; et al. Importance of hedgehog interacting protein and other lung function genes in asthma. J. Allergy Clin. Immunol. 2011, 127, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Baron, R.M.; Hardin, M.; Cho, M.H.; Zielinski, J.; Hawrylkiewicz, I.; Sliwinski, P.; Hersh, C.P.; Mancini, J.D.; Lu, K.; et al. Identification of a chronic obstructive pulmonary disease genetic determinant that regulates HHIP. Hum. Mol. Genet. 2011, 21, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Wilk, J.B.; Chen, T.-H.; Gottlieb, D.J.; Walter, R.; Nagle, M.; Brandler, B.J.; Myers, R.; Borecki, I.B.; Silverman, E.K.; Weiss, S.T.; et al. A Genome-Wide Association Study of Pulmonary Function Measures in the Framingham Heart Study. PLoS Genet. 2009, 5, e1000429. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.A. Epithelial-Mesenchymal Interactions in Pulmonary Fibrosis. Annu. Rev. Physiol. 2011, 73, 413–435. [Google Scholar] [CrossRef]
- Moeller, A.; Ask, K.; Warburton, D.; Gauldie, J.; Kolb, M. The bleomycin animal model: A useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem. Cell Biol. 2008, 40, 362–382. [Google Scholar] [CrossRef]
- Moshai, E.F.; Wémeau-Stervinou, L.; Cigna, N.; Brayer, S.; Sommé, J.M.; Crestani, B.; Mailleux, A.A. Targeting the Hedgehog–Glioma-Associated Oncogene Homolog Pathway Inhibits Bleomycin-Induced Lung Fibrosis in Mice. Am. J. Respir. Cell Mol. Biol. 2014, 51, 11–25. [Google Scholar] [CrossRef]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M.; NASH CRN. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar] [CrossRef]
- Choi, S.S.; Bradrick, S.; Qiang, G.; Mostafavi, A.; Chaturvedi, G.; Weinman, S.A.; Diehl, A.M.; Jhaveri, R. Up-regulation of Hedgehog pathway is associated with cellular permissiveness for hepatitis C virus replication. Hepatology 2011, 54, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, B.; Syn, W.-K.; Delgado, I.; Karaca, G.F.; Jung, Y.; Wang, J.; Zubiaga, A.M.; Fresnedo, O.; Omenetti, A.; Zdanowicz, M.; et al. Hedgehog signaling is critical for normal liver regeneration after partial hepatectomy in mice. Hepatology 2010, 51, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.-K.; Choi, S.S.; Liaskou, E.; Karaca, G.F.; Agboola, K.M.; Oo, Y.H.; Mi, Z.; Pereira, T.A.; Zdanowicz, M.; Malladi, P.; et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2010, 53, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Smelkinson, M.G. The Hedgehog Signaling Pathway Emerges as a Pathogenic Target. J. Dev. Biol. 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Zhou, D.; Hao, S.; Zhou, L.; He, W.; Nie, J.; Hou, F.F.; Liu, Y. Sonic Hedgehog Signaling Mediates Epithelial–Mesenchymal Communication and Promotes Renal Fibrosis. J. Am. Soc. Nephrol. 2012, 23, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.-I.; Yánez, D.C.; Papaioannou, E.; Ross, S.; Crompton, T. Sonic Hedgehog Signalling in the Regulation of Barrier Tissue Homeostasis and Inflammation. FEBS J. 2021, 1, 1–22. [Google Scholar] [CrossRef]
- Furmanski, A.L.; Saldana, J.I.; Ono, M.; Sahni, H.; Paschalidis, N.; D’Acquisto, F.; Crompton, T. Tissue-Derived Hedgehog Proteins Modulate Th Differentiation and Disease. J. Immunol. 2013, 190, 2641–2649. [Google Scholar] [CrossRef]
- Elias, J.A.; Lee, C.G.; Zheng, T.; Ma, B.; Homer, R.; Zhu, Z. New insights into the pathogenesis of asthma. J. Clin. Investig. 2003, 111, 291–297. [Google Scholar] [CrossRef]
- Hogg, J.C.; Timens, W. The Pathology of Chronic Obstructive Pulmonary Disease. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 435–459. [Google Scholar] [CrossRef]
- Holgate, S.T.; Wenzel, S.; Postma, D.S.; Weiss, S.T.; Renz, H.; Sly, P. Asthma. Nat. Rev.-Dis. Primers 2015, 1, 15025. [Google Scholar] [CrossRef]
- Wang, X.-Z.; Zhang, H.-H.; Qian, Y.-L.; Tang, L.-F. Sonic Hedgehog (Shh) and CC Chemokine Ligand 2 Signaling Pathways in Asthma. J. Chin. Med. Assoc. 2019, 82, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Michel, K.D.; Uhmann, A.; Dressel, R.; van den Brandt, J.; Hahn, H.; Reichardt, H.M. The Hedgehog Receptor Patched1 in T Cells Is Dispensable for Adaptive Immunity in Mice. PLoS ONE 2013, 8, e61034. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Wang, X.; Zhang, Y.; Hao, J.; Xing, C.; Chu, Q.; Wang, G.; Zhao, J.; Wang, J.; Dong, Q.; et al. Interleukin-20 Promotes Airway Remodeling in Asthma. Inflammation 2014, 37, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Bai, C.; Yang, J.; Lou, G.; Li, Q.; Chen, R. Effect of mesenchymal stem cells on inhibiting airway remodeling and airway inflammation in chronic asthma. J. Cell. Biochem. 2013, 114, 1595–1605. [Google Scholar] [CrossRef]
- Radzikinas, K.; Aven, L.; Jiang, Z.; Tran, T.; Paez-Cortez, J.; Boppidi, K.; Lu, J.; Fine, A.; Ai, X. A Shh/miR-206/BDNF cascade coordinates innervation and formation of airway smooth muscle. J. Neurosci. 2011, 31, 15407–15415. [Google Scholar] [CrossRef]
- Thompson, M.A.; Britt, R.D.; Kuipers, I.; Stewart, A.; Thu, J.; Pandya, H.C.; MacFarlane, P.; Pabelick, C.M.; Martin, R.J.; Prakash, Y. cAMP-mediated secretion of brain-derived neurotrophic factor in developing airway smooth muscle. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 2506–2514. [Google Scholar] [CrossRef]
- Aravamudan, B.; Thompson, M.; Pabelick, C.; Prakash, Y.S. Brain-derived neurotrophic factor induces proliferation of human airway smooth muscle cells. J. Cell. Mol. Med. 2012, 16, 812–823. [Google Scholar] [CrossRef]
- De la Roche, M.; Ritter, A.T.; Angus, K.L.; Dinsmore, C.; Earnshaw, C.H.; Reiter, J.F.; Griffiths, G.M. Hedgehog Signaling Controls T Cell Killing at the Immunological Synapse. Science 2013, 342, 1247–1250. [Google Scholar] [CrossRef]
- Wang, Y.; Davidow, L.; Arvanites, A.C.; Blanchard, J.; Lam, K.; Xu, K.; Oza, V.; Yoo, J.W.; Ng, J.M.; Curran, T.; et al. Glucocorticoid Compounds Modify Smoothened Localization and Hedgehog Pathway Activity. Chem. Biol. 2012, 19, 972–982. [Google Scholar] [CrossRef]
- Burgess, J.; Ge, Q.; Poniris, M.H.; Boustany, S.; Twigg, S.M.; Black, J.L.; Johnson, P.R.A. Connective tissue growth factor and vascular endothelial growth factor from airway smooth muscle interact with the extracellular matrix. Am. J. Physiol. Cell. Mol. Physiol. 2006, 290, L153–L161. [Google Scholar] [CrossRef]
- Bermudez, O.; Hennen, E.; Koch, I.; Lindner, M.; Eickelberg, O. Gli1 Mediates Lung Cancer Cell Proliferation and Sonic Hedgehog-Dependent Mesenchymal Cell Activation. PLoS ONE 2013, 8, e63226. [Google Scholar] [CrossRef] [PubMed]
- Royce, S.G.; Tan, L.; Koek, A.A.; Tang, M.L. Effect of extracellular matrix composition on airway epithelial cell and fibroblast structure: Implications for airway remodeling in asthma. Ann. Allergy Asthma Immunol. 2009, 102, 238–246. [Google Scholar] [CrossRef]
- Rogers, N.K.; Clements, D.; Dongre, A.; Harrison, T.W.; Shaw, D.; Johnson, S.R. Extra-Cellular Matrix Proteins Induce Matrix Metalloproteinase-1 (MMP-1) Activity and Increase Airway Smooth Muscle Contraction in Asthma. PLoS ONE 2014, 9, e90565. [Google Scholar] [CrossRef]
- Dennler, S.; André, J.; Verrecchia, F.; Mauviel, A. Cloning of the Human GLI2 Promoter Transcriptional Activation by Transforming Growth Factor-Β via Smad3/Β-Catenin Cooperation. J. Biol. Chem. 2009, 284, 31523–31531. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Pierrat, M.-J.; Mauviel, A. Crosstalk between TGF-β and hedgehog signaling in cancer. FEBS Lett. 2012, 586, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Ijaz, T.; Pazdrak, K.; Kalita, M.; Konig, R.; Choudhary, S.; Tian, B.; Boldogh, I.; Brasier, A. Systems biology approaches to understanding Epithelial Mesenchymal Transition (EMT) in mucosal remodeling and signaling in asthma. World Allergy Organ. J. 2014, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hackett, T.-L. Epithelial–mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 53–59. [Google Scholar] [CrossRef]
- Borthwick, L.A.; Parker, S.M.; Brougham, K.A.; Johnson, G.E.; Gorowiec, M.R.; Ward, C.; Lordan, J.L.; Corris, P.A.; Kirby, J.A.; Fisher, A.J. Epithelial to mesenchymal transition (EMT) and airway remodelling after human lung transplantation. Thorax 2009, 64, 770–777. [Google Scholar] [CrossRef]
- Horn, A.; Kireva, T.; Palumbo-Zerr, K.; Dees, C.; Tomcik, M.; Cordazzo, C.; Zerr, P.; Akhmetshina, A.; Ruat, M.; Distler, O.; et al. Inhibition of hedgehog signalling prevents experimental fibrosis and induces regression of established fibrosis. Ann. Rheum. Dis. 2012, 71, 785–789. [Google Scholar] [CrossRef]
- Caldwell, S.; Park, S.H. The epidemiology of hepatocellular cancer: From the perspectives of public health problem to tumor biology. J. Gastroenterol. 2009, 44, 96–101. [Google Scholar] [CrossRef]
- Pereira, T.D.A.; Witek, R.P.; Syn, W.-K.; Choi, S.S.; Bradrick, S.; Karaca, G.F.; Agboola, K.M.; Jung, Y.; Omenetti, A.; Moylan, C.A.; et al. Viral factors induce Hedgehog pathway activation in humans with viral hepatitis, cirrhosis, and hepatocellular carcinoma. Lab. Investig. 2010, 90, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Zompetta, C.; Vescarelli, E.; Rizzello, C.; Cardi, A.; Valia, S.; Antonelli, G.; Marchese, C.; Torrisi, M.R.; Faggioni, A.; et al. HCV derived from sera of HCV-infected patients induces pro-fibrotic effects in human primary fibroblasts by activating GLI. Sci. Rep. 2016, 6, 30649. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Cho, H.K.; Hong, S.P.; Cheong, J. Hepatitis B virus X protein stimulates the Hedgehog–Gli activation through protein stabilization and nuclear localization of Gli1 in liver cancer cells. Cancer Lett. 2011, 309, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Port, R.J.; Pinheiro-Maia, S.; Hu, C.; Arrand, J.R.; Wei, W.; Young, L.S.; Dawson, C.W. Epstein-Barr virus induction of the Hedgehog signalling pathway imposes a stem cell phenotype on human epithelial cells. J. Pathol. 2013, 231, 367–377. [Google Scholar] [CrossRef]
- Pal, A.D.; Banerjee, S. Epstein–Barr virus latent membrane protein 2A mediated activation of Sonic Hedgehog pathway induces HLA class Ia downregulation in gastric cancer cells. Virology 2015, 484, 22–32. [Google Scholar] [CrossRef]
- Fuccio, L.; Eusebi, L.H.; Bazzoli, F. Gastric cancer, Helicobacter pylori infection and other risk factors. World J. Gastrointest. Oncol. 2010, 2, 342. [Google Scholar] [CrossRef]
- Schumacher, M.A.; Donnelly, J.M.; Engevik, A.C.; Xiao, C.; Yang, L.; Kenny, S.; Varro, A.; Hollande, F.; Samuelson, L.C.; Zavros, Y. Gastric Sonic Hedgehog Acts as a Macrophage Chemoattractant During the Immune Response to Helicobacter pylori. Gastroenterology 2012, 142, 1150–1159.e6. [Google Scholar] [CrossRef]
- Mutoh, H.; Hayakawa, H.; Sashikawa, M.; Sakamoto, H.; Sugano, K. Direct repression of Sonic Hedgehog expression in the stomach by Cdx2 leads to intestinal transformation. Biochem. J. 2010, 427, 423–434. [Google Scholar] [CrossRef]
- Barros, R.; Marcos, N.; Reis, C.A.; de Luca, A.; David, L.; Almeida, R. CDX2 expression is induced by Helicobacter pylori in AGS cells. Scand. J. Gastroenterol. 2009, 44, 124–125. [Google Scholar] [CrossRef]
- Matsuda, K.; Yamauchi, K.; Matsumoto, T.; Sano, K.; Yamaoka, Y.; Ota, H. Quantitative analysis of the effect of Helicobacter pylori on the expressions of SOX2, CDX2, MUC2, MUC5AC, MUC6, TFF1, TFF2, and TFF3 mRNAs in human gastric carcinoma cells. Scand. J. Gastroenterol. 2008, 43, 25–33. [Google Scholar] [CrossRef]
- Zheng, L.-L.; Li, C.; Ping, J.; Zhou, Y.; Li, Y.; Hao, P. The Domain Landscape of Virus-Host Interactomes. BioMed Res. Int. 2014, 2014, 867235. [Google Scholar] [CrossRef] [PubMed]
- Pogach, M.S.; Cao, Y.; Millien, G.; Ramirez, M.I.; Williams, M.C. Key developmental regulators change during hyperoxia-induced injury and recovery in adult mouse lung. J. Cell. Biochem. 2006, 100, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Asai, J.; Takenaka, H.; Kusano, K.F.; Ii, M.; Luedemann, C.; Curry, C.; Eaton, E.; Iwakura, A.; Tsutsumi, Y.; Hamada, H.; et al. Topical Sonic Hedgehog Gene Therapy Accelerates Wound Healing in Diabetes by Enhancing Endothelial Progenitor Cell–Mediated Microvascular Remodeling. Circulation 2006, 113, 2413–2424. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef]
- Luo, J.-D.; Hu, T.-P.; Wang, L.; Chen, M.-S.; Liu, S.-M.; Chen, A.F. Sonic hedgehog improves delayed wound healing via enhancing cutaneous nitric oxide function in diabetes. Am. J. Physiol. Metab. 2009, 297, E525–E531. [Google Scholar] [CrossRef]
- Le, H.; Kleinerman, R.; Lerman, O.Z.; Brown, D.; Galiano, R.; Gurtner, G.C.; Warren, S.M.; Levine, J.P.; Saadeh, P.B. Hedgehog signaling is essential for normal wound healing. Wound Repair Regen. 2008, 16, 768–773. [Google Scholar] [CrossRef]
- Lowrey, J.A.; Stewart, G.A.; Lindey, S.; Hoyne, G.F.; Dallman, M.J.; Howie, S.E.M.; Lamb, J.R. Sonic hedgehog promotes cell cycle progression in activated peripheral CD4(+) T lymphocytes. J. Immunol. 2002, 169, 1869–1875. [Google Scholar] [CrossRef]
- Stewart, G.A.; Lowrey, J.A.; Wakelin, S.J.; Fitch, P.M.; Lindey, S.; Dallman, M.J.; Lamb, J.R.; Howie, S.E.M. Sonic Hedgehog Signaling Modulates Activation of and Cytokine Production by Human Peripheral CD4+ T Cells. J. Immunol. 2002, 169, 5451–5457. [Google Scholar] [CrossRef]
- Lee, K.-A.; Kim, B.; Bhin, J.; Kim, D.H.; You, H.; Kim, E.-K.; Kim, S.-H.; Ryu, J.-H.; Hwang, D.; Lee, W.-J. Bacterial Uracil Modulates Drosophila DUOX-Dependent Gut Immunity via Hedgehog-Induced Signaling Endosomes. Cell Host Microbe 2015, 17, 191–204. [Google Scholar] [CrossRef]
- Bier, E.; Nizet, V. Hedgehog: Linking Uracil to Innate Defense. Cell Host Microbe 2015, 17, 146–148. [Google Scholar] [CrossRef]
- Lee, K.-A.; Kim, B.; You, H.; Lee, W.-J. Uracil-induced signaling pathways for DUOX-dependent gut immunity. Fly 2015, 9, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Crompton, T.; Outram, S.V.; Hager-Theodorides, A.L. Sonic hedgehog signalling in T-cell development and activation. Nat. Rev. Immunol. 2007, 7, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Borowski, C.; Martin, C.; Gounari, F.; Haughn, L.; Aifantis, I.; Grassi, F.; von Boehmer, H. On the brink of becoming a T cell. Curr. Opin. Immunol. 2002, 14, 200–206. [Google Scholar] [CrossRef]
- Wakelin, S.J.; Forsythe, J.L.; Garden, O.J.; Howie, S.E. Commercially available recombinant sonic hedgehog up-regulates Ptc and modulates the cytokine and chemokine expression of human macrophages: An effect mediated by endotoxin contamination? Immunobiology 2008, 213, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Marwaha, S.; Schumacher, M.A.; Zavros, Y.; Eghbalnia, H.R. Crosstalks between Cytokines and Sonic Hedgehog in Helicobacter pylori Infection: A Mathematical Model. PLoS ONE 2014, 9, e111338. [Google Scholar] [CrossRef]
- Nhu, Q.M.; Aceves, S.S. Tissue Remodeling in Chronic Eosinophilic Esophageal Inflammation: Parallels in Asthma and Therapeutic Perspectives. Front. Med. 2017, 4, 128. [Google Scholar] [CrossRef]
- Smelkinson, M.G.; Guichard, A.; Teijaro, J.R.; Malur, M.; Loureiro, M.E.; Jain, P.; Ganesan, S.; Zúñiga, E.I.; Krug, R.M.; Oldstone, M.B.; et al. Influenza NS1 directly modulates Hedgehog signaling during infection. PLoS Pathog. 2017, 13, e1006588. [Google Scholar] [CrossRef]
- Dagklis, A.; Pauwels, D.; Lahortiga, I.; Geerdens, E.; Bittoun, E.; Cauwelier, B.; Tousseyn, T.; Uyttebroeck, A.; Maertens, J.; Verhoef, G.; et al. Hedgehog pathway mutations in T-cell acute lymphoblastic leukemia. Haematologica 2014, 100, e102–e105. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, L.; Polverino, F.; Guo, F.; Hao, Y.; Lao, T.; Xu, S.; Li, L.; Pham, B.; Owen, C.A.; et al. Hedgehog interacting protein (HHIP) represses airway remodeling and metabolic reprogramming in COPD-derived airway smooth muscle cells. Sci. Rep. 2021, 11, 9074. [Google Scholar] [CrossRef]
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma Growth Inhibition by Hedgehog Pathway Blockade. Science 2002, 297, 1559–1561. [Google Scholar] [CrossRef]
- Heretsch, P.; Tzagkaroulaki, L.; Giannis, A. Cyclopamine and Hedgehog Signaling: Chemistry, Biology, Medical Perspectives. Angew. Chem. Int. Ed. 2010, 49, 3418–3427. [Google Scholar] [CrossRef] [PubMed]
- Shindo, N.; Sakai, A.; Arai, D.; Matsuoka, O.; Yamasaki, Y.; Higashinakagawa, T. The ESC–E(Z) complex participates in the hedgehog signaling pathway. Biochem. Biophys. Res. Commun. 2005, 327, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.Z. Synthetic miRNAs targeting the GLI-1 transcription factor inhibit division and induce apoptosis in pancreatic tumor cells. Cancer Res. 2006, 66, 639. [Google Scholar]
- Belgacemi, R.; Luczka, E.; Ancel, J.; Diabasana, Z.; Perotin, J.-M.; Germain, A.; Lalun, N.; Birembaut, P.; Dubernard, X.; Mérol, J.-C. Airway epithelial cell differentiation relies on deficient Hedgehog signalling in COPD. EBioMedicine 2020, 51, 102572. [Google Scholar] [CrossRef]
- Yánez, D.C.; Papaioannou, E.; Chawda, M.M.; Rowell, J.; Ross, S.; Lau, C.-I.; Crompton, T. Systemic Pharmacological Smoothened Inhibition Reduces Lung T-Cell Infiltration and Ameliorates Th2 Inflammation in a Mouse Model of Allergic Airway Disease. Front. Immunol. 2021, 12, 3653. [Google Scholar] [CrossRef]
- Königshoff, M.; Balsara, N.; Pfaff, E.-M.; Kramer, M.; Chrobak, I.; Seeger, W.; Eickelberg, O. Functional Wnt Signaling Is Increased in Idiopathic Pulmonary Fibrosis. PLoS ONE 2008, 3, e2142. [Google Scholar] [CrossRef]
- Mucenski, M.L.; Nation, J.M.; Thitoff, A.R.; Besnard, V.; Xu, Y.; Wert, S.E.; Harada, N.; Taketo, M.M.; Stahlman, M.T.; Whitsett, J.A. β-Catenin regulates differentiation of respiratory epithelial cells in vivo. Am. J. Physiol. Cell. Mol. Physiol. 2005, 289, L971–L979. [Google Scholar] [CrossRef]
- Sun, B.-C.; Zhao, X.-L.; Zhang, S.-W.; Liu, Y.-X.; Wang, L.; Wang, X. Sulindac induces apoptosis and protects against colon carcinoma in mice. World J. Gastroenterol. 2005, 11, 2822–2826. [Google Scholar] [CrossRef]
- Lan, X.; Wen, H.; Cheng, K.; Plagov, A.; Shoshtari, S.S.M.; Malhotra, A.; Singhal, P.C. Hedgehog pathway plays a vital role in HIV-induced epithelial-mesenchymal transition of podocyte. Exp. Cell Res. 2017, 352, 193–201. [Google Scholar] [CrossRef]
- Singh, V.B.; Singh, M.V.; Gorantla, S.; Poluektova, L.; Maggirwar, S.B. Smoothened Agonist Reduces Human Immunodeficiency Virus Type-1-Induced Blood-Brain Barrier Breakdown in Humanized Mice. Sci. Rep. 2016, 6, 26876. [Google Scholar] [CrossRef]
- Singh, V.B.; Singh, M.V.; Piekna-Przybylska, D.; Gorantla, S.; Poluektova, L.Y.; Maggirwar, S.B. Sonic Hedgehog mimetic prevents leukocyte infiltration into the CNS during acute HIV infection. Sci. Rep. 2017, 7, 9578. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Phenotype | Remarks | Refs. |
|---|---|---|---|
| Shh−/− | Hypoplastic lungs (single lobe) with diminished epithelium; trachea and trachea-esophageal fistula to be malfunctioned. | Lethal at birth | [53,62] |
| Shh+/− | There is not any reported abnormality. | Not lethal | [63] |
| Ptch1−/− | It is lethal before the beginning of lung development. | Lethal at E8.5–E9.5 | [34,64] |
| Ptch1+/− | There is not any reported abnormality. | Not lethal | [64] |
| GLI-1−/− | Normal appearance. | Not lethal | [58] |
| GLI-2−/− | Hypoplastic lungs along with severe patterning abnormalities in single-lobed right lung and decreased mesenchyme, a little hypoplastic trachea and esophagus | Lethal at birth | [65] |
| GLI-2+/− | Normal appearance. | Not lethal | [58] |
| GLI-3−/− | Hypoplastic lungs with reduced size and abnormal appearance of lobes. | Lethal at E14.5 | [66] |
| GLI-3+/− | Normal appearance. | Not lethal | [67] |
| GLI-1−/−, GLI-2+/− | Hypoplastic lungs with diminished size and less severe than GLI1−/−; GLI2+/+ | 50% lethal until P21 | [58] |
| GLI-1−/−, GLI-2−/− | Two lobes are severely hypoplastic | Lethal at birth | [34,58] |
| GLI-2−/−, GLI-3+/− | Hypoplastic lungs with development of abnormal proximal lung that is failed to perform left and right lung separation, consequently, tracheobronchial fistula and distal lung partially formation occur | Lethal at birth | [65] |
| GLI-2−/−, GLI-3−/− | Severe form of phenotype and shows failure performance in order for the formation of the trachea, lung, and esophagus | Lethal at E10.5 but some embryos survive until E13.5 | [65] |
| Hhip−/− | Hypoplastic lung (single lobe) with abnormal formation of the second generation of lung buds | Lethal at birth | [61] |
| Hhip+/− | There is not any reported abnormality. | Not lethal | [61] |
| Hhip−/−, Ptch1+/− | It is a form of hyoplasia more severe than Hhip+/− lungs; thickened epithelium. | Lethal at birth | [61] |
| Disease Type | Technique | Outcome | Refs. |
|---|---|---|---|
| Parencymal lung disease | |||
| IPF | Microarray | Gene expression of Ptch1 is altered in IPF lungs disease. | [46] |
| ISH | Shh gene is highly expressed in the epithelium of the fibrotic zone | [48,74] | |
| IHC | Smo, GLI1, and Ptch1 are expressed in the fibroblastic zone of IPF lung disease. | [17] | |
| IHC | Shh is expressed in hyperplastic alveolar type II cells in the fibrotic area. | [47,68] | |
| qRT-PCR | Shh, GLI1, and Ptch1 are upregulated in IPF lungs disease. | [17] | |
| ELISA | Shh is high in BALF from IPF lungs. | [48] | |
| NSIP | IHC | Shh is expressed in epithelial cells of the thickened alveolar wall. | [47,68] |
| ISH | Shh is minimally expressed in the epithelium but higher than in normal lungs. | [74] | |
| COP | IHC | Shh is expressed in buds of organizing exudates. | [47] |
| Airway diseases | |||
| Asthma | GWAS | SNPs in Hhip regions on 4q31 and Ptch1 on 9Q22-31 is associated with decreased lung function (FEV1/FVC ratio) and asthma-related phenotypes. | [75] |
| COPD | qRT-PCR | Hhip is decreased in COPD lungs disease. | [76] |
| WB | Hhip is decreased in COPD lungs disease. | [76] | |
| GWAS | SNP in region close Hhip gene on 4q31 related to diminished lung function (FEV1/FVC ratio) and COPD-related phenotypes. | [76,77] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, L.-H.; Barkat, M.Q.; Syed, S.K.; Shah, S.; Abbas, G.; Xu, C.; Mahdy, A.; Hussain, N.; Hussain, L.; Majeed, A.; et al. Hedgehog Signaling: Linking Embryonic Lung Development and Asthmatic Airway Remodeling. Cells 2022, 11, 1774. https://doi.org/10.3390/cells11111774
Zeng L-H, Barkat MQ, Syed SK, Shah S, Abbas G, Xu C, Mahdy A, Hussain N, Hussain L, Majeed A, et al. Hedgehog Signaling: Linking Embryonic Lung Development and Asthmatic Airway Remodeling. Cells. 2022; 11(11):1774. https://doi.org/10.3390/cells11111774
Chicago/Turabian StyleZeng, Ling-Hui, Muhammad Qasim Barkat, Shahzada Khurram Syed, Shahid Shah, Ghulam Abbas, Chengyun Xu, Amina Mahdy, Nadia Hussain, Liaqat Hussain, Abdul Majeed, and et al. 2022. "Hedgehog Signaling: Linking Embryonic Lung Development and Asthmatic Airway Remodeling" Cells 11, no. 11: 1774. https://doi.org/10.3390/cells11111774
APA StyleZeng, L.-H., Barkat, M. Q., Syed, S. K., Shah, S., Abbas, G., Xu, C., Mahdy, A., Hussain, N., Hussain, L., Majeed, A., Khan, K.-u.-R., Wu, X., & Hussain, M. (2022). Hedgehog Signaling: Linking Embryonic Lung Development and Asthmatic Airway Remodeling. Cells, 11(11), 1774. https://doi.org/10.3390/cells11111774