
Reducing PDK1/Akt Activity: An Effective Therapeutic Target in the Treatment of Alzheimer’s Disease

Abstract
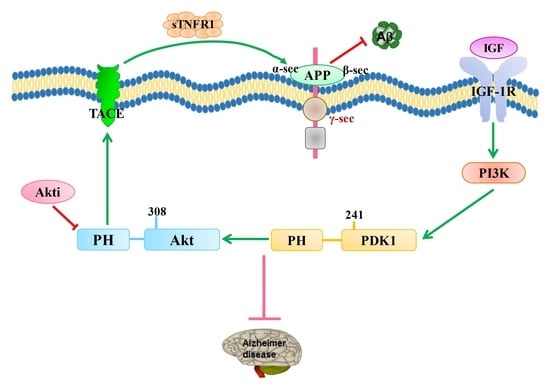
:
1. Introduction
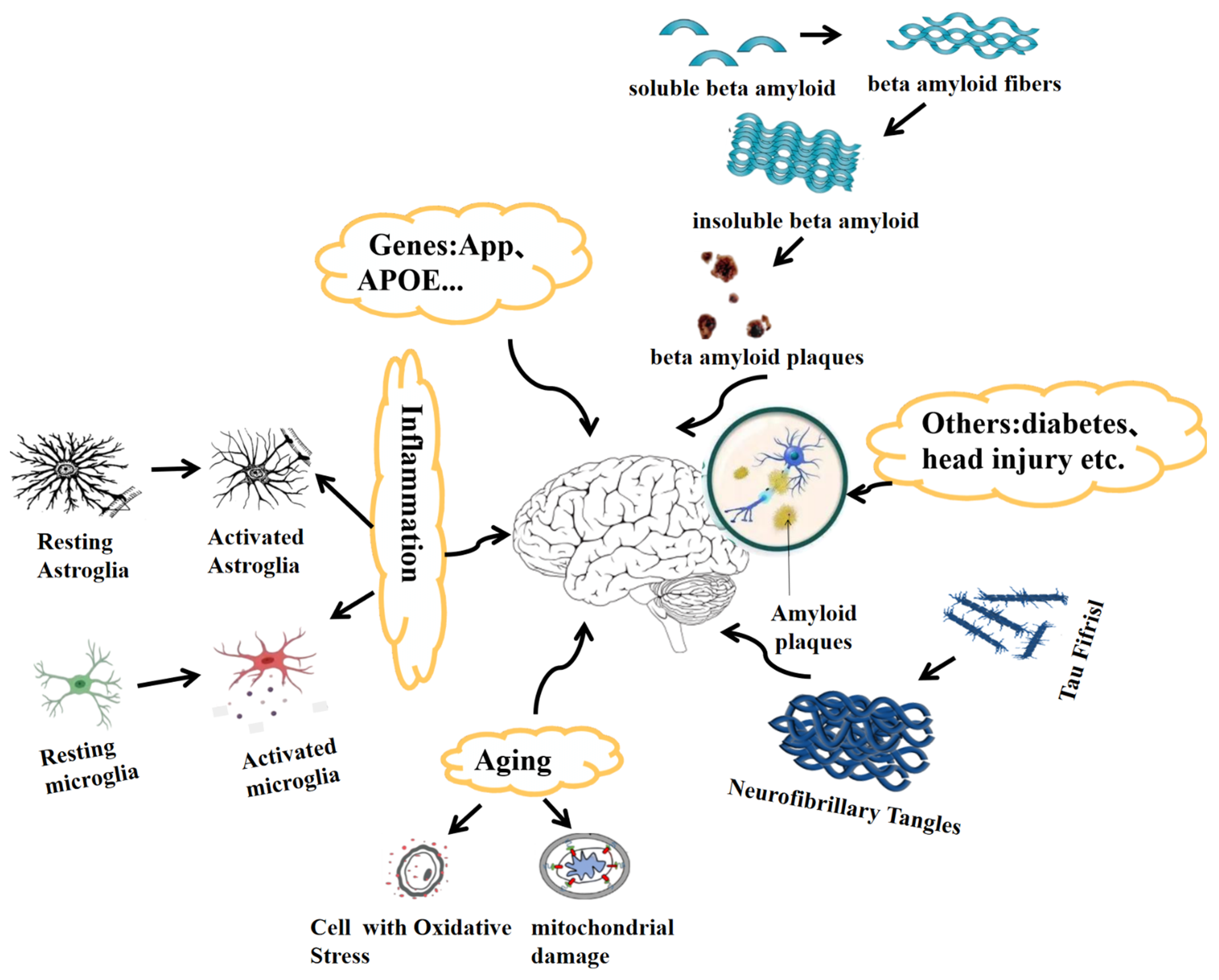
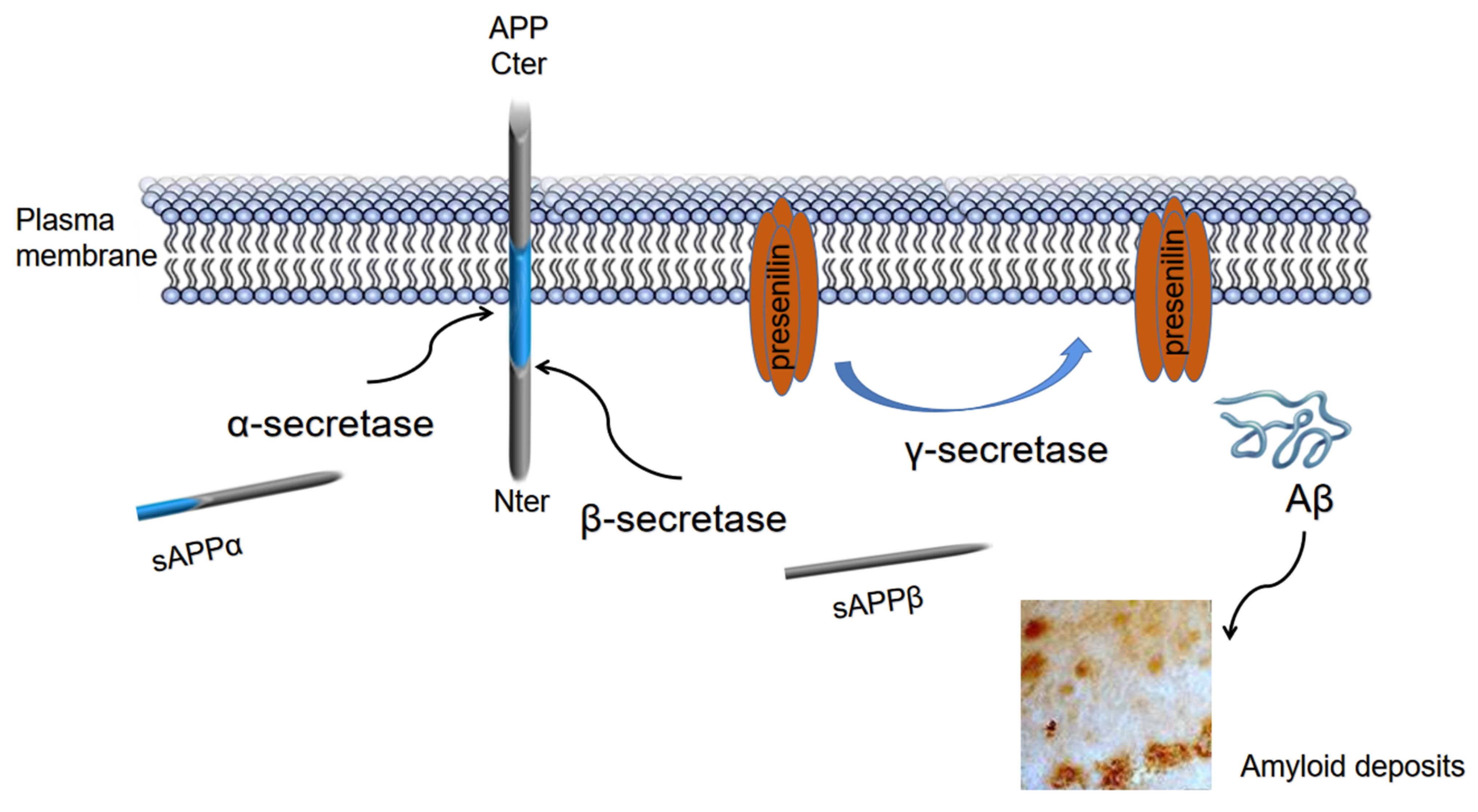
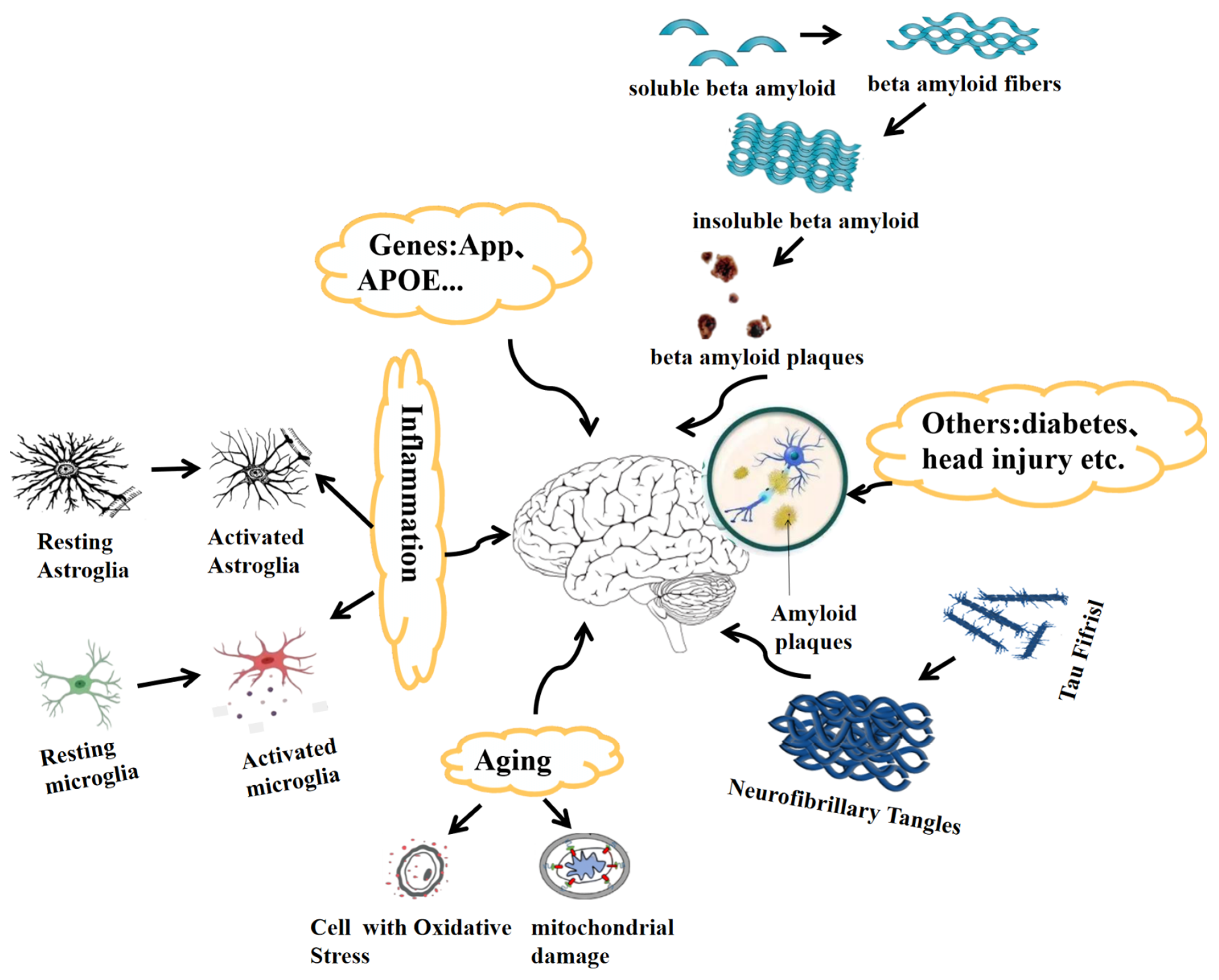
2. Pathological Features of Alzheimer’s Disease
2.1. Genetics of Alzheimer’s Disease
2.2. Tau in Alzheimer’s Disease
2.3. Aging as a Factor in Alzheimer’s Disease
2.4. Neuroinflammation in Alzheimer’s Disease
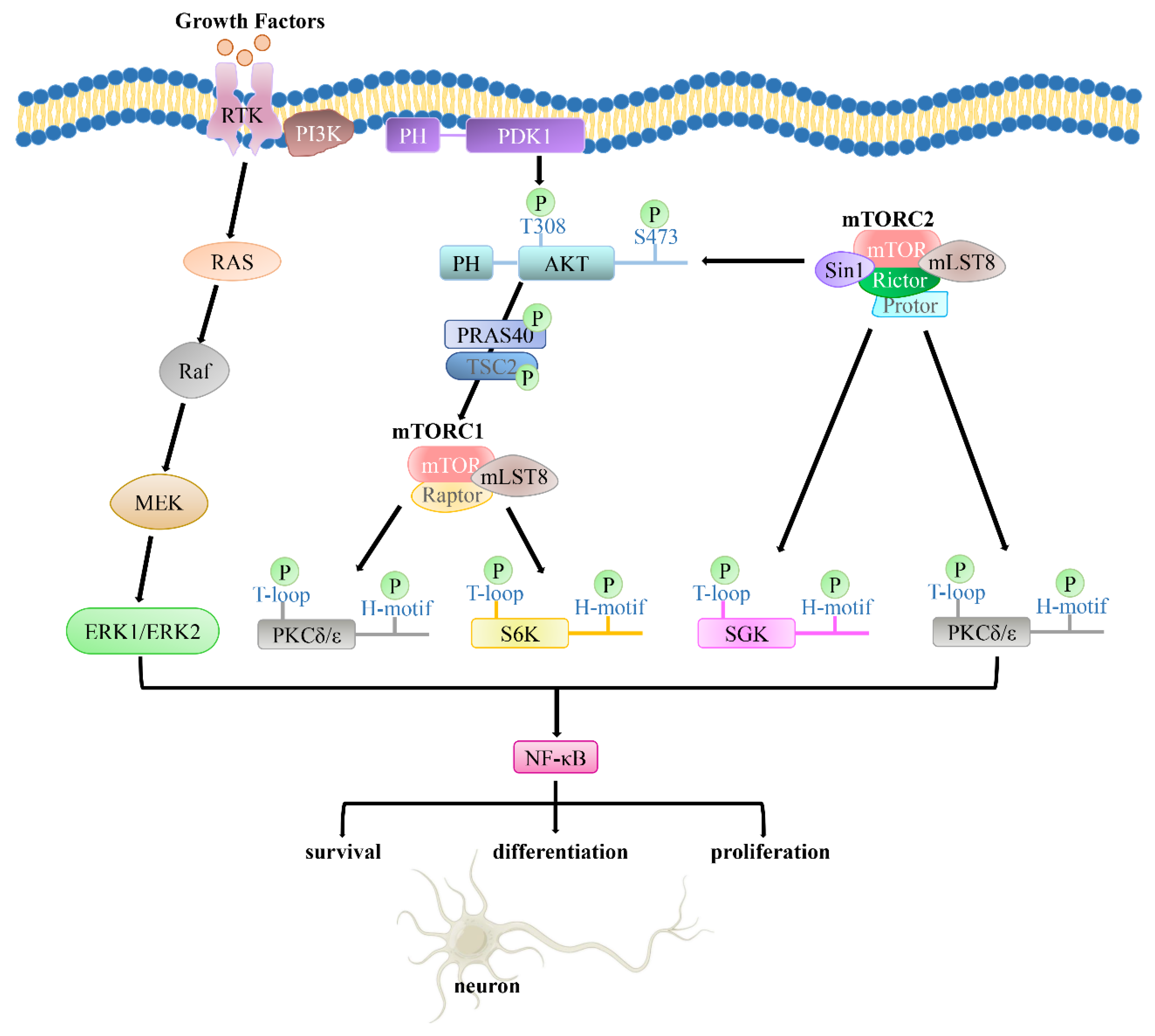
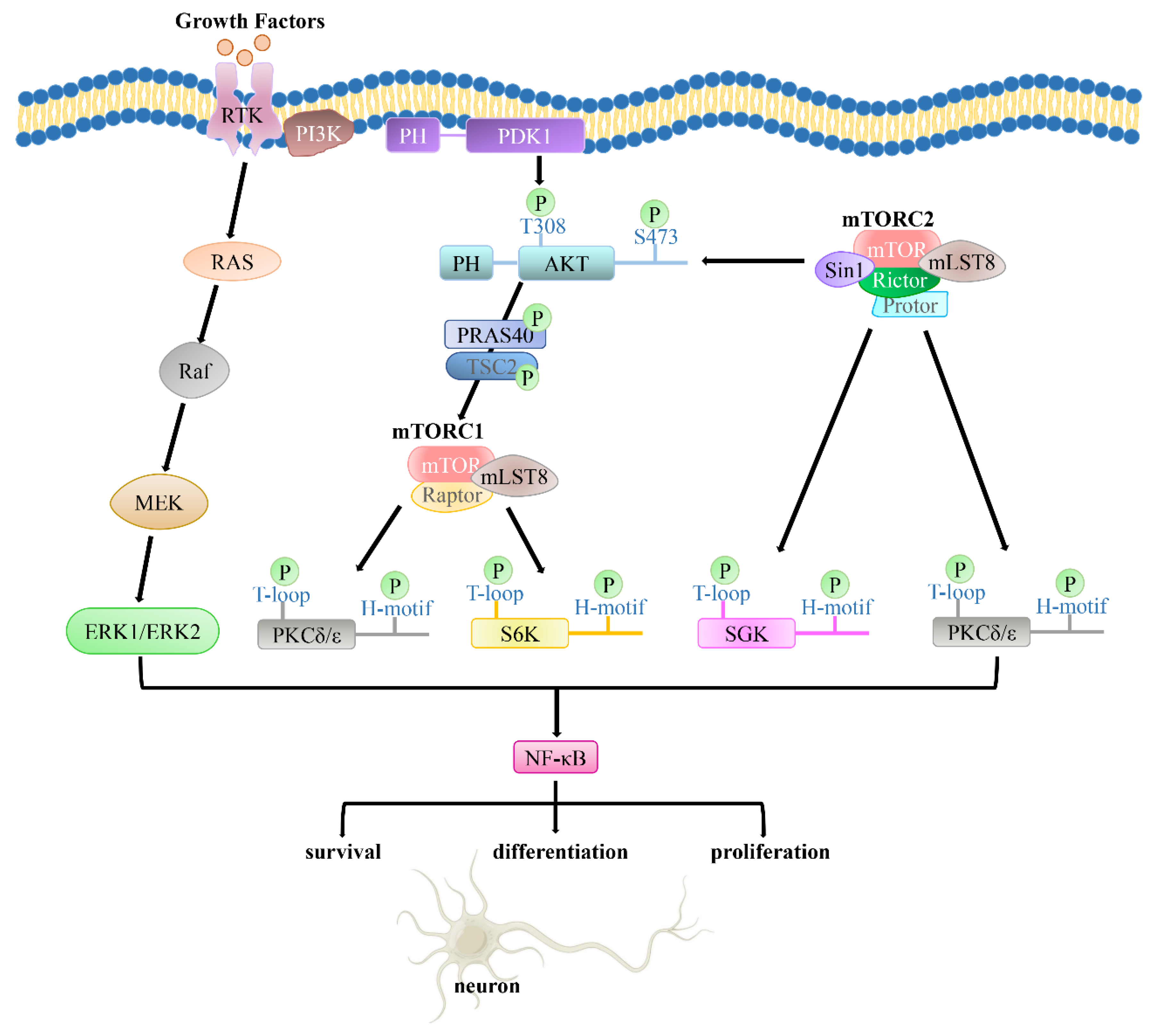
3. Structure and Function of the PI3K/PDK1/Akt Signaling Pathway
3.1. Structure and Function of PI3K
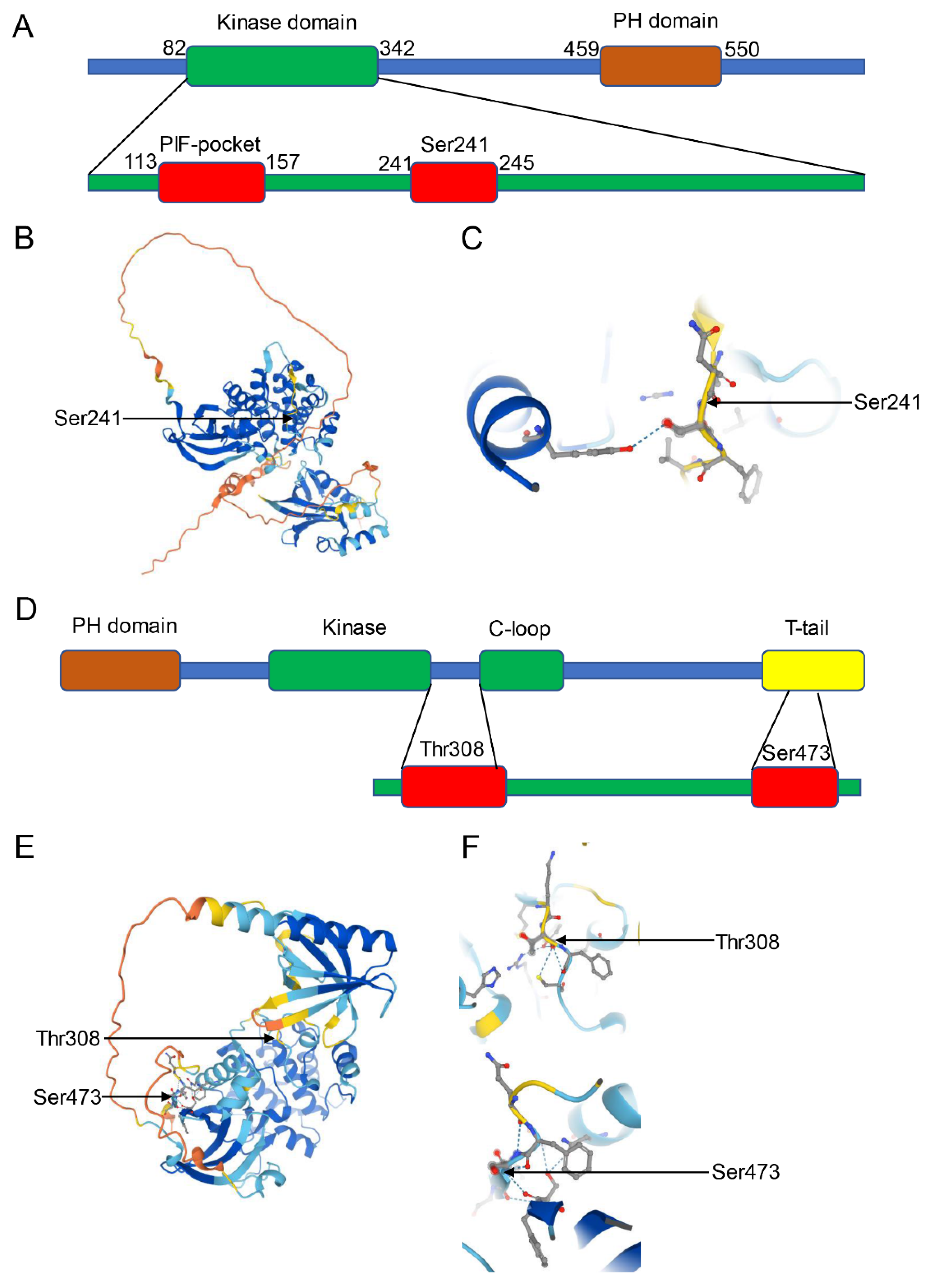
3.2. Structure and Function of PDK1
3.3. Structure and Function of Akt
4. The PI3K/PDK1/Akt Pathway in Normal and AD Brains
4.1. The PI3K/PDK1/Akt Pathway in Normal Brains
4.2. The PI3K/PDK1/Akt Pathway in AD Brains
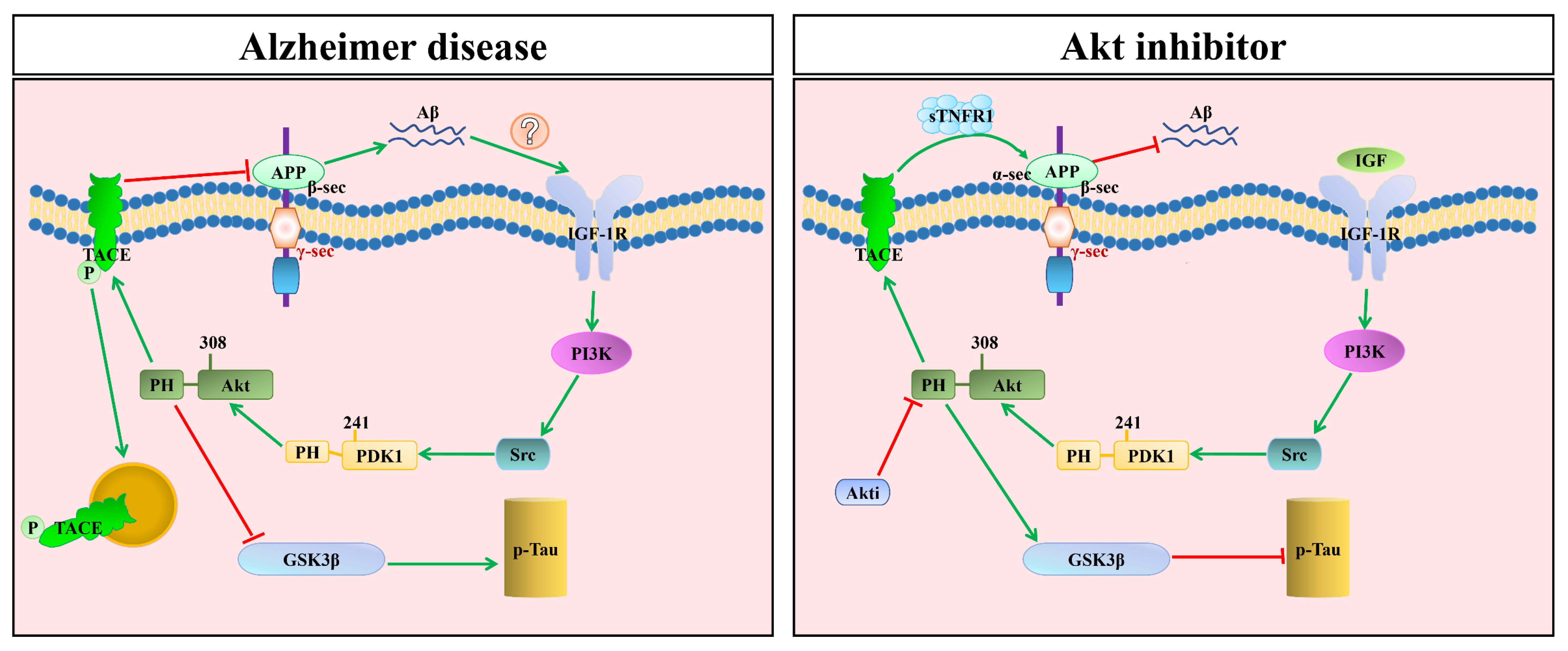
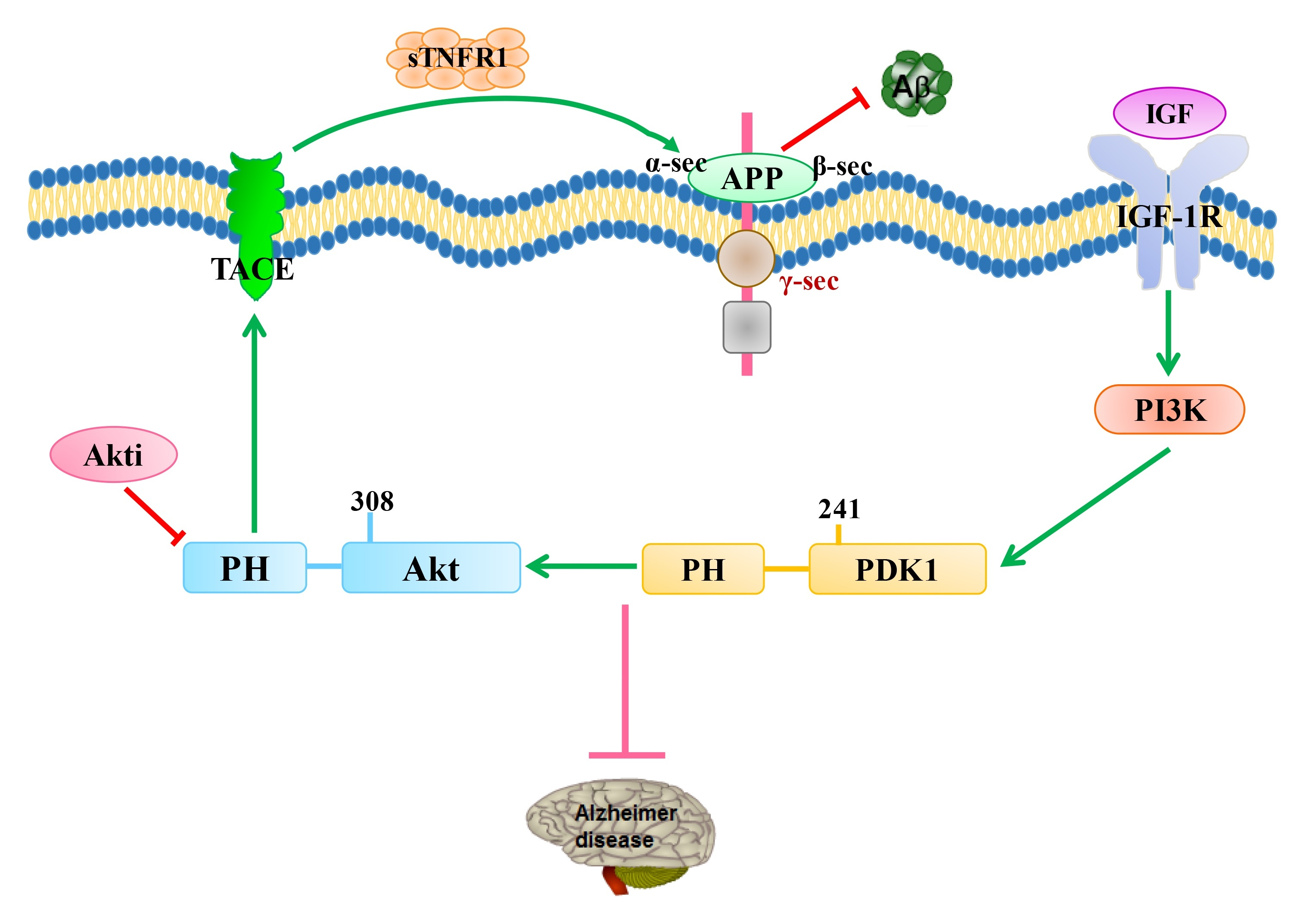
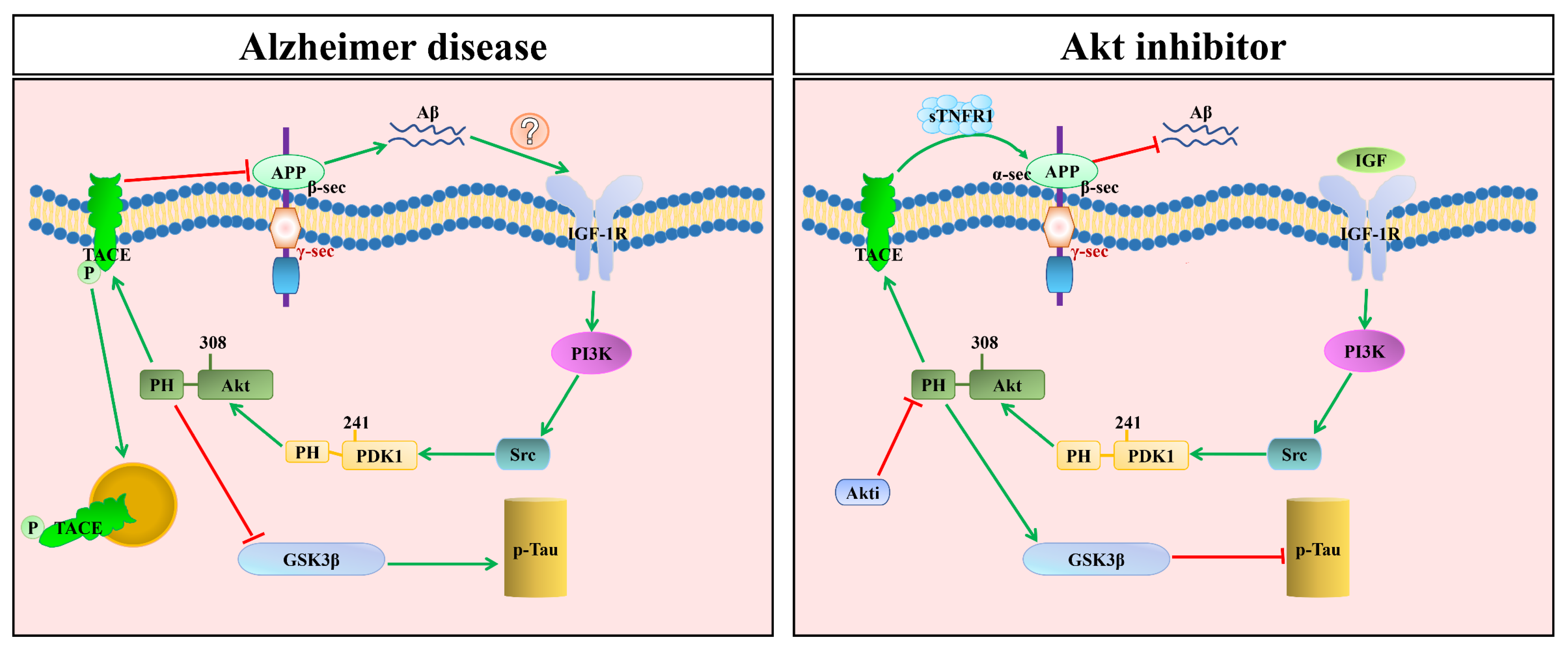
5. The Therapeutic Possibilities of the Modulation of PDK1/Akt in AD
5.1. The Therapeutic Possibility of Mutation of the PDK1 PH-Domain in AD
5.2. The Therapeutic Possibility of Mutating PDK1’s PH-Domain Dependence with TACE in AD
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ijaopo, E.O. Dementia-related agitation: A review of non-pharmacological interventions and analysis of risks and benefits of pharmacotherapy. Transl. Psychiatry 2017, 7, e1250. [Google Scholar] [CrossRef] [Green Version]
- Dementia Statistics. Alzheimer’s Disease International (ADI). Available online: https://www.alzint.org/about/dementia-factsfigures/dementia-statistics/ (accessed on 3 August 2021).
- Murphy, M.P.; Le Vine, H., III. Alzheimer’s disease and the amyloid-peptide. J. Alzheimer’s Dis. 2010, 19, 311. [Google Scholar] [CrossRef] [Green Version]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R. The diagnosis of dementia due to alzheimer’s disease: Recommendations from the national institute on aging- alzheimer’s association workgroups on diagnostic guidelines for alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460.
- Auld, D.S.; Kornecook, T.J.; Bastianetto, S.; Quirion, R. Alzheimer’s disease and the basal forebrain cholinergic system: Re-lations to β-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 2002, 68, 209–245. [Google Scholar] [CrossRef]
- Farlow, M.R.; Miller, M.L.; Pejovic, V. Treatment options in Alzheimer’s disease: Maximizing benefit, managing expectations. Dement. Geriatr. Cogn. Disord. 2008, 25, 408–422. [Google Scholar] [CrossRef]
- Bayascas, J.R. PDK1: The Major Transducer of PI 3-Kinase Actions. Tuberculosis 2010, 346, 9–29. [Google Scholar] [CrossRef]
- Alessi, D.R.; Deak, M.; Casamayor, A.; Caudwell, F.B.; Morrice, N.; Norman, D.G.; Gaffney, P.; Reese, C.B.; MacDougall, C.N.; Harbison, D.; et al. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): Structural and functional homology with the Drosophila DSTPK61 kinase. Curr. Biol. 1997, 7, 776–789. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Wang, X.; Cai, C.; Zeng, S.; Bai, J.; Guo, K.; Fang, M.; Hu, J.; Liu, H.; Zhu, L.; et al. FGF21 promotes functional recovery after hypoxic-ischemic brain injury in neonatal rats by activating the PI3K/Akt signaling pathway via FGFR1/β-klotho. Exp. Neurol. 2019, 317, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of iso-form-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Wei, Y.; Han, X.; Zhao, C. PDK1 regulates the survival of the developing cortical interneurons. Mol. Brain 2020, 13, 14–65. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, J.; Cho, Y. PDK1 is a negative regulator of axon regeneration. Mol. Brain 2021, 14, 31. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Yu, L.; Hou, J.; Jackson, R.J.; Wang, H.; Huang, C.; Liu, T.; Wang, Q.; Zou, X.; Morris, R.G.; et al. Conditional Deletion of PDK1 in the Forebrain Causes Neuron Loss and Increased Apoptosis during Cortical Development. Front. Cell. Neurosci. 2017, 11, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietri, M.; Dakowski, C.; Hannaoui, S.; Alleaume-Butaux, A.; Hernandez-Rapp, J.; Ragagnin, A.; Mouil-let-Richard, S.; Haik, S.; Bailly, Y.; Peyrin, J.M.; et al. PDK1 decreases TACE-mediated α-secretase activity and promotes disease progression in prion and Alzheimer’s diseases. Nat. Med. 2013, 19, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Pascual-Guiral, S.; Ponce, R.; Gimenez-Llort, L.; Baltrons, M.; Arancio, O.; Palacio, J.R.; Clos, V.M.; Yuste, V.J.; Bayascas, J.R. Reducing the levels of Akt activation by PDK1 knock-in mutation protects neuronal cultures against synthetic amyloid-beta peptides. Front. Aging Neurosic. 2018, 9, 435. [Google Scholar] [CrossRef] [Green Version]
- Rao, C.V.; Farooqui, M.; Madhavaram, A.; Zhang, Y.; Asch, A.S.; Yamada, H.Y. GSK3-ARC/Arg3.1 and GSK3-Wnt signaling axes trigger amyloid-β accumulation and neuroinflammation in middle-aged Shugoshin 1 mice. Aging Cell 2020, 19, 13221. [Google Scholar] [CrossRef]
- Caccamo, A.; Maldonado, M.A.; Majumder, S.; Medina, D.X.; Holbein, W.; Magrí, A.; Oddo, S. Naturally secreted amyloid-beta increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J. Biol. Chem. 2011, 286, 8924–8932. [Google Scholar] [CrossRef] [Green Version]
- Velazquez, R.; Shaw, D.M.; Caccamo, A.; Oddo, S. Pim1 inhibition as a novel therapeutic strategy for Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 52. [Google Scholar] [CrossRef] [Green Version]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 11. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, N.Q.; Yan, F.; Jin, H.; Zhou, S.Y.; Shi, J.S.; Jin, F. Diabetes mellitus and Alzheimer’s disease: GSK-3beta as a potential link. Behav. Brian Res. 2018, 339, 57–65. [Google Scholar] [CrossRef]
- Campion, D.; Dumanchin, C.; Hannequin, D.; Dubois, B.; Belliard, S.; Puel, M.; Thomas-Anterion, C.; Michon, A.; Martin, C.; Charbonnier, F.; et al. Early-Onset Autosomal Dominant Alzheimer Disease: Prevalence, Genetic Heterogeneity, and Mutation Spectrum. Am. J. Hum. Genet. 1999, 65, 664–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertram, L.; Lill, C.M.; Tanzi, R.E. The Genetics of Alzheimer Disease: Back to the Future. Neuron 2010, 68, 270–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and thera-peutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular basis of familial and sporadic Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.; Coban, M.; Shahar, A.; Sankaran, B.; Hockla, A.; Lacham, S.; Caulfield, T.R.; Radisky, E.S.; Papo, N. Disulfide engineering of human Kunitz-type serine protease inhibitors enhances proteolytic stability and target affinity toward mesotrypsin. J. Biol. Chem. 2019, 294, 5105–5120. [Google Scholar] [CrossRef] [Green Version]
- Wasco, W.; Gurubhagavatula, S.; Paradis, M.D.; Romano, D.M.; Sisodia, S.S.; Hyman, B.T.; Neve, R.L.; Tanzi, R.E. Isolation and characterization of APLP2 encoding a homologue of the Alzheimer’s associated amyloid beta protein precursor. Nat. Genet. 1993, 5, 95–100. [Google Scholar] [CrossRef]
- Coulson, E.J.; Paliga, K.; Beyreuther, K.; Masters, C.L. What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem. Int. 2000, 36, 175–184. [Google Scholar] [CrossRef]
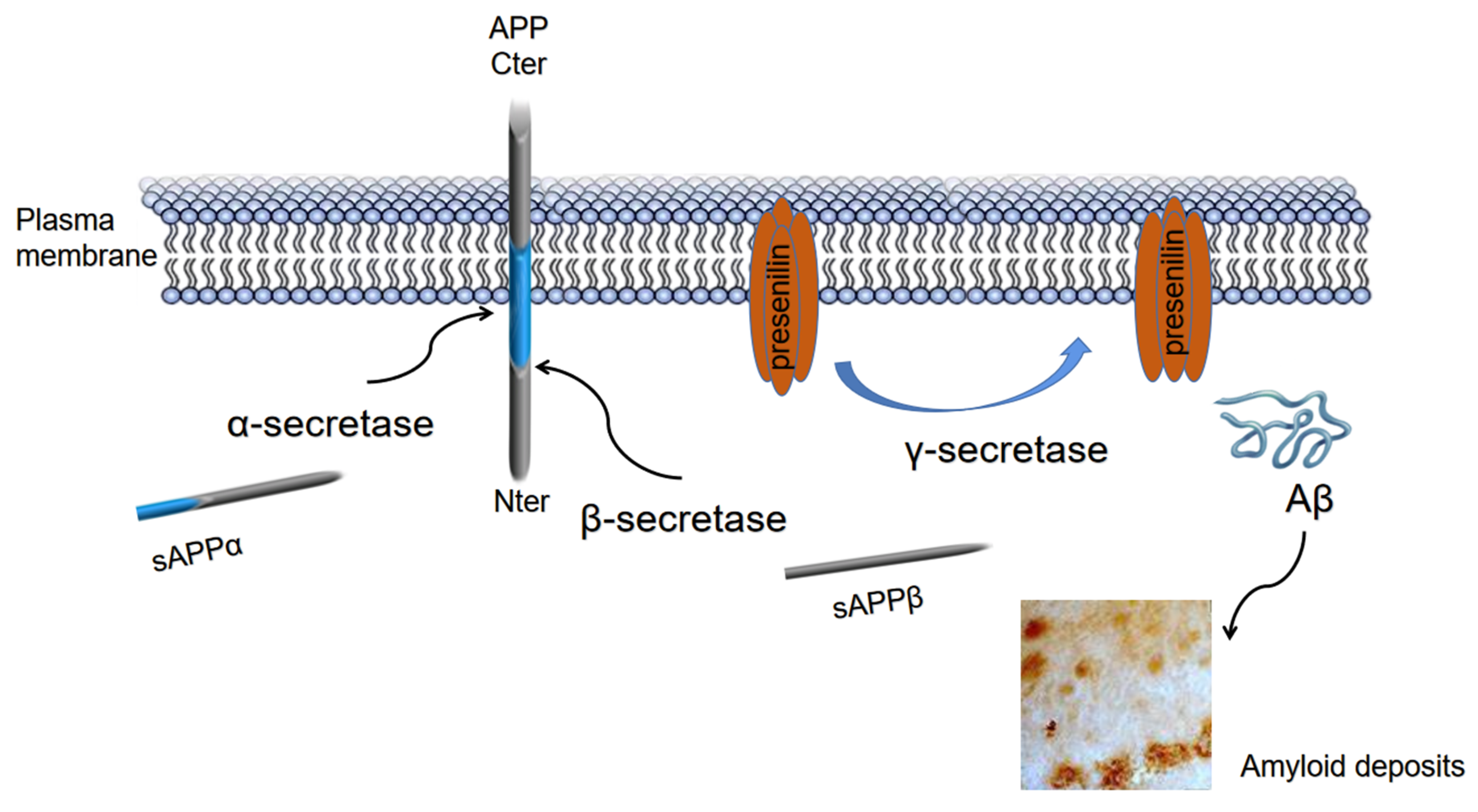
- Checler, F.; Afram, E.; Pardossi-Piquard, R.; Lauritzen, I. Is γ-secretase a beneficial inactivating enzyme of the toxic APP C-terminal fragment C99? J. Biol. Chem. 2021, 296, 100489. [Google Scholar] [CrossRef]
- Mattson, M.P. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol. Rev. 1997, 77, 1081–1132. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Wang, Y.; McCarthy, D.; Wen, H.; Borchelt, D.R.; Price, D.L.; Wong, P.C. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 2001, 4, 233–234. [Google Scholar] [CrossRef]
- Miranda, A.; Montiel, E.; Ulrich, H.; Paz, C. Selective Secretase Targeting for Alzheimer’s Disease Therapy. J. Alzheimer’s Dis. 2021, 81, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The beta-Secretase BACE1 in Alz-heimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Walsh, D.M. Alzheimer’s disease: Synaptic dysfunction and α-beta. Mol. Neurodegener. 2009, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Prà, I.; Chiarini, A.; Gui, L.; Chakravarthy, B.; Pacchiana, R.; Gardenal, E. Do astrocytes collaborate with neurons in spreading the “infectious” Aβ and tau drivers of Alzheimer’s disease? Neuroscientist 2015, 21, 9–29. [Google Scholar] [CrossRef]
- Kurz, A.; Perneczky, R. Novel insights for the treatment of Alzheimer’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 373–379. [Google Scholar] [CrossRef]
- Gillardon, F.; Rist, W.; Kussmaul, L.; Vogel, J.; Berg, M.; Danzer, K.; Kraut, N.; Hengerer, B. Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics 2007, 7, 605–616. [Google Scholar] [CrossRef]
- Fedeli, C.; Filadi, R.; Rossi, A.; Mammucari, C.; Pizzo, P. PSEN2 (presenilin 2) mutants linked to familial Alzheimer disease impair autophagy by altering Ca2+ homeostasis. Autophagy 2019, 15, 2044–2062. [Google Scholar] [CrossRef]
- Fung, S.; Smith, C.L.; Prater, K.E.; Case, A.; Green, K.; Osnis, L.; Winston, C.; Kinoshita, Y.; Sopher, B.; Morrison, R.S.; et al. Early-onset familial Alzheimer disease variant PSEN2 N141I heterozygosity is associated with altered microglia Phenotype. J. Alzhemers Dis. 2020, 77, 675–688. [Google Scholar] [CrossRef]
- Lanoiselée, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Q.; Jia, L.; Wang, Q.; Zhao, L.; Jin, H.; Li, T.; Quan, M.; Xu, L.; Li, B.; Li, Y.; et al. Identification of a novel PSEN1 Gly111Val missense mutation in a Chinese pedigree with early-onset Alzheimer’s disease. Neurobiol. Aging 2020, 85, 155. [Google Scholar] [CrossRef] [PubMed]
- Pembroke, W.G.; Hartl, C.L.; Geschwind, D.H. Evolutionary conservation and divergence of the human brain transcriptome. Genome Biol. 2021, 22, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Takami, K.; Terai, K.; Matsuo, A.; Walker, D.G.; McGeer, P.L. Expression of presenilin-1 and -2 mRNAs in rat and Alzheimer’s disease brains. Brain Res. 1997, 748, 122–130. [Google Scholar] [CrossRef]
- Lee, M.; Slunt, H.H.; Martin, L.J.; Thinakaran, G.; Kim, G.; Gandy, S.E.; Seeger, M.; Koo, E.; Price, D.L.; Sisodia, S.S. Expression of Presenilin 1 and 2 (PS1 and PS2) in Human and Murine Tissues. J. Neurosci. 1996, 16, 7513–7525. [Google Scholar] [CrossRef] [Green Version]
- Belloy, M.E.; Napolioni, V.; Greicius, M.D. A Quarter Century of APOE and Alzheimer’s Disease: Progress to Date and the Path Forward. Neuron 2019, 101, 820–838. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Liu, C.C.; Qiao, W.; Bu, G. Apolipoprotein E, receptors, and modulation of Alzheimer’s disease. Biol. Psychiatry 2018, 83, 347–357. [Google Scholar] [CrossRef]
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Share Apolipoprotein E: Structural insights and links to Alzheimer disease pathogenesis. Neuron 2021, 109, 205–221. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.-C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Huebbe, P.; Rimbach, G. Evolution of human apolipoprotein E (APOE) isoforms: Gene structure, protein function and interaction with dietary factors. Aging Res. Rev. 2017, 37, 146–161. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Liu, C.-C.; Yamazaki, A.; Shue, F.; Martens, Y.A.; Chen, Y.; Qiao, W.; Kurti, A.; Oue, H.; Ren, Y.; et al. Vascular ApoE4 Impairs Behavior by Modulating Gliovascular Function. Neuron 2020, 109, 438–447.e6. [Google Scholar] [CrossRef]
- Lindahl-Jacobsen, R.; Tan, Q.; Mengel-From, J.; Christensen, K.; Nebel, A.; Christiansen, L. Effects of the APOE epsilon2 allele on mortality and cognitive function in the oldest old. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Xiong, M.; Gratuze, M.; Bao, X.; Shi, Y.; Andhey, P.S.; Manis, M.; Schroeder, C.; Yin, Z.; Madore, C.; et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 2021, 109, 1657–1674.e7. [Google Scholar] [CrossRef] [PubMed]
- Michaelson, D.M. APOE ε4: The most prevalent yet understudied risk factor for Alzheimer’s disease. Alzheimers Dement 2014, 10, 861–868. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and Apolipoprotein E Receptors: Normal Biology and Roles in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Morillo, E.; Hansson, O.; Atagi, Y.; Bu, G.; Minthon, L.; Diamandis, E.P.; Nielsen, H.M. Total apolipo-protein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathol. 2014, 127, 633–643. [Google Scholar] [CrossRef]
- Getz, G.S.; Reardon, C.A. Apoprotein E and Reverse Cholesterol Transport. Int. J. Mol. Sci. 2018, 19, 3479. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.; Korczyn, A.D.; Karussis, D.M.; Michaelson, D.M. The effects of APOE genotype on age at onset and progression of neurodegenerative diseases. Neurology 2001, 57, 1482–1485. [Google Scholar] [CrossRef]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Tan, L.; Yu, J.T.; Tan, L. Tau in Alzheimer’s disease: Mechanisms and therapeutic strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef]
- Drummond, E.; Pires, G.; MacMurray, C.; Askenazi, M.; Nayak, S.; Bourdon, M.; Safar, J.; Ueberheide, B.; Wisniewski, T. Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain 2020, 143, 2803–2817. [Google Scholar] [CrossRef]
- Chong, F.P.; Ng, K.Y.; Koh, R.Y.; Chye, S.M. Tau Proteins and Tauopathies in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2018, 38, 965–980. [Google Scholar] [CrossRef]
- Horie, K.; Barthélemy, N.R.; Sato, C.; Bateman, R.J. CSF tau microtubule binding region identifies tau tangle and clinical stages of Alzheimer’s disease. Brain 2020, 144, 515–527. [Google Scholar] [CrossRef]
- Vossel, K.A.; Xu, J.C.; Fomenko, V.; Miyamoto, T.; Suberbielle, E.; Knox, J.A.; Ho, K.; Kim, D.H.; Yu, G.Q.; Mucke, L. Tau reduction prevents Abeta-induced axonal transport deficits by blocking activation of GSK3beta. J. Cell. Biol. 2015, 209, 419–433. [Google Scholar] [CrossRef] [Green Version]
- Vossel, K.A.; Zhang, K.; Brodbeck, J.; Daub, A.C.; Sharma, P.; Finkbeiner, S.; Cui, B.; Mucke, L. Tau reduction prevents Abeta-induced defects in axonal transport. Science 2010, 330, 198. [Google Scholar] [CrossRef] [Green Version]
- Elie, A.; Prezel, E.; Guérin, C.; Denarier, E.; Ramirez-Rios, S.; Serre, L.; Andrieux, A.; Fourest-Lieuvin, A.; Blanchoin, L.; Arnal, I. Tau co-organizes dynamic microtubule and actin networks. Sci. Rep. 2015, 5, 9964. [Google Scholar] [CrossRef] [Green Version]
- Prezel, E.; Elie, A.; Delaroche, J.; Stoppin-Mellet, V.; Bosc, C.; Serre, L.; Fourest-Lieuvin, A.; Andrieux, A.; Vantard, M.; Arnal, I. Tau can switch microtubule network organizations: From random networks to dynamic and stable bundles. Mol. Biol. Cell 2018, 29, 154–165. [Google Scholar] [CrossRef]
- Chang, C.W.; Shao, E.; Mucke, L. Tau: Enabler of diverse brain disorders and target of rapidly evolving thera-peutic strategies. Science 2021, 371, 8255. [Google Scholar] [CrossRef]
- Sohn, P.D.; Huang, C.T.-L.; Yan, R.; Fan, L.; Tracy, T.E.; Camargo, C.M.; Montgomery, K.M.; Arhar, T.; Mok, S.-A.; Freilich, R.; et al. Pathogenic Tau Impairs Axon Initial Segment Plasticity and Excitability Homeostasis. Neuron 2019, 104, 458–470.e5. [Google Scholar] [CrossRef]
- Milà-Alomà, M.; Salvadó, G.; Gispert, J.D.; Vilor-Tejedor, N.; Grau-Rivera, O.; Sala-Vila, A.; Sánchez-Benavides, G.; Arenaza-Urquijo, E.M.; Crous-Bou, M.; González-de-Echávarri, J.M.; et al. Amyloid beta, tau, synaptic, neurodegeneration, and glial biomarkers in the preclinical stage of the Alzheimer’s continuum. Alzheimers Dement 2020, 16, 1358–1371. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2015, 12, 15–27. [Google Scholar] [CrossRef]
- Sánchez, M.P.; García-Cabrero, A.M.; Sánchez-Elexpuru, G.; Burgos, D.F.; Serratosa, J.M. Tau-induced pathology in epilepsy and dementia: Notions from patients and animal models. Int. J. Mol. Sci. 2018, 19, 1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmqvist, S.; Tideman, P.; Cullen, N.; Zetterberg, H.L.; Blennow, K.; Alzheimer’s Disease Neuroimaging Initiative; Dage, J.L.; Stomrud, E.; Janelidze, S.; Mattsson-Carlgren, N.; et al. Prediction of future Alzheimer’s disease dementia using plasma phosphotau combined with other accessible measures. Nat. Med. 2021, 27, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Korman, D.; Baker, S.L.; Landau, S.M.; Jagust, W.J. Longitudinal Cognitive and Biomarker Measurements Support a Unidirectional Pathway in Alzheimer’s Disease Pathophysiology. Biol. Psychiatry 2020, 89, 786–794. [Google Scholar] [CrossRef]
- Cheng, Y.; Bai, F. The association of tau with mitochondrial dysfunction in Alzheimer’s disease. Front Neurosci. 2018, 12, 163. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Johnson, G.V.W.; Nehrke, K. The crosstalk between pathological tau phosphorylation and mitochon-drial dysfunction as a key to understanding and treating Alzheimer’s disease. Mol. Neurobiol. 2020, 57, 5103–5120. [Google Scholar] [CrossRef] [PubMed]
- Cisternas, P.; Taylor, X.; Lasagna-Reeves, C.A. The amyloid-tau-neuroinflammation axis in the context of cerebral amyloid angiopathy. Int. J. Mol. Sci. 2019, 20, 6319. [Google Scholar] [CrossRef] [Green Version]
- Bryant, A.G.; Manhard, M.K.; Salat, D.H.; Rosen, B.R.; Hyman, B.T.; Johnson, K.A.; Huang, S.; Bennett, R.E.; Yen, Y.-F. Heterogeneity of Tau Deposition and Microvascular Involvement in MCI and AD. Curr. Alzheimer Res. 2021, 18, 711–720. [Google Scholar] [CrossRef]
- Ashford, J.W. The dichotomy of Alzheimer’s disease pathology: Amyloid-beta and Tau. J. Alzheimers Dis. 2019, 68, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Sengoku, R. Aging and Alzheimer’s disease pathology. Neuropathology 2020, 40, 22–29. [Google Scholar] [CrossRef]
- Soldan, A.; Pettigrew, C.; Cai, Q.; Wang, J.; Wang, M.C.; Moghekar, A.; Miller, M.I.; Albert, M.; BIOCARD Research Team. Cognitive reserve and long-term change in cognition in aging and preclinical Alzheimer’s disease. Neurobiol Aging 2017, 60, 164–172. [Google Scholar] [CrossRef]
- Mecocci, P.; Boccardi, V.; Cecchetti, R.; Bastiani, P.; Scamosci, M.; Ruggiero, C.; Baroni, M. A long journey into aging, brain aging, and Alzheimer’s disease following the oxidative stress tracks. J. Alzheimers Dis. 2018, 62, 1319–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X.; Jiang, Q.; McDermott, J.; Han, J.-D.J. Aging and Alzheimer’s disease: Comparison and associations from molecular to system level. Aging Cell 2018, 17, e12802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, H.M.; Swerdlow, R.H. Mitochondrial links between brain aging and Alzheimer’s disease. Transl. Neurodegener. 2021, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Puli, L.; Patil, C.R. Role of reactive oxygen species in the progression of Alzheimer’s disease. Drug Discov. Today 2020, 26, 794–803. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Moulton, M.J.; Barish, S.; Ralhan, I.; Chang, J.; Goodman, L.D.; Harland, J.G.; Marcogliese, P.C.; Johansson, J.O.; Ioannou, M.S.; Bellen, H.J. Neuronal ROS-induced glial lipid droplet formation is altered by loss of Alzheimer’s dis-ease-associated genes. Proc. Natl. Acad. Sci. USA 2021, 118, e2112095118. [Google Scholar] [CrossRef]
- Nesi, G.; Sestito, S.; Digiacomo, M.; Rapposelli, S. Oxidative Stress, mitochondrial abnormalities and proteins deposition: Multitarget approaches in Alzheimer’s disease. Curr. Top Med. Chem. 2017, 17, 3062–3079. [Google Scholar]
- Luque-Contreras, D.; Carvajal, K.; Toral-Rios, D.; Franco-Bocanegra, D.; Campos-Peña, V. Oxidative stress and metabolic syndrome: Cause or consequence of Alzheimer’s disease? Oxid. Med. Cell Longev. 2014, 20, 11. [Google Scholar] [CrossRef]
- Dansokho, C.; Heneka, M.T. Neuroinflammatory responses in Alzheimer’s disease. J. Neural Transm. 2017, 125, 771–779. [Google Scholar] [CrossRef]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T.; Jalouli, M.; Rahman, M.A.; Jeandet, P.; Behl, T.; Alexiou, A.; Albadrani, G.M.; Ab-del-Daim, M.M.; Perveen, A.; et al. Neuroinflammatory signaling in the pathogenesis of Alzheimer’s disease. Curr. Neuropharmacol. 2022, 20, 126–146. [Google Scholar] [CrossRef] [PubMed]
- Bagyinszky, E.; Youn, Y.C.; An, S.S.A.; Kim, S.Y. The genetics of Alzheimer’s disease. Clin. Interv. Aging 2014, 9, 535–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Rathakrishnan, P.; Xiao, H.; Gao, T.; Duong, D.M.; Pennington, M.W.; Lah, J.J.; Seyfried, N.T.; et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemeyer, N.; Hanft, K.-M.; Akriditou, M.-A.; Unger, N.; Park, E.S.; Stanley, E.R.; Staszewski, O.; Dimou, L.; Prinz, M. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 2017, 134, 441–458. [Google Scholar] [CrossRef] [Green Version]
- Cserép, C.; Pósfai, B.; Dénes, Á. Shaping neuronal fate: Functional heterogeneity of direct microglia-neuron interactions. Neuron 2021, 109, 222–240. [Google Scholar] [CrossRef]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Ren, S.; Breuillaud, L.; Yao, W.; Yin, T.; Norris, K.A.; Zehntner, S.P.; D’Adamio, L. TNF-alpha-mediated reduc-tion in in-hibitory neurotransmission precedes sporadic Alzheimer’s disease pathology in young Trem2(R47H) rats. J. Biol. Chem. 2021, 296, 100089. [Google Scholar] [CrossRef]
- Carter, S.F.; Herholz, K.; Rosa-Neto, P.; Pellerin, L.; Nordberg, A.; Zimmer, E.R. Astrocyte Biomarkers in Alzheimer’s Disease. Trends Mol. Med. 2019, 25, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Fakhoury, M. Microglia and astrocytes in Alzheimer’s disease: Implications for therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhang, D.; Liu, Q.; Zhang, J.; Mu, K.; Gao, X.; Zhang, K.; Li, H.; Wang, Q.; Zheng, Y.; et al. Alpha-asarone Improves Cognitive Function of APP/PS1 Mice and Reducing Abeta42, P-tau and neuroinflammation, and promoting neuron survival in the hippocampus. Neuroscience 2021, 458, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Pineda, J.A.; Vera-Lopez, K.J.; Shrivastava, P.; Chávez-Fumagalli, M.A.; Nieto-Montesinos, R.; Alvarez-Fernandez, K.L.; Mamani, L.D.G.; Del-Carpio, G.D.; Gomez-Valdez, B.; Miller, C.L.; et al. Vascular smooth muscle cell dysfunction contribute to neuroinflammation and Tau hyperphosphorylation in Alzheimer disease. iScience 2021, 24, 102993. [Google Scholar] [CrossRef]
- Oliveira, J.D.; Kucharska, E.; Garcez, M.L.; Rodrigues, M.S.; Quevedo, J.; Moreno-Gonzalez, I.; Budni, J. Inflammatory cascade in Alzheimer’s disease pathogenesis: A review of experimental findings. Cells 2021, 10, 2581. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Frey, R.S.; Gao, X.; Javaid, K.; Siddiqui, S.S.; Rahman, A.; Malik, A.B. Phosphatidylinositol 3-kinase gamma signaling through protein kinase Czeta induces NADPH oxidase-mediated oxidant generation and NF-kappaB activation in endothelial cells. J. Biol. Chem. 2006, 281, 16128–16138. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Vogt, P.K. Class I PI3K in oncogenic cellular transformation. Oncogene 2008, 27, 5486–5496. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, K. Class II phosphatidylinositol 3-kinase isoforms in vesicular trafficking. Biochem. Soc. Trans. 2021, 49, 893–901. [Google Scholar] [CrossRef]
- Fyffe, C.; Buus, R.; Falasca, M. Genetic and epigenetic regulation of phosphoinositide 3-kinase isoforms. Curr. Pharm. Des. 2013, 19, 680–686. [Google Scholar] [CrossRef]
- Iacono, A.; Pompa, A.; De Marchis, F.; Panfili, E.; Greco, F.A.; Coletti, A.; Orabona, C.; Volpi, C.; Belladonna, M.L.; Mon-danelli, G.; et al. Class IA PI3Ks regulate subcellular and functional dynamics of IDO1. EMBO Rep. 2020, 21, 49756. [Google Scholar] [CrossRef] [PubMed]
- Geering, B.; Cutillas, P.R.; Vanhaesebroeck, B. Regulation of class IA PI3Ks: Is there a role for monomeric PI3K subunits? Biochem. Soc. Trans. 2007, 35, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Qiu, Y.; Kong, D. Class I phosphatidylinositol 3-kinase inhibitors for cancer therapy. Acta Pharm. Sin. B 2016, 7, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Xu, Y.; Li, M.; Chen, Z. Inhibition Sequence of miR-205 Hinders the Cell Proliferation and Migration of Lung Cancer Cells by Regulating PETN-Mediated PI3K/AKT Signal Pathway. Mol. Biotechnol. 2021, 63, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Chu, N.; Viennet, T.; Bae, H.; Salguero, A.; Boeszoermenyi, A.; Arthanari, H.; Cole, P.A. The structural determinants of PH domain-mediated regulation of Akt revealed by segmental labeling. elife 2020, 9, 59151. [Google Scholar] [CrossRef]
- Guo, J.P.; Coppola, D.; Cheng, J.Q. IKBKE protein activates Akt independent of phosphatidylinositol 3-kinase/PDK1/mTORC2 and the pleckstrin homology domain to sustain malignant transformation. J. Biol. Chem. 2016, 291, 22853. [Google Scholar] [CrossRef] [Green Version]
- Thapa, N.; Chen, M.; Horn, H.T.; Choi, S.; Wen, T.; Anderson, R.A. Phosphatidylinositol-3-OH kinase signalling is spatially organized at endosomal compartments by microtubule-associated protein 4. Nat. Cell Biol. 2020, 22, 1357–1370. [Google Scholar] [CrossRef]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef]
- Gagliardi, P.A.; Puliafito, A.; Primo, L. PDK1: At the crossroad of cancer signaling pathways. Semin. Cancer Biol. 2017, 48, 27–35. [Google Scholar] [CrossRef]
- Mora, A.; Komander, D.; van Aalten, D.; Alessi, D.R. PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol. 2004, 15, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Hossen, M.J.; Kim, S.C.; Yang, S.; Kim, H.G.; Jeong, D.; Yi, Y.-S.; Sung, N.Y.; Lee, J.-O.; Kim, J.-H.; Cho, J.Y. PDK1 disruptors and modulators: A patent review. Expert Opin. Ther. Patents 2015, 25, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Falasca, M. Targeting PDK1 in cancer. Curr. Med. Chem. 2011, 18, 2763–2769. [Google Scholar] [CrossRef] [PubMed]
- Bayascas, J.R. Dissecting the role of the 3-phosphoinositide-dependent protein kinase-1 (PDK1) signalling pathways. Cell Cycle 2008, 7, 2978–2982. [Google Scholar] [CrossRef]
- Komander, D.; Kular, G.; Deak, M.; Alessi, D.R.; van Aalten, D.M. Role of T-loop phosphorylation in PDK1 activation, stability, and substrate binding. J. Biol. Chem. 2005, 280, 18797–18802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, M.J.; Ramos, F.J.; Chen, H.; Quon, M.; Dong, L.Q.; Liu, F. Mouse 3-Phosphoinositide-dependent Protein Kinase-1 Undergoes Dimerization and trans-Phosphorylation in the Activation Loop. J. Biol. Chem. 2003, 278, 42913–42919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Suárez, M.; Gutiérrez-Martínez, I.Z.; Hernández-Trejo, J.A.; Hernández-Ruiz, M.; Suárez-Pérez, D.; Candelario, A.; Kamekura, R.; Medina-Contreras, O.; Schnoor, M.; Ortiz-Navarrete, V.; et al. 14-3-3 Proteins regulate Akt Thr308 phosphorylation in intestinal epithelial cells. Cell Death Differ. 2016, 23, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dan, H.C.; Antonia, R.; Baldwin, A.S. PI3K/Akt promotes feedforward mTORC2 activation through IKKα. Oncotarget 2016, 7, 21064–21075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busschots, K.; Lopez-Garcia, L.A.; Lammi, C.; Stroba, A.; Zeuzem, S.; Piiper, A.; Alzari, P.M.; Neimanis, S.; Arencibia, J.M.; Engel, M.; et al. Substrate-Selective Inhibition of Protein Kinase PDK1 by Small Compounds that Bind to the PIF-Pocket Allosteric Docking Site. Chem. Biol. 2012, 19, 1152–1163. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Harris, T.K. Steady-state Kinetic Mechanism of PDK1. J. Biol. Chem. 2006, 281, 21670–21681. [Google Scholar] [CrossRef] [Green Version]
- Balendran, A.; Casamayor, A.; Deak, M.; Paterson, A.; Gaffney, P.; Currie, R.; Downes, C.; Alessi, D.R. PDK1 acquires PDK2 activity in the presence of a synthetic peptide derived from the carboxyl terminus of PRK2. Curr. Biol. 1999, 9, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Jafari, M.; Ghadami, E.; Dadkhah, T.; Akhavan-Niaki, H. PI3k/AKT signaling pathway: Erythropoiesis and beyond. J. Cell. Physiol. 2018, 234, 2373–2385. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, S.; Mauro, A.; Cellini, V.; Patacchiola, F. The role of Akt signalling in the mammalian ovary. Int. J. Dev. Biol. 2012, 56, 809–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degan, S.E.; Gelman, I.H. Emerging Roles for AKT Isoform Preference in Cancer Progression Pathways. Mol. Cancer Res. 2021, 19, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Calleja, V.; Laguerre, M.; Larijani, B. Role of the C-terminal regulatory domain in the allosteric inhibition of PKB/Akt. Adv. Biol. Regul. 2012, 52, 46–57. [Google Scholar] [CrossRef]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef] [Green Version]
- Garza, J.C.; Guo, M.; Zhang, W.; Lu, X.-Y. Leptin restores adult hippocampal neurogenesis in a chronic unpre-dictable stress model of depression and reverses glucocorticoid-induced inhibition of GSK-3β/β-catenin signaling. Mol. Psychiatry 2012, 17, 790–808. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Chen, J.; Chen, H.; Duan, Z.; Xu, Q.; Wei, M.; Wang, L.; Zhong, M. Leptin regulates proliferation and apoptosis of colorectal carcinoma through PI3K/Akt/mTOR signalling pathway. J. Biosci. 2011, 37, 91–101. [Google Scholar] [CrossRef]
- Cowan, C.M.; Bossing, T.; Page, A.; Shepherd, D.; Mudher, A. Soluble hyperphosphorylated tau causes micro-tubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol. 2010, 120, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Lippens, G.; Sillen, A.; Landrieu, I.; Amniai, L.; Sibille, N.; Barbier, P.; Leroy, A.; Hanoulle, X.; Wieruszeski, J.M. Tau aggregation in Alzheimer’s disease: What role for phosphorylation? Prion 2007, 1, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Huang, H. Interplay Among PI3K/AKT, PTEN/FOXO and AR Signaling in Prostate Cancer. Adv. Exp. Med. Biol. 2019, 1210, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Farhan, M.; Wang, H.; Gaur, U.; Little, P.; Xu, J.; Zheng, W. FOXO Signaling Pathways as Therapeutic Targets in Cancer. Int. J. Biol. Sci. 2017, 13, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Kitagishi, Y.; Nakanishi, A.; Ogura, Y.; Matsuda, S. Dietary regulation of PI3K/AKT/GSK-3β pathway in Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaworski, J.; Spangler, S.; Seeburg, D.P.; Hoogenraad, C.; Sheng, M. Control of Dendritic Arborization by the Phosphoinositide-3′-Kinase-Akt-Mammalian Target of Rapamycin Pathway. J. Neurosci. 2005, 25, 11300–11312. [Google Scholar] [CrossRef]
- Kumar, V.; Zhang, M.-X.; Swank, M.W.; Kunz, J.; Wu, G.-Y. Regulation of Dendritic Morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK Signaling Pathways. J. Neurosci. 2005, 25, 11288–11299. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, L.; Pei, L.; Ju, W.; Ahmadian, G.; Lu, J.; Wang, Y.; Liu, F.; Wang, Y.T. Control of Synaptic Strength, a Novel Function of Akt. Neuron 2003, 38, 915–928. [Google Scholar] [CrossRef] [Green Version]
- Horwood, J.M.; Dufour, F.; Laroche, S.; Davis, S. Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur. J. Neurosci. 2006, 23, 3375–3384. [Google Scholar] [CrossRef]
- Griffin, R.J.; Moloney, A.; Kelliher, M.; Johnston, J.A.; Ravid, R.; Dockery, P.; O’Connor, R.; O’Neill, C. Activa-tion of Akt/AKT, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J. Neurochem. 2005, 93, 105–117. [Google Scholar] [CrossRef]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signaling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef]
- Sonoda, Y.; Mukai, H.; Matsuo, K.; Takahashi, M.; Ono, Y.; Maeda, K.; Akiyama, H.; Kawamata, T. Accumula-tion of tu-mor-suppressor PTEN in Alzheimer neurofibrillary tangles. Neurosci. Lett. 2010, 471, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, L.; Fernandez, F.; Johnson, J.B.; Naiker, M.; Owoola, A.G.; Broszczak, D.A. Oxidative stress in alzheimer’s disease: A review on emergent natural polyphenolic therapeutics. Complement Ther. Med. 2020, 49, 102294. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.; Kiely, A.P.; Coakley, M.F.; Manning, S.; Long-Smith, C.M. Insulin and IGF-1 signaling: Longevity, protein homoeostasis and Alzheimer’s disease. Biochem. Soc. Trans. 2012, 40, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin Invest. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.-X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef]
- Neill, C.O. PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp. Gerontol. 2013, 48, 647–653. [Google Scholar] [CrossRef]
- Willette, A.A.; Johnson, S.C.; Birdsill, A.C.; Sager, M.A.; Christian, B.; Baker, L.D.; Craft, S.; Oh, J.; Statz, E.; Hermann, B.P.; et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimer’s Dement 2015, 11, 504–510. [Google Scholar] [CrossRef] [Green Version]
- Razani, E.; Pourbagheri-Sigaroodi, A.; Safaroghli-Azar, A.; Zoghi, A.; Shanaki-Bavarsad, M.; Bashash, D. The PI3K/Akt signaling axis in Alzheimer’s disease: A valuable target to stimulate or suppress? Cell Stress Chaperones. 2021, 26, 871–887. [Google Scholar] [CrossRef]
- Gao, X.; Harris, T.K. Role of the PH domain in regulating in vitro autophosphorylation events required for reconstitution of PDK1 catalytic activity. Bioorg. Chem. 2006, 34, 200–223. [Google Scholar] [CrossRef]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 Is an Insulin-Regulated Inhibitor of the mTORC1 Protein Kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Y.; Hsieh, T.; Wang, C.; Cheng, K.; Wang, L.; Lin, T.; Cheung, C.H.A.; Wu, C.; Chiang, H. Aging-induced Akt activation involves in aging-related pathologies and Aβ-induced toxicity. Aging Cell 2019, 18, e12989. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Mei, Z.; Wu, J.-W.; Wang, Z.-X. Enzymatic Activity and Substrate Specificity of Mitogen-activated Protein Kinase p38α in Different Phosphorylation States. J. Biol. Chem. 2008, 283, 26591–26601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.H.; de Pablo, Y.; Lee, H.P.; Lee, H.G.; Smith, M.A.; Shah, K. Cdk5 is a major regulator of p38 cascade: Relevance to neurotoxicity in Alzheimer’s disease. J. Neurochem. 2010, 113, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Teng, Y.; Zhang, X.; Lv, X.; Yin, Y. Metformin Alleviated Aβ-Induced Apoptosis via the Suppression of JNK MAPK Signaling Pathway in Cultured Hippocampal Neurons. BioMed Res. Int. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Kim, N.J. Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of Alzheimer’s disease. Molecules 2017, 22, 1287. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.Q.; Sun, X.B.; Xu, Y.X.; Zhao, H.; Zhu, Q.Y.; Zhu, C.Q. Astaxanthin upregulates heme oxygenase-1 expression through ERK1/2 pathway and its protective effect against β-amyloid-induced cytotoxicity in SH-SY5Y cells. Brain Res. 2010, 1360, 159–167. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, J.L.; Wang, Y.R.; Fa, X.Z. Apigenin attenuates copper-mediated β-amyloid neurotoxicity through antioxidation, mitochondrion protection and mapk signal inactivation in an ad cell model. Brain Res. 2013, 1492, 33–45. [Google Scholar] [CrossRef]
- Dresselhaus, E.C.; Boersma, M.C.H.; Meffert, M.K. Targeting of NF-κB to dendritic spines is required for synaptic signaling and spine development. J. Neurosci. 2018, 38, 4093–4103. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Zhou, D.; Bruce-Keller, A.J.; Kindy, M.S.; Mattson, M.P. Lack of the p50 subunit of nuclear factor-kappaB increases the vulnerability of hippocampal neurons to excitotoxic injury. J. Neurosci. 1999, 19, 8856–8865. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Hong, Y.; Zhang, K.; Wang, J.; Zheng, L.; Zhang, Z.; Hu, Z.; Han, X.; Han, Y.; Chen, T.; et al. BAG-1M coactivates BACE1 transcription through NF-kappaB and accelerates Abeta production and memory deficit in Alzheimer’s disease mouse model. Biochim. Biophys. Acta Mol. Basis. Dis. 2017, 9, 2398–2407. [Google Scholar] [CrossRef]
- Cheng, F.; Fransson, L.; Mani, K. Proinflammatory cytokines induce accumulation of glypican-1-derived heparan sulfate and the C-terminal fragment of β-cleaved APP in autophagosomes of dividing neuronal cells. Glycobiology 2020, 30, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-J.; Koh, S.-H. The role of PI3K/AKT pathway and its therapeutic possibility in Alzheimer’s disease. Hanyang Med. Rev. 2017, 37, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Oomura, Y.; Hori, N.; Shiraishi, T.; Fukunaga, K.; Takeda, H.; Tsuji, M.; Matsumiya, T.; Ishibashi, M.; Aou, S.; Li, X.; et al. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides 2006, 27, 2738–2749. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.; Solovyova, N.; Irving, A. Leptin and its role in hippocampal synaptic plasticity. Prog. Lipid Res. 2006, 45, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Duvall, A.; Gallicchio, V. Lithium treatment in clinical medicine: History, current status and future use. J. Cell. Sci. Ther. 2017, 8, 3. [Google Scholar] [CrossRef]
- Komander, D.; Fairservice, A.; Deak, M.; Kular, G.S.; Prescott, A.; Downes, C.P.; Safrany, S.; Alessi, D.; van Aalten, D. Structural insights into the regulation of PDK1 by phosphoinositides and inositol phosphates. EMBO J. 2004, 23, 3918–3928. [Google Scholar] [CrossRef] [Green Version]
- Bayascas, J.R.; Wullschleger, S.; Sakamoto, K.; García-Martínez, J.M.; Clacher, C.; Komander, D.; van Aalten, D.M.F.; Boini, K.M.; Lang, F.; Lipina, C.; et al. Mutation of the PDK1 PH Domain Inhibits Protein Kinase B/Akt, Leading to Small Size and Insulin Resistance. Mol. Cell. Biol. 2008, 28, 3258–3272. [Google Scholar] [CrossRef] [Green Version]
- Zurashvili, T.; Cordon-Barris, L.; Ruiz-Babot, G.; Zhou, X.; Gomez, N.; Gimenez-Llort, L.; Bayascas, J.R. Inter-action of PDK1 with phosphoinositides is essential for neuronal differentiation but dispensable for neuronal survival. Mol. Cell Biol. 2013, 33, 1027–1040. [Google Scholar] [CrossRef] [Green Version]
- Najafov, A.; Shpiro, N.; Alessi, D.R. Akt is efficiently activated by PIF-pocket- and PtdIns(3,4,5)P3-dependent mechanisms leading to resistance to PDK1 inhibitors. Biochem. J. 2012, 448, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Cordon-Barris, L.; Zurashvili, T.; Bayascas, J.R. Fine-tuning the intensity of the AKT/Akt signal enables diverse physiological responses. Cell Cycle 2014, 13, 3164–3168. [Google Scholar] [CrossRef] [Green Version]
- Giménez-Llort, L.; Santana-Santana, M.; Bayascas, J.R. The impact of the PI3K/Akt signaling pathway in anxi-ety and working memory in young and middle-aged PDK1 K465E knock-in mice. Front Behav. Neurosci. 2020, 14, 61. [Google Scholar] [CrossRef] [PubMed]
- Akhondzadeh, S.; Noroozian, M.; Mohammadi, M.; Ohadinia, S.; Jamshidi, A.H.; Khani, M. Salvia officinalis extract in the treatment of patients with mild to moderate Alzheimer’s disease: A double blind, randomized and placebo-controlled trial. J. Clin. Pharm. Ther. 2003, 28, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, X.; Teng, Z.; Zhang, T.; Li, Y. Downregulation of PI3K/Akt/mTOR signaling pathway in curcumin-induced autophagy in APP/PS1 double transgenic mice. Eur. J. Pharmacol. 2014, 740, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Xiong, N.; Jia, M.; Chen, C.; Xiong, J.; Zhang, Z.; Huang, J.; Hou, L.; Yang, H.; Cao, X.; Liang, Z.; et al. Potential autophagy enhancers attenuate rotenone-induced toxicity in SH-SY5Y. Neuroscience 2011, 199, 292–302. [Google Scholar] [CrossRef]
- Zhang, L.; Fang, Y.; Cheng, X.; Lian, Y.J.; Xu, H.L.; Zeng, Z.S.; Zhu, H.C. TRPML1 participates in the progression of Alzheimer’s disease by regulating the PPARγ/AMPK/Mtor signalling pathway. Cell Physiol. Biochem. 2017, 43, 2446–2456. [Google Scholar] [CrossRef]
- Tramutola, A.; Lanzillotta, C.; Di Domenico, F. Targeting mTOR to reduce Alzheimer-related cognitive decline: From current hits to future therapies. Expert Rev. Neurother. 2016, 17, 33–45. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhang, J.; Zhu, Y.G.; Zhang, J.; Shen, H.; Lu, J.P.; Pan, X.D.; Lin, N.; Dai, X.M.; Zhou, M.; et al. Tripchlorolide improves cognitive deficits by reducing amyloid β and upregulating synapse-related proteins in a transgenic model of Alzheimer’s Disease. J. Neurochem. 2015, 133, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.T.; Zhang, Q.; Gao, X.R.; Ding, F. An active fraction of Achyranthes bidentata polypeptides prevents apoptosis in-duced by serum deprivation in SH-SY5Y cells through activation of PI3K/AKT/Gsk3β pathways. Neurochem. Res. 2011, 36, 2186–2194. [Google Scholar] [CrossRef]
- Zaytseva, Y.Y.; Valentino, J.D.; Gulhati, P.; Evers, B.M. mTOR inhibitors in cancer therapy. Cancer Lett. 2012, 319, 1–7. [Google Scholar] [CrossRef]
- Ahmed, A.R.; Candeo, A.; D’Abrantes, S.; Needham, S.R.; Yadav, R.B.; Botchway, S.W.; Parker, A.W. Directly imaging the localisation and photosensitization properties of the panmTOR inhibitor, AZD2014, in living cancer cells. J. Photochem. Photobiol. B 2020, 213, 112055. [Google Scholar] [CrossRef]
- Manterola, L.; Hernando-Rodríguez, M.; Ruiz, A.; Apraiz, A.; Arrizabalaga, O.; Vellón, L.; Alberdi, E.; Cavaliere, F.; Lacerda, H.M.; Jimenez, S.; et al. 1-42 β-amyloid peptide requires PDK1/nPKC/Rac 1 pathway to induce neuronal death. Transl. Psychiatry 2013, 3, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santana-Santana, M.; Bayascas, J.-R.; Giménez-Llort, L. Fine-Tuning the PI3K/Akt Signaling Pathway Intensity by Sex and Genotype-Load: Sex-Dependent Homozygotic Threshold for Somatic Growth but Feminization of Anxious Phenotype in Middle-Aged PDK1 K465E Knock-In and Heterozygous Mice. Biomedicines 2021, 9, 747. [Google Scholar] [CrossRef] [PubMed]
- Gooz, M. ADAM-17: The enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 146–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef] [PubMed]
- Ezpeleta, J.; Baudouin, V.; Arellano-Anaya, Z.E.; Boudet-Devaud, F.; Pietri, M.; Baudry, A.; Haeberlé, A.-M.; Bailly, Y.; Kellermann, O.; Launay, J.-M.; et al. Production of seedable Amyloid-β peptides in model of prion diseases upon PrP Sc-induced PDK1 overactivation. Nat. Commun. 2019, 10, 3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumbo, S.; Paterson, C.; Yang, F.; Hood, V.L.; Law, A.J. PKBbeta/AKT2 deficiency impacts brain mTOR sig-naling, pre-frontal cortical physiology, hippocampal plasticity and select murine behaviors. Mol. Psychiatry 2021, 26, 411–428. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Molecular Mechanisms | References |
|---|---|---|
| Salvia officinalis | Activates PI3K/Akt | [183] |
| Curcumin | Activates PI3K/Akt/mTOR | [184] |
| Leptin | Activates PI3K/Akt | [138,139] |
| Tripchlorolide | Activates PI3K/Akt/mTOR | [188] |
| Achyranthes | Activates PI3K/Akt | [189] |
| Lithium | Activates the PI3K/Akt axis | [185] |
| Rapamycin | Inhibits mTORC1 | [186] |
| PI103 | Inhibits mTORC1 | [190] |
| AZD8055 | Inhibits mTORC1 and mTORC2 | [187] |
| INK128 | Inhibits mTORC1 and mTORC2 | [191] |
| BX912 | Inhibits PDK1 | [15] |
| OSU03012 | Inhibits PDK1 | [192] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Du, Y.; Zhao, X.; Wu, C.; Yu, P. Reducing PDK1/Akt Activity: An Effective Therapeutic Target in the Treatment of Alzheimer’s Disease. Cells 2022, 11, 1735. https://doi.org/10.3390/cells11111735
Yang S, Du Y, Zhao X, Wu C, Yu P. Reducing PDK1/Akt Activity: An Effective Therapeutic Target in the Treatment of Alzheimer’s Disease. Cells. 2022; 11(11):1735. https://doi.org/10.3390/cells11111735
Chicago/Turabian StyleYang, Shaobin, Yaqin Du, Xiaoqian Zhao, Chendong Wu, and Peng Yu. 2022. "Reducing PDK1/Akt Activity: An Effective Therapeutic Target in the Treatment of Alzheimer’s Disease" Cells 11, no. 11: 1735. https://doi.org/10.3390/cells11111735
APA StyleYang, S., Du, Y., Zhao, X., Wu, C., & Yu, P. (2022). Reducing PDK1/Akt Activity: An Effective Therapeutic Target in the Treatment of Alzheimer’s Disease. Cells, 11(11), 1735. https://doi.org/10.3390/cells11111735