
Artificial Intelligence and Circulating Cell-Free DNA Methylation Profiling: Mechanism and Detection of Alzheimer’s Disease


 ,
, 
 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Methylation Profiling
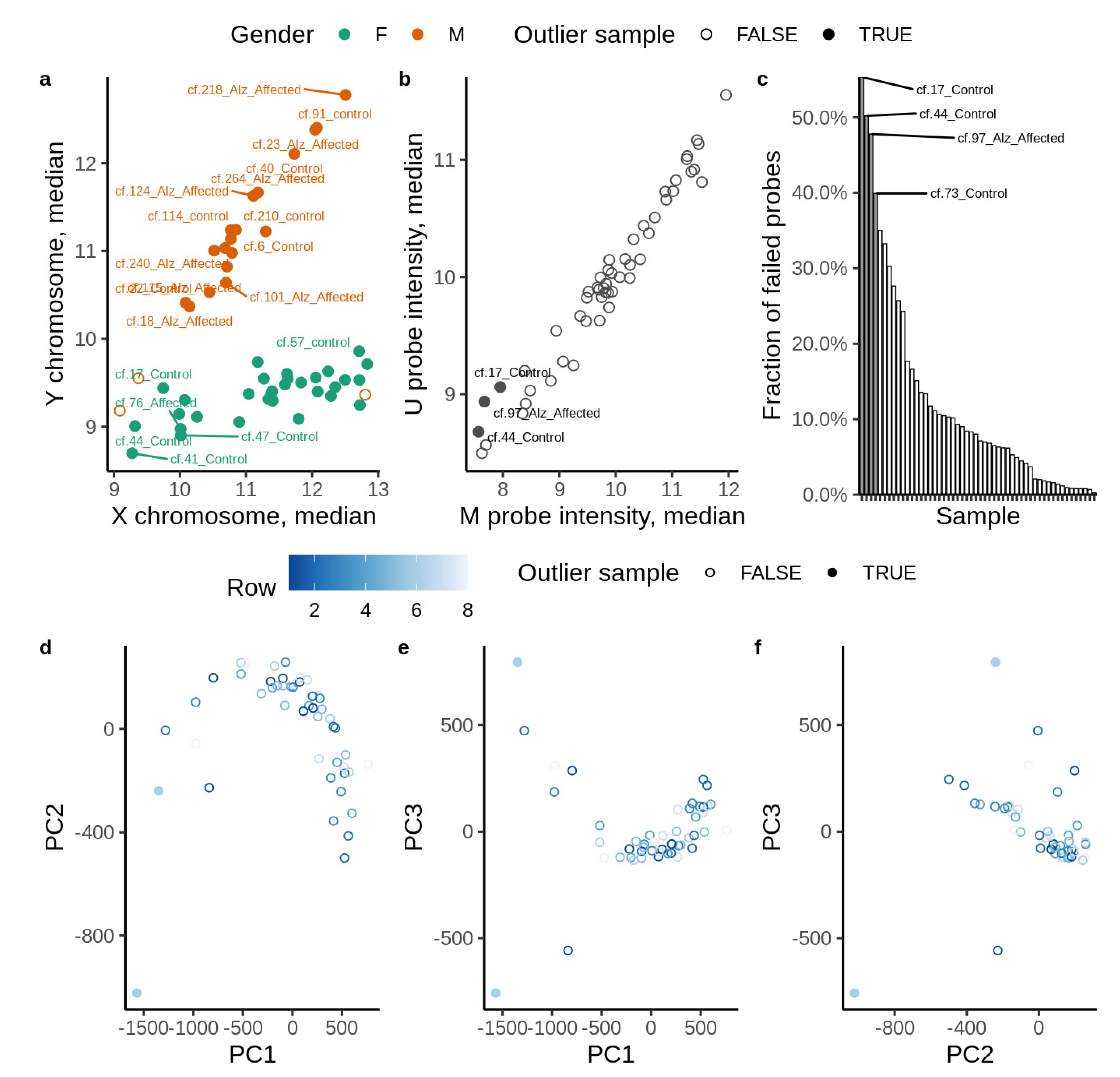
2.2. Data Preparation
2.3. Statistical and Bioinformatic Analysis
2.4. Enrichment Analysis
2.5. Artificial Intelligence/Deep Learning (AI/DL) Analysis
- hyper_params <- list(
- activation = c(“Rectifier”,“Tanh”),
- hidden = list(c(100),c(200),c(10,10),c(20,20),c(50,50),c(30,30,30),c(25,25,25,25)),
- input_dropout_ratio = c(0,0.05,0.1),
- hidden_dropout_ratios = c(0.6,0.5,0.6,0.6),
- l1 = seq(0,1e-4,1e-6),
- l2 = seq(0,1e-4,1e-6),
- train_samples_per_iteration = c(0,-2),
- epochs = c(500),
- momentum_start = c(0,0.5),
- rho = c(0.5,0.99),
- quantile_alpha = c(0,1),
- huber_alpha = seq(0,1))
2.6. Validation
2.7. Model Uncertainties
3. Results
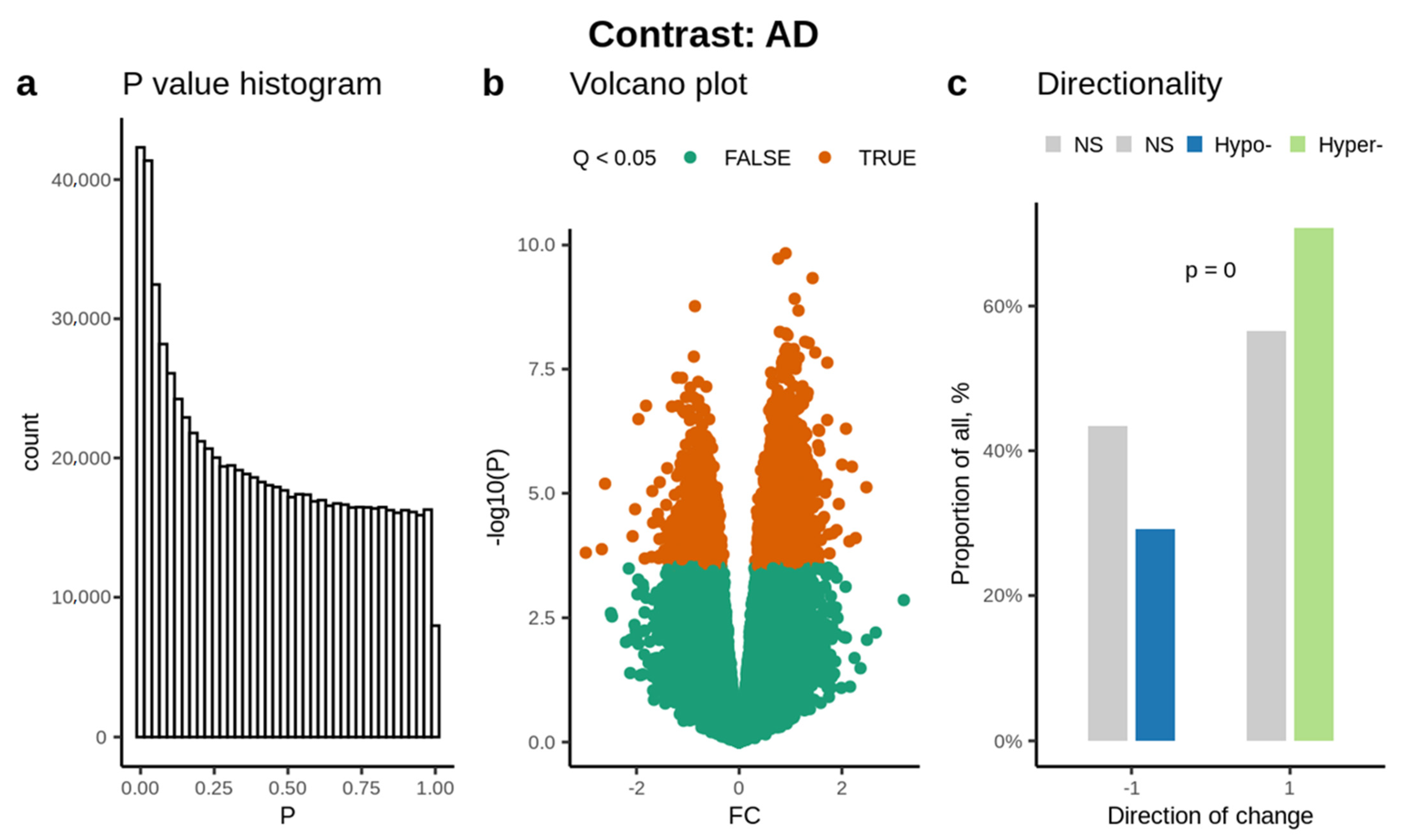
3.1. Abundance of Significantly Methylated Cytosines
3.2. Enrichment Analysis
3.3. Disease and Functional Enrichment
3.4. AI Prediction of AD
4. Discussion
Disease and Functional Enrichment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hampel, H.; Toschi, N.; Baldacci, F.; Zetterberg, H.; Blennow, K.; Kilimann, I.; Teipel, S.J.; Cavedo, E.; Melo Dos Santos, A.; Epelbaum, S.; et al. Alzheimer’s disease biomarker-guided diagnostic workflow using the added value of six combined cerebrospinal fluid candidates: Abeta1-42, total-tau, phosphorylated-tau, NFL, neurogranin, and YKL-40. Alzheimers Dement. 2018, 14, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef] [Green Version]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Kurdyukov, S.; Bullock, M. DNA Methylation Analysis: Choosing the Right Method. Biology 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.; Sherr, G.L. Epigenetic Modifications in Alzheimer’s Neuropathology and Therapeutics. Front Neurosci. 2019, 13, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finotti, A.; Allegretti, M.; Gasparello, J.; Giacomini, P.; Spandidos, D.A.; Spoto, G.; Gambari, R. Liquid biopsy and PCR-free ultrasensitive detection systems in oncology (Review). Int. J. Oncol. 2018, 53, 1395–1434. [Google Scholar] [CrossRef]
- Tadimety, A.; Closson, A.; Li, C.; Yi, S.; Shen, T.; Zhang, J.X.J. Advances in liquid biopsy on-chip for cancer management: Technologies, biomarkers, and clinical analysis. Crit. Rev. Clin. Lab. Sci. 2018, 55, 140–162. [Google Scholar] [CrossRef]
- Liu, Q.; Ma, J.; Deng, H.; Huang, S.J.; Rao, J.; Xu, W.B.; Huang, J.S.; Sun, S.Q.; Zhang, L. Cardiac-specific methylation patterns of circulating DNA for identification of cardiomyocyte death. BMC Cardiovasc. Disord. 2020, 20, 310. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Ungerer, V.; Diehl, F.; Anker, P.; Dor, Y.; Fleischhacker, M.; Gahan, P.B.; Hui, L.; Holdenrieder, S.; Thierry, A.R. Towards systematic nomenclature for cell-free DNA. Hum. Genet. 2021, 140, 565–578. [Google Scholar] [CrossRef]
- Garg, N.; Hidalgo, L.G.; Aziz, F.; Parajuli, S.; Mohamed, M.; Mandelbrot, D.A.; Djamali, A. Use of Donor-Derived Cell-Free DNA for Assessment of Allograft Injury in Kidney Transplant Recipients During the Time of the Coronavirus Disease 2019 Pandemic. Transplant. Proc. 2020, 52, 2592–2595. [Google Scholar] [CrossRef]
- Knight, S.R.; Thorne, A.; Faro, M.L.L. Donor-specific cell-free DNA as a biomarker in solid organ transplantation. A systematic review. Transplantation 2019, 103, 273–283. [Google Scholar] [CrossRef]
- Pai, M.C.; Kuo, Y.M.; Wang, I.F.; Chiang, P.M.; Tsai, K.J. The Role of Methylated Circulating Nucleic Acids as a Potential Biomarker in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 2440–2449. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, G.; Seshadri, S. Circulating biomarkers that predict incident dementia. Alzheimers Res. Ther. 2014, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Goetzl, E.J.; Kapogiannis, D.; Lista, S.; Vergallo, A. Biomarker-Drug and Liquid Biopsy Co-development for Disease Staging and Targeted Therapy: Cornerstones for Alzheimer’s Precision Medicine and Pharmacology. Front. Pharm. 2019, 10, 310. [Google Scholar] [CrossRef] [PubMed]
- Bahado-Singh, R.O.; Sonek, J.; McKenna, D.; Cool, D.; Aydas, B.; Turkoglu, O.; Bjorndahl, T.; Mandal, R.; Wishart, D.; Friedman, P.; et al. Artificial Intelligence and amniotic fluid multiomics analysis: The prediction of perinatal outcome in asymptomatic short cervix. Ultrasound Obs. Gynecol. 2019, 54, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Bahado-Singh, R.O.; Yilmaz, A.; Bisgin, H.; Turkoglu, O.; Kumar, P.; Sherman, E.; Mrazik, A.; Odibo, A.; Graham, S.F. Artificial intelligence and the analysis of multi-platform metabolomics data for the detection of intrauterine growth restriction. PLoS ONE 2019, 14, e0214121. [Google Scholar] [CrossRef] [PubMed]
- Alpay Savasan, Z.; Yilmaz, A.; Ugur, Z.; Aydas, B.; Bahado-Singh, R.O.; Graham, S.F. Metabolomic Profiling of Cerebral Palsy Brain Tissue Reveals Novel Central Biomarkers and Biochemical Pathways Associated with the Disease: A Pilot Study. Metabolites 2019, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Aydas, B.; Mishra, N.K.; Guda, C.; Radhakrishna, U. Deep Learning/Artificial Intelligence and Blood-Based DNA Epigenomic Prediction of Cerebral Palsy. Int. J. Mol. Sci. 2019, 20, 2075. [Google Scholar] [CrossRef] [Green Version]
- Hung, T.N.K.; Le, N.Q.K.; Le, N.H.; Tuan, L.V.; Nguyen, T.P.; Thi, C.; Kang, J.H. An AI-based Prediction Model for Drug-drug Interactions in Osteoporosis and Paget’s Diseases from SMILES. Mol. Inf. 2022, e2100264. [Google Scholar] [CrossRef]
- Tng, S.S.; Le, N.Q.K.; Yeh, H.Y.; Chua, M.C.H. Improved Prediction Model of Protein Lysine Crotonylation Sites Using Bidirectional Recurrent Neural Networks. J. Proteome Res. 2022, 21, 265–273. [Google Scholar] [CrossRef]
- Costello, Z.; Martin, H.G. A machine learning approach to predict metabolic pathway dynamics from time-series multiomics data. NPJ Syst. Biol. Appl. 2018, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Jo, T.; Nho, K.; Saykin, A.J. Deep Learning in Alzheimer’s Disease: Diagnostic Classification and Prognostic Prediction Using Neuroimaging Data. Front. Aging Neurosci. 2019, 11, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirbandi, K.; Khalafi, M.; Mirza-Aghazadeh-Attari, M.; Tahmasbi, M.; Kiani Shahvandi, H.; Javanmardi, P.; Rahim, F. Accuracy of deep learning model-assisted amyloid positron emission tomography scan in predicting Alzheimer’s disease: A Systematic Review and meta-analysis. Inform. Med. Unlocked 2021, 25, 100710. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, X.; Wang, Y.; Kim, Y. Applied machine learning in Alzheimer’s disease research: Omics, imaging, and clinical data. Emerg. Top. Life Sci. 2021, 5, 765–777. [Google Scholar] [CrossRef]
- Finney, C.A.; Delerue, F.; Shvetcov, A. Artificial intelligence-driven meta-analysis of brain gene expression data identifies novel gene candidates in Alzheimer’s Disease. medRxiv 2022. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Bartak, B.K.; Kalmar, A.; Galamb, O.; Wichmann, B.; Nagy, Z.B.; Tulassay, Z.; Dank, M.; Igaz, P.; Molnar, B. Blood Collection and Cell-Free DNA Isolation Methods Influence the Sensitivity of Liquid Biopsy Analysis for Colorectal Cancer Detection. Pathol. Oncol. Res. 2019, 25, 915–923. [Google Scholar] [CrossRef]
- Sheinerman, K.S.; Toledo, J.B.; Tsivinsky, V.G.; Irwin, D.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Chen-Plotkin, A.; Wolk, D.A.; McCluskey, L.F.; et al. Circulating brain-enriched microRNAs as novel biomarkers for detection and differentiation of neurodegenerative diseases. Alzheimers Res. 2017, 9, 89. [Google Scholar] [CrossRef]
- Hardy, T.; Zeybel, M.; Day, C.P.; Dipper, C.; Masson, S.; McPherson, S.; Henderson, E.; Tiniakos, D.; White, S.; French, J.; et al. Plasma DNA methylation: A potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut 2017, 66, 1321–1328. [Google Scholar] [CrossRef]
- Ramirez, K.; Fernández, R.; Collet, S.; Kiyar, M.; Delgado-Zayas, E.; Gómez-Gil, E.; Van Den Eynde, T.; T’Sjoen, G.; Guillamon, A.; Mueller, S.C.; et al. Epigenetics Is Implicated in the Basis of Gender Incongruence: An Epigenome-Wide Association Analysis. Front Neurosci. 2021, 15, 701017. [Google Scholar] [CrossRef]
- Mansell, G.; Gorrie-Stone, T.J.; Bao, Y.; Kumari, M.; Schalkwyk, L.S.; Mill, J.; Hannon, E. Guidance for DNA methylation studies: Statistical insights from the Illumina EPIC array. BMC Genom. 2019, 20, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alakwaa, F.M.; Chaudhary, K.; Garmire, L.X. Deep Learning Accurately Predicts Estrogen Receptor Status in Breast Cancer Metabolomics Data. J. Proteome Res. 2018, 17, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- O’Connell, G.C.; Alder, M.L. Large-scale informatic analysis to algorithmically identify blood biomarkers of neurological damage. Proc. Natl. Acad. Sci. USA 2020, 117, 20764–20775. [Google Scholar] [CrossRef] [PubMed]
- Shigemizu, D.; Mori, T.; Akiyama, S.; Higaki, S.; Watanabe, H.; Sakurai, T.; Niida, S.; Ozaki, K. Identification of potential blood biomarkers for early diagnosis of Alzheimer’s disease through RNA sequencing analysis. Alzheimers Res.Ther. 2020, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M. DNA hypermethylation in disease: Mechanisms and clinical relevance. Epigenetics 2019, 14, 1141–1163. [Google Scholar] [CrossRef] [Green Version]
- Borgel, J.; Tyl, M.; Schiller, K.; Pusztai, Z.; Dooley, C.M.; Deng, W.; Wooding, C.; White, R.J.; Warnecke, T.; Leonhardt, H.; et al. KDM2A integrates DNA and histone modification signals through a CXXC/PHD module and direct interaction with HP1. Nucleic Acids Res. 2017, 45, 1114–1129. [Google Scholar] [CrossRef] [Green Version]
- Mastroeni, D.; Delvaux, E.; Nolz, J.; Tan, Y.; Grover, A.; Oddo, S.; Coleman, P.D. Aberrant intracellular localization of H3k4me3 demonstrates an early epigenetic phenomenon in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 3121–3129. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Seo, J.; Chun, Y.S. Targeted Downregulation of kdm4a Ameliorates Tau-engendered Defects in Drosophila melanogaster. J. Korean Med. Sci. 2019, 34, e225. [Google Scholar] [CrossRef]
- Nielsen, J.B.; Rom, O.; Surakka, I.; Graham, S.E. Loss-of-function genomic variants highlight potential therapeutic targets for cardiovascular disease. Nat. Commun. 2020, 11, 6417. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Liang, Y.; Zhang, X.; Xu, J.; Lin, J.; Zhang, R.; Kang, K.; Liu, C.; Zhao, C.; Zhao, M. Low-Density Lipoprotein Cholesterol and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Front. Aging Neurosci. 2020, 12, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konar, A.; Kalra, R.S.; Chaudhary, A.; Nayak, A.; Guruprasad, K.P.; Satyamoorthy, K.; Ishida, Y.; Terao, K.; Kaul, S.C.; Wadhwa, R. Identification of Caffeic Acid Phenethyl Ester (CAPE) as a Potent Neurodifferentiating Natural Compound That Improves Cognitive and Physiological Functions in Animal Models of Neurodegenerative Diseases. Front. Aging Neurosci. 2020, 12, 561925. [Google Scholar] [CrossRef] [PubMed]
- Asada, K.; Kaneko, S.; Takasawa, K.; Machino, H.; Takahashi, S.; Shinkai, N.; Shimoyama, R.; Komatsu, M.; Hamamoto, R. Integrated Analysis of Whole Genome and Epigenome Data Using Machine Learning Technology: Toward the Establishment of Precision Oncology. Front. Oncol. 2021, 11, 666937. [Google Scholar] [CrossRef]
- Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Li, W.; Tam, K.M.V.; Chan, W.W.R.; Koon, A.C.; Ngo, J.C.K.; Chan, H.Y.E.; Lau, K.-F. Neuronal adaptor FE65 stimulates Rac1-mediated neurite outgrowth by recruiting and activating ELMO1. J. Biol. Chem. 2018, 293, 7674–7688. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, M.; Sekiya, M.; Hara, N.; Miyashita, A.; Kuwano, R.; Ikeuchi, T.; Iijima, K.M.; Nakaya, A. Disruption of a RAC1-centred network is associated with Alzheimer’s disease pathology and causes age-dependent neurodegeneration. Hum. Mol. Genet. 2020, 29, 817–833. [Google Scholar] [CrossRef]
- Huang, Y.; Sun, X.; Jiang, H.; Yu, S.; Robins, C.; Armstrong, M.J.; Li, R.; Mei, Z.; Shi, X.; Gerasimov, E.S.; et al. A machine learning approach to brain epigenetic analysis reveals kinases associated with Alzheimer’s disease. Nat. Commun. 2021, 12, 4472. [Google Scholar] [CrossRef]
- Watson, C.T.; Roussos, P.; Garg, P.; Ho, D.J.; Azam, N.; Katsel, P.L.; Haroutunian, V.; Sharp, A.J. Genome-wide DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med. 2016, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Mahendran, N.; Duraj, R.V. A deep learning framework with an embedded-based feature selection approach for the early detection of the Alzheimer’s disease. Comput. Biol. Med. 2022, 141, 105056. [Google Scholar] [CrossRef]
- Hannon, E.; Lunnon, K.; Schalkwyk, L.; Mill, J. Interindividual methylomic variation across blood, cortex, and cerebellum: Implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics 2015, 10, 1024–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Zhang, B.; Wei, D.; Zhang, Z. Identification of Methylated Gene Biomarkers in Patients with Alzheimer’s Disease Based on Machine Learning. BioMed Res. Int. 2020, 2020, 8348147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Giese, K.P. Calcium/calmodulin-dependent kinase II and Alzheimer’s disease. Mol. Brain 2015, 8, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Lee, H.G.; Raina, A.K.; Perry, G.; Smith, M.A. The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neuro-Signals 2002, 11, 270–281. [Google Scholar] [CrossRef]
- Saura, C.A.; Valero, J. The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Rev. Neurosci. 2011, 22, 153–169. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium signalling and Alzheimer’s disease. Neurochem. Res. 2011, 36, 1149–1156. [Google Scholar] [CrossRef]
- Tong, B.C.-K.; Wu, A.J.; Li, M.; Cheung, K.-H. Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2018, 1865, 1745–1760. [Google Scholar]
- Koran, M.E.I.; Hohman, T.J.; Thornton-Wells, T.A. Genetic interactions found between calcium channel genes modulate amyloid load measured by positron emission tomography. Hum. Genet. 2014, 133, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Li, C.; Du, G.; Pan, M.; Liu, G.; Pan, W.; Li, X. High glucose upregulates myosin light chain kinase to induce microfilament cytoskeleton rearrangement in hippocampal neurons. Mol. Med. Rep. 2018, 18, 216–222. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Braithwaite, S.P.; Stock, J.B.; Lombroso, P.J.; Nairn, A.C. Protein phosphatases and Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2012, 106, 343–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriques, A.G.; Müller, T.; Oliveira, J.M.; Cova, M.; da Cruz e Silva, C.B.; da Cruz e Silva, O.A.B. Altered protein phosphorylation as a resource for potential AD biomarkers. Sci. Rep. 2016, 6, 30319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [Green Version]
- Ayka, A.; Sehirli, A.O. The Role of the SLC Transporters Protein in the Neurodegenerative Disorders. Clin. Psychopharmacol. Neurosci. 2020, 18, 174–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Sun, H.; Chen, Z.; Xu, H.; Bu, G.; Zheng, H. Implications of GABAergic Neurotransmission in Alzheimer’s Disease. Front Aging Neurosci. 2016, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Qi, Y.; Sun, Z. The Role of Sonic Hedgehog Pathway in the Development of the Central Nervous System and Aging-Related Neurodegenerative Diseases. Front. Mol. Biosci. 2021, 8, 711710. [Google Scholar] [CrossRef]
- Vorobyeva, A.G.; Saunders, A.J. Amyloid-β interrupts canonical Sonic hedgehog signaling by distorting primary cilia structure. Cilia 2018, 7, 5. [Google Scholar] [CrossRef] [Green Version]
- Bocharova, A.; Vagaitseva, K.; Marusin, A.; Zhukova, N.; Zhukova, I.; Minaycheva, L.; Makeeva, O.; Stepanov, V. Association and Gene-Gene Interactions Study of Late-Onset Alzheimer’s Disease in the Russian Population. Genes 2021, 12, 1647. [Google Scholar] [CrossRef]
- Liu, D.; Dai, S.X.; He, K.; Li, G.H.; Liu, J.; Liu, L.G.; Huang, J.F.; Xu, L.; Li, W.X. Identification of hub ubiquitin ligase genes affecting Alzheimer’s disease by analyzing transcriptome data from multiple brain regions. Sci. Prog. 2021, 104, 368504211001146. [Google Scholar] [CrossRef]
- Deters, K.D.; Nho, K.; Risacher, S.L.; Kim, S.; Ramanan, V.K.; Crane, P.K.; Apostolova, L.G.; Saykin, A.J.; Alzheimer’s Disease Neuroimaging, I. Genome-wide association study of language performance in Alzheimer’s disease. Brain Lang. 2017, 172, 22–29. [Google Scholar] [CrossRef]
- Lee, W.S.; Lee, W.-H.; Bae, Y.C.; Suk, K. Axon Guidance Molecules Guiding Neuroinflammation. Exp. Neurobiol. 2019, 28, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Placzek, M.; Xu, H. Axon guidance effect of classical morphogens Shh and BMP7 in the hypothalamo-pituitary system. Neurosci. Lett. 2013, 553, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Cheng, X.; Lu, J.; Li, X. Exogenous BMP-7 Facilitates the Recovery of Cardiac Function after Acute Myocardial Infarction through Counteracting TGF-beta1 Signaling Pathway. Tohoku J. Exp. Med. 2018, 244, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowery, J.W.; de Caestecker, M.P. BMP signaling in vascular development and disease. Cytokine Growth Factor Rev. 2010, 21, 287–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licastro, F.; Chiappelli, M.; Caldarera, C.M.; Porcellini, E.; Carbone, I.; Caruso, C.; Lio, D.; Corder, E.H. Sharing pathogenetic mechanisms between acute myocardial infarction and Alzheimer’s disease as shown by partially overlapping of gene variant profiles. J. Alzheimers Dis. 2011, 23, 421–431. [Google Scholar] [CrossRef]
- Akila Parvathy Dharshini, S.; Taguchi, Y.h.; Michael Gromiha, M. Exploring the selective vulnerability in Alzheimer disease using tissue specific variant analysis. Genomics 2019, 111, 936–949. [Google Scholar] [CrossRef]
- Amparan, D.; Avram, D.; Thomas, C.G.; Lindahl, M.G.; Yang, J.; Bajaj, G.; Ishmael, J.E. Direct interaction of myosin regulatory light chain with the NMDA receptor. J. Neurochem. 2005, 92, 349–361. [Google Scholar] [CrossRef]
- Balabanski, L.; Serbezov, D.; Atanasoska, M.; Karachanak-Yankova, S.; Hadjidekova, S.; Nikolova, D.; Boyanova, O.; Staneva, R.; Vazharova, R.; Mihailova, M.; et al. Rare genetic variants prioritize molecular pathways for semaphorin interactions in Alzheimer’s disease patients. Biotechnol. Biotechnol. Equip. 2021, 35, 1256–1262. [Google Scholar] [CrossRef]
- Dibattista, M.; Pifferi, S.; Menini, A.; Reisert, J. Alzheimer’s Disease: What Can We Learn From the Peripheral Olfactory System? Front. Neurosci. 2020, 14, 440. [Google Scholar] [CrossRef]
- Upadhyay, A.; Hosseinibarkooie, S.; Schneider, S.; Kaczmarek, A.; Torres-Benito, L.; Mendoza-Ferreira, N.; Overhoff, M.; Rombo, R.; Grysko, V.; Kye, M.J.; et al. Neurocalcin Delta Knockout Impairs Adult Neurogenesis Whereas Half Reduction Is Not Pathological. Front. Mol. Neurosci. 2019, 12, 19. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.A.; Woltjer, R.L.; Goodenbour, J.M.; Horvath, S.; Geschwind, D.H. Genes and pathways underlying regional and cell type changes in Alzheimer’s disease. Genome Med. 2013, 5, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Er, A.; Aydas, B.; Turkoglu, O.; Taskin, B.D.; Duman, M.; Yilmaz, D.; Radhakrishna, U. Artificial Intelligence and the detection of pediatric concussion using epigenomic analysis. Brain Res. 2020, 1726, 146510. [Google Scholar] [CrossRef]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Aydas, B.; Yilmaz, A.; Metpally, R.P.; Carey, D.J.; Crist, R.C.; Berrettini, W.H.; Wilson, G.D.; Imam, K.; et al. Artificial intelligence and leukocyte epigenomics: Evaluation and prediction of late-onset Alzheimer’s disease. PLoS ONE 2021, 16, e0248375. [Google Scholar] [CrossRef]
- Huang, J.H.; Xie, H.L.; Yan, J.; Lu, H.M.; Xu, Q.S.; Liang, Y.Z. Using random forest to classify T-cell epitopes based on amino acid properties and molecular features. Anal. Chim. Acta 2013, 804, 70–75. [Google Scholar] [CrossRef]
- Mahadevan, S.; Shah, S.L.; Marrie, T.J.; Slupsky, C.M. Analysis of metabolomic data using support vector machines. Anal. Chem. 2008, 80, 7562–7570. [Google Scholar] [CrossRef]
- Liland, K.H. Multivariate methods in metabolomics—From pre-processing to dimension reduction and statistical analysis. TrAC Trends Anal. Chem. 2011, 30, 827–841. [Google Scholar] [CrossRef]
- Candel, A.; Parmar, V.; Ledell, E.; Arora, A. Deep Learning with H2O; H2O. ai Inc.: Mountain View, CA, USA, 2016; pp. 1–21. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| SVM | GLM | PAM | RF | LDA | DL | |
|---|---|---|---|---|---|---|
| AUC 95% CI | 0.9760 (0.8900–1) | 0.9773 (0.8500–1) | 0.9886 (0.9000–1) | 0.9874 (0.9000–1) | 0.9493 (0.9500–1) | 0.9988 (0.9500–1) |
| Sensitivity | 0.9200 | 0.9200 | 0.9200 | 0.9300 | 0.9350 | 0.9450 |
| Specificity | 0.9220 | 0.9090 | 0.9080 | 0.9100 | 0.9250 | 0.9450 |
| SVM | GLM | PAM | RF | LDA | DL | |
|---|---|---|---|---|---|---|
| AUC 95% CI | 0.9700 (0.8700–1) | 0.9673 (0.8800–1) | 0.9786 (0.8800–1) | 0.9774 (0.8800–1) | 0.9393 (0.8700–1) | 0.9840 (0.9250–1) |
| Sensitivity | 0.9100 | 0.9100 | 0.9100 | 0.9200 | 0.9250 | 0.9250 |
| Specificity | 0.9120 | 0.8990 | 0.8980 | 0.9000 | 0.9150 | 0.9350 |
| SVM | GLM | PAM | RF | LDA | DL | |
|---|---|---|---|---|---|---|
| AUC 95% CI | 0.9770 (0.8900–1) | 0.9744 (0.8500–1) | 0.9856 (0.9000–1) | 0.9860 (0.9000–1) | 0.9488 (0.9500–1) | 0.9988 (0.9500–1) |
| Sensitivity | 0.9200 | 0.9200 | 0.9200 | 0.9300 | 0.9350 | 0.9450 |
| Specificity | 0.9220 | 0.9090 | 0.9080 | 0.9100 | 0.9250 | 0.9450 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahado-Singh, R.O.; Radhakrishna, U.; Gordevičius, J.; Aydas, B.; Yilmaz, A.; Jafar, F.; Imam, K.; Maddens, M.; Challapalli, K.; Metpally, R.P.; et al. Artificial Intelligence and Circulating Cell-Free DNA Methylation Profiling: Mechanism and Detection of Alzheimer’s Disease. Cells 2022, 11, 1744. https://doi.org/10.3390/cells11111744
Bahado-Singh RO, Radhakrishna U, Gordevičius J, Aydas B, Yilmaz A, Jafar F, Imam K, Maddens M, Challapalli K, Metpally RP, et al. Artificial Intelligence and Circulating Cell-Free DNA Methylation Profiling: Mechanism and Detection of Alzheimer’s Disease. Cells. 2022; 11(11):1744. https://doi.org/10.3390/cells11111744
Chicago/Turabian StyleBahado-Singh, Ray O., Uppala Radhakrishna, Juozas Gordevičius, Buket Aydas, Ali Yilmaz, Faryal Jafar, Khaled Imam, Michael Maddens, Kshetra Challapalli, Raghu P. Metpally, and et al. 2022. "Artificial Intelligence and Circulating Cell-Free DNA Methylation Profiling: Mechanism and Detection of Alzheimer’s Disease" Cells 11, no. 11: 1744. https://doi.org/10.3390/cells11111744
APA StyleBahado-Singh, R. O., Radhakrishna, U., Gordevičius, J., Aydas, B., Yilmaz, A., Jafar, F., Imam, K., Maddens, M., Challapalli, K., Metpally, R. P., Berrettini, W. H., Crist, R. C., Graham, S. F., & Vishweswaraiah, S. (2022). Artificial Intelligence and Circulating Cell-Free DNA Methylation Profiling: Mechanism and Detection of Alzheimer’s Disease. Cells, 11(11), 1744. https://doi.org/10.3390/cells11111744