
Interleukin-1 and Nuclear Factor Kappa B Signaling Promote Breast Cancer Progression and Treatment Resistance


Abstract
1. Introduction
2. IL-1 Expression and Signaling Is Associated with Breast Cancer (BCa) Disease Progression
2.1. IL-1 Accumulation Is Elevated in BCa
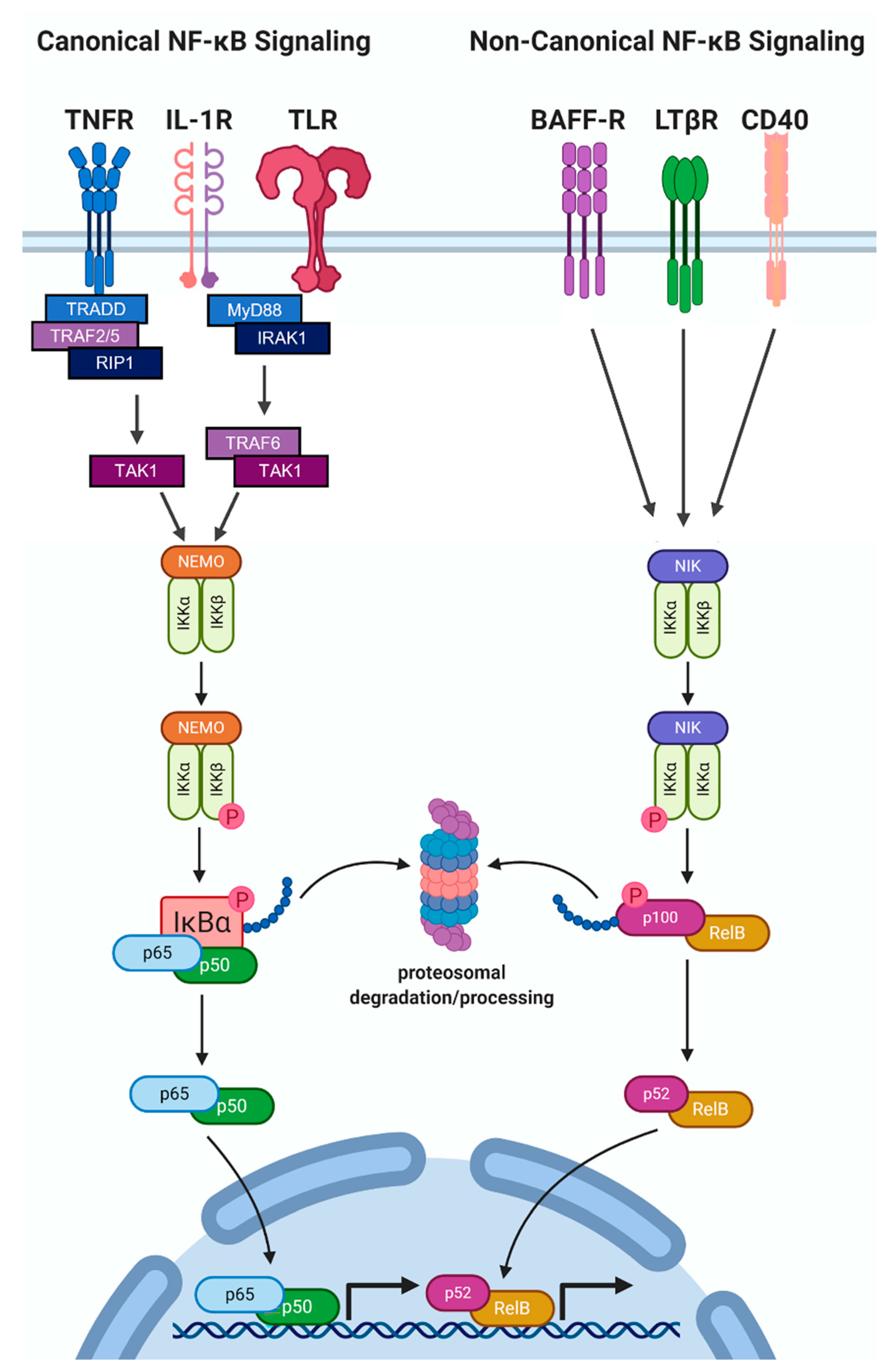
2.2. NF-κB Protein Accumulation and Activity Are High in Hormone Receptor Negative BCa
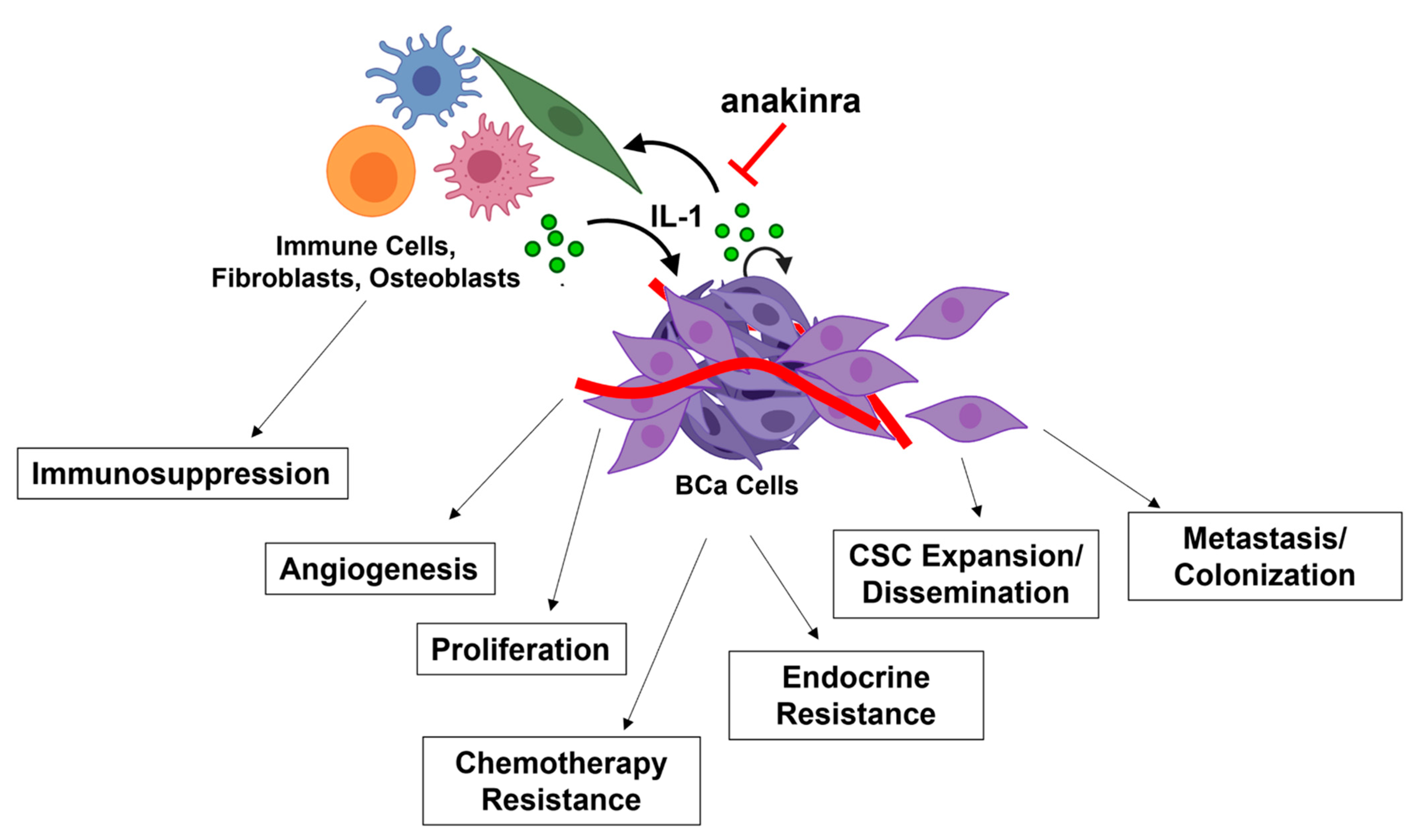
3. The Role of IL-1-NF-κB in BCa Progression
3.1. Proliferation
3.2. Clonogenicity/Stemness
3.3. Angiogenesis
3.4. Metastasis
4. The Role of IL-1 in BCa Therapeutic Resistance
4.1. Endocrine Resistance
4.2. Chemotherapy Resistance
4.3. Immunotherapy and Immunosuppression
5. Anakinra Clinical Trials
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed]
- Verstrepen, L.; Bekaert, T.; Chau, T.-L.; Tavernier, J.; Chariot, A.; Beyaert, R. TLR-4, IL-1R and TNF-R signaling to NF-kappaB: Variations on a common theme. Cell. Mol. Life Sci. 2008, 65, 2964–2978. [Google Scholar] [CrossRef] [PubMed]
- Wesche, H.; Henzel, W.J.; Shillinglaw, W.; Li, S.; Cao, Z. MyD88: An Adapter That Recruits IRAK to the IL-1 Receptor Complex. Immunity 1997, 7, 837–847. [Google Scholar] [CrossRef]
- Pyrillou, K.; Burzynski, L.C.; Clarke, M.C.H. Alternative Pathways of IL-1 Activation, and Its Role in Health and Disease. Front. Immunol. 2020, 11, 613170. [Google Scholar] [CrossRef] [PubMed]
- Munn, L.L. Cancer and inflammation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2017, 9, e1370. [Google Scholar] [CrossRef] [PubMed]
- Vilsmaier, T.; Rack, B.; König, A.; Friese, K.; Janni, W.; Jeschke, U.; Acher, T.W. Influence of Circulating Tumour Cells on Production of IL-1α, IL-1β and IL-12 in Sera of Patients with Primary Diagnosis of Breast Cancer before Treatment. Anticancer Res. 2016, 36, 5227–5236. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, S.; Lee, J.S.; Jie, C.; Park, M.H.; Iwakura, Y.; Patel, Y.; Soni, M.; Reisman, D.; Chen, H. HER2 Overexpression Triggers an IL1α Proinflammatory Circuit to Drive Tumorigenesis and Promote Chemotherapy Resistance. Cancer Res. 2018, 78, 2040–2051. [Google Scholar] [CrossRef]
- Al-Hassan, A.A.; Al-Ghurabi, B.; Al-Karkhi, I. Prognostic Value of Proinflammatory Cytokines in Breast Cancer. J. Biomol. Res. Ther. 2012, 1, 2. [Google Scholar] [CrossRef]
- Miller, L.J.; Kurtzman, S.H.; Anderson, K.; Wang, Y.; Stankus, M.; Renna, M.; Lindquist, R.; Barrows, G.; Kreutzer, D.L. Interleukin-1 family expression in human breast cancer: Interleukin-1 receptor antagonist. Cancer Investig. 2000, 18, 293–302. [Google Scholar] [CrossRef]
- Singer, C.F.; Hudelist, G.; Gschwantler-Kaulich, D.; Fink-Retter, A.; Mueller, R.; Walter, I.; Czerwenka, K.; Kubista, E. Interleukin-1alpha protein secretion in breast cancer is associated with poor differentiation and estrogen receptor alpha negativity. Int. J. Gynecol. Cancer 2006, 16 (Suppl. S2), 556–559. [Google Scholar] [CrossRef]
- Wu, T.-C.; Xu, K.; Martinek, J.; Young, R.R.; Banchereau, R.; George, J.; Turner, J.; Kim, K.I.; Zurawski, S.; Wang, X.; et al. IL1 Receptor Antagonist Controls Transcriptional Signature of Inflammation in Patients with Metastatic Breast Cancer. Cancer Res. 2018, 78, 5243–5258. [Google Scholar] [CrossRef] [PubMed]
- Tulotta, C.; Lefley, D.V.; Freeman, K.; Gregory, W.M.; Hanby, A.M.; Heath, P.R.; Nutter, F.; Wilkinson, J.M.; Spicer-Hadlington, A.R.; Liu, X.; et al. Endogenous Production of IL1B by Breast Cancer Cells Drives Metastasis and Colonization of the Bone Microenvironment. Clin. Cancer Res. 2019, 25, 2769–2782. [Google Scholar] [CrossRef] [PubMed]
- Chavey, C.; Bibeau, F.; Gourgou-Bourgade, S.; Burlinchon, S.; Boissière, F.; Laune, D.; Roques, S.; Lazennec, G. Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 2007, 9, R15. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Yuan, R.Q.; Fuchs, A.; Yao, Y.; Joseph, A.; Schwall, R.; Schnitt, S.J.; Guida, A.; Hastings, H.M.; Andres, J.; et al. Expression of interleukin-1beta in human breast carcinoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1997, 80, 421–434. [Google Scholar]
- Soria, G.; Ofri-Shahak, M.; Haas, I.; Yaal-Hahoshen, N.; Leider-Trejo, L.; Leibovich-Rivkin, T.; Weitzenfeld, P.; Meshel, T.; Shabtai, E.; Gutman, M.; et al. Inflammatory mediators in breast cancer: Coordinated expression of TNFα & IL-1β with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer 2011, 11, 130. [Google Scholar]
- Uscanga-Perales, G.I.; Santuario-Facio, S.K.; Ortiz-López, R. Triple negative breast cancer: Deciphering the biology and heterogeneity. Med. Univ. 2016, 18, 105–114. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Cordle, S.R.; Donald, R.; Read, M.A.; Hawiger, J. Lipopolysaccharide induces phosphorylation of MAD3 and activation of c-Rel and related NF-kappa B proteins in human monocytic THP-1 cells. J. Biol. Chem. 1993, 268, 11803–11810. [Google Scholar] [CrossRef]
- Derudder, E.; Dejardin, E.; Pritchard, L.L.; Green, D.R.; Körner, M.; Baud, V. RelB/p50 Dimers Are Differentially Regulated by Tumor Necrosis Factor-α and Lymphotoxin-β Receptor Activation. J. Biol. Chem. 2003, 278, 23278–23284. [Google Scholar] [CrossRef]
- Hiscott, J.; Marois, J.; Garoufalis, J.; D’Addario, M.; Roulston, A.; Kwan, I.; Pepin, N.; Lacoste, J.; Nguyen, H.; Bensi, G. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: Evidence for a positive autoregulatory loop. Mol. Cell. Biol. 1993, 13, 6231–6240. [Google Scholar]
- Shi, J.-H.; Sun, S.-C. Tumor Necrosis Factor Receptor-Associated Factor Regulation of Nuclear Factor κB and Mitogen-Activated Protein Kinase Pathways. Front. Immunol. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Claudio, E.; Brown, K.; Park, S.; Wang, H.; Siebenlist, U. BAFF-induced NEMO-independent processing of NF-κB2 in maturing B cells. Nat. Immunol. 2002, 3, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Coope, H.J. CD40 regulates the processing of NF-kappaB2 p100 to p52. EMBO J. 2002, 21, 5375–5385. [Google Scholar] [CrossRef]
- Dejardin, E.; Droin, N.M.; Delhase, M.; Haas, E.; Cao, Y.; Makris, C.; Li, Z.-W.; Karin, M.; Ware, C.F.; Green, D.R. The Lymphotoxin-β Receptor Induces Different Patterns of Gene Expression via Two NF-κB Pathways. Immunity 2002, 17, 525–535. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Zheng, L.H.; Zhao, Y.H.; Feng, H.L.; Liu, Y.J. Endocrine resistance in breast cancer. Climacteric 2014, 17, 522–528. [Google Scholar] [CrossRef]
- Biswas, D.K.; Shi, Q.; Baily, S.; Strickland, I.; Ghosh, S.; Pardee, A.B.; Iglehart, J.D. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10137–10142. [Google Scholar] [CrossRef]
- Zhou, Y.; Eppenberger-Castori, S.; Eppenberger, U.; Benz, C.C. The NFkappaB pathway and endocrine-resistant breast cancer. Endocr. Relat. Cancer 2005, 12 (Suppl. S1), S37–S46. [Google Scholar] [CrossRef]
- Rojo, F.; González-Pérez, A.; Furriol, J.; Nicolau, M.J.; Ferrer, J.; Burgués, O.; Sabbaghi, M.; González-Navarrete, I.; Cristobal, I.; Serrano, L.; et al. Non-canonical NF-κB pathway activation predicts outcome in borderline oestrogen receptor positive breast carcinoma. Br. J. Cancer 2016, 115, 322–331. [Google Scholar] [CrossRef]
- Oida, K.; Matsuda, A.; Jung, K.; Xia, Y.; Jang, H.; Amagai, Y.; Ahn, G.; Nishikawa, S.; Ishizaka, S.; Jensen-Jarolim, E.; et al. Nuclear factor-ĸB plays a critical role in both intrinsic and acquired resistance against endocrine therapy in human breast cancer cells. Sci. Rep. 2014, 4, 4057. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Belguise, K.; O’Neill, C.F.; Sánchez-Morgan, N.; Romagnoli, M.; Eddy, S.F.; Mineva, N.D.; Yu, Z.; Min, C.; Trinkaus-Randall, V.; et al. RelB NF-kappaB represses estrogen receptor alpha expression via induction of the zinc finger protein Blimp1. Mol. Cell. Biol. 2009, 29, 3832–3844. [Google Scholar] [CrossRef] [PubMed]
- Nawas, A.F.; Mistry, R.; Narayanan, S.; Thomas-Jardin, S.E.; Ramachandran, J.; Ravichandran, J.; Neduvelil, E.; Luangpanh, K.; Delk, N.A. IL-1 induces p62/SQSTM1 and autophagy in ERα+/PR+ BCa cell lines concomitant with ERα and PR repression, conferring an ERα−/PR− BCa-like phenotype. J. Cell. Biochem. 2018, 120, 1477–1491. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.A.A.; Patel, V.; Gwede, M.; Morgado, M.; Tomasevich, K.; Fong, E.L.L.; Farach-Carson, M.C.C.; Delk, N.A. IL-1β Induces p62/SQSTM1 and Represses Androgen Receptor Expression in Prostate Cancer Cells. J. Cell Biochem. 2014, 115, 2188–2197. [Google Scholar] [CrossRef] [PubMed]
- Thomas-Jardin, S.E.; Dahl, H.; Kanchwala, M.S.; Ha, F.; Jacob, J.; Soundharrajan, R.; Bautista, M.; Nawas, A.F.; Robichaux, D.; Mistry, R.; et al. RELA is sufficient to mediate interleukin-1 repression of androgen receptor expression and activity in an LNCaP disease progression model. Prostate 2020, 80, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Korniluk, A.; Koper, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Irish J. Med. Sci. 2017, 186, 57–62. [Google Scholar] [CrossRef]
- Danforth, D.N.; Sgagias, M.K. Interleukin 1 alpha blocks estradiol-stimulated growth and down-regulates the estrogen receptor in MCF-7 breast cancer cells in vitro. Cancer Res. 1991, 51, 1488–1493. [Google Scholar]
- Sgagias, M.K.; Kasid, A.; Danforth, D.N. Interleukin-1 alpha and tumor necrosis factor-alpha (TNF alpha) inhibit growth and induce TNF messenger RNA in MCF-7 human breast cancer cells. Mol. Endocrinol. 1991, 5, 1740–1747. [Google Scholar] [CrossRef][Green Version]
- Dahl, H.C.; Kanchwala, M.; Thomas-jardin, S.E.; Sandhu, A.; Kanumuri, P.; Xing, C.; Delk, N.A.; Nawas, A.F.; Xing, C.; Lin, C.; et al. Chronic IL-1 exposure drives LNCaP cells to evolve androgen and AR independence. PLoS ONE 2020, 15, e0242970. [Google Scholar] [CrossRef]
- Culig, Z.; Hobisch, A.; Herold, M.; Hittmair, A.; Thurnher, M.; Eder, I.E.; Cronauer, M.V.; Rieser, C.; Ramoner, C.; Bartsch, G.; et al. Interleukin 1beta mediates the modulatory effects of monocytes on LNCaP human prostate cancer cells. Br. J. Cancer 1998, 78, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Dagenais, M.; Dupaul-Chicoine, J.; Douglas, T.; Champagne, C.; Morizot, A.; Saleh, M. The Interleukin (IL)-1R1 pathway is a critical negative regulator of PyMT-mediated mammary tumorigenesis and pulmonary metastasis. Oncoimmunology 2017, 6, e1287247. [Google Scholar] [CrossRef] [PubMed]
- Tulotta, C.; Lefley, D.V.; Moore, C.K.; Amariutei, A.E.; Spicer-Hadlington, A.R.; Quayle, L.A.; Hughes, R.O.; Ahmed, K.; Cookson, V.; Evans, C.A.; et al. IL-1B drives opposing responses in primary tumours and bone metastases; harnessing combination therapies to improve outcome in breast cancer. NPJ Breast Cancer 2021, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, M.K.; Alspach, E. Senescence and Immunoregulation in the Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 754069. [Google Scholar] [CrossRef]
- Perrott, K.M.; Wiley, C.D.; Desprez, P.-Y.; Campisi, J. Apigenin suppresses the senescence-associated secretory phenotype and paracrine effects on breast cancer cells. GeroScience 2017, 39, 161–173. [Google Scholar] [CrossRef]
- Sosnoski, D.M.; Norgard, R.J.; Grove, C.D.; Foster, S.J.; Mastro, A.M. Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin. Exp. Metastasis 2015, 32, 335–344. [Google Scholar] [CrossRef]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef]
- Eyre, R.; Alférez, D.G.; Santiago-Gómez, A.; Spence, K.; McConnell, J.C.; Hart, C.; Simões, B.M.; Lefley, D.; Tulotta, C.; Storer, J.; et al. Microenvironmental IL1β promotes breast cancer metastatic colonisation in the bone via activation of Wnt signalling. Nat. Commun. 2019, 10, 5016. [Google Scholar] [CrossRef]
- Sarmiento-Castro, A.; Caamaño-Gutiérrez, E.; Sims, A.H.; Hull, N.J.; James, M.I.; Santiago-Gómez, A.; Eyre, R.; Clark, C.; Brown, M.E.; Brooks, M.D.; et al. Increased Expression of Interleukin-1 Receptor Characterizes Anti-estrogen-Resistant ALDH + Breast Cancer Stem Cells. Stem Cell Rep. 2020, 15, 307–316. [Google Scholar] [CrossRef]
- Zhou, W.; Guo, S.; Gonzalez-Perez, R.R. Leptin pro-angiogenic signature in breast cancer is linked to IL-1 signalling. Br. J. Cancer 2011, 104, 128–137. [Google Scholar] [CrossRef]
- Fan, X.; He, L.; Dai, Q.; He, J.; Chen, X.; Dai, X.; Zhang, C.; Sun, D.; Meng, X.; Sun, S.; et al. Interleukin-1β augments the angiogenesis of endothelial progenitor cells in an NF-κB/CXCR7-dependent manner. J. Cell. Mol. Med. 2020, 24, 5605–5614. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, G.; Saarinen, N.; Abrahamsson, A.; Dabrosin, C. Tamoxifen, Flaxseed, and the Lignan Enterolactone Increase Stroma- and Cancer Cell–Derived IL-1Ra and Decrease Tumor Angiogenesis in Estrogen-Dependent Breast Cancer. Cancer Res. 2011, 71, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Holen, I.; Lefley, D.V.; Francis, S.E.; Rennicks, S.; Bradbury, S.; Coleman, R.E.; Ottewell, P. IL-1 drives breast cancer growth and bone metastasis in vivo. Oncotarget 2016, 7, 75571–75584. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef]
- Lefley, D.; Howard, F.; Arshad, F.; Bradbury, S.; Brown, H.; Tulotta, C.; Eyre, R.; Alférez, D.; Wilkinson, J.M.; Holen, I.; et al. Development of clinically relevant in vivo metastasis models using human bone discs and breast cancer patient-derived xenografts. Breast Cancer Res. 2019, 21, 130. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef]
- You, D.; Jeong, Y.; Yoon, S.Y.; AKim, S.; Kim, S.W.; Nam, S.J.; Lee, J.E.; Kim, S. Celastrol attenuates the inflammatory response by inhibiting IL-1β expression in triple-negative breast cancer cells. Oncol. Rep. 2021, 45, 89. [Google Scholar] [CrossRef]
- Jeon, M.; Han, J.; Nam, S.J.; Lee, J.E.; Kim, S. Elevated IL-1β expression induces invasiveness of triple negative breast cancer cells and is suppressed by zerumbone. Chem. Biol. Interact. 2016, 258, 126–133. [Google Scholar] [CrossRef]
- Templeton, Z.S.; Lie, W.-R.; Wang, W.; Rosenberg-Hasson, Y.; Alluri, R.V.; Tamaresis, J.S.; Bachmann, M.H.; Lee, K.; Maloney, W.J.; Contag, C.H.; et al. Breast Cancer Cell Colonization of the Human Bone Marrow Adipose Tissue Niche. Neoplasia 2015, 17, 849–861. [Google Scholar] [CrossRef]
- Filippi, I.; Carraro, F.; Naldini, A. Interleukin-1 β Affects MDAMB231 Breast Cancer Cell Migration under Hypoxia: Role of HIF-1 α and NF κ B Transcription Factors. Mediators Inflamm. 2015, 2015, 789414. [Google Scholar] [CrossRef]
- Oh, K.; Lee, O.-Y.; Park, Y.; Seo, M.W.; Lee, D.-S. IL-1β induces IL-6 production and increases invasiveness and estrogen-independent growth in a TG2-dependent manner in human breast cancer cells. BMC Cancer 2016, 16, 724. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.; Ko, E.; Kim, H.S.; Park, A.K.; Moon, H.-G.; Noh, D.-Y.; Lee, D.-S. Transglutaminase 2 facilitates the distant hematogenous metastasis of breast cancer by modulating interleukin-6 in cancer cells. Breast Cancer Res. 2011, 13, R96. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Cai, X.; Wang, Y.; Wang, D.; Wang, T.; Gong, H.; Sun, H.; Jia, Q.; Zhou, W.; Wu, Z.; et al. NAT1 promotes osteolytic metastasis in luminal breast cancer by regulating the bone metastatic niche via NF-κB/IL-1B signaling pathway. Am. J. Cancer Res. 2020, 10, 2464–2479. [Google Scholar] [PubMed]
- Popeda, M.; Stokowy, T.; Bednarz-Knoll, N.; Jurek, A.; Niemira, M.; Bielska, A.; Kretowski, A.; Kalinowski, L.; Szade, J.; Markiewicz, A.; et al. NF-kappa B Signaling-Related Signatures Are Connected with the Mesenchymal Phenotype of Circulating Tumor Cells in Non-Metastatic Breast Cancer. Cancers 2019, 11, 1961. [Google Scholar] [CrossRef] [PubMed]
- Pein, M.; Insua-Rodríguez, J.; Hongu, T.; Riedel, A.; Meier, J.; Wiedmann, L.; Decker, K.; Essers, M.A.G.; Sinn, H.-P.; Spaich, S.; et al. Metastasis-initiating cells induce and exploit a fibroblast niche to fuel malignant colonization of the lungs. Nat. Commun. 2020, 11, 1494. [Google Scholar] [CrossRef]
- Mon, N.N.; Senga, T.; Ito, S. Interleukin-1β activates focal adhesion kinase and Src to induce matrix metalloproteinase-9 production and invasion of MCF-7 breast cancer cells. Oncol. Lett. 2017, 13, 955–960. [Google Scholar] [CrossRef]
- Franco-Barraza, J.; Valdivia-Silva, J.E.; Zamudio-Meza, H.; Castillo, A.; García-Zepeda, E.A.; Benítez-Bribiesca, L.; Meza, I. Actin cytoskeleton participation in the onset of IL-1beta induction of an invasive mesenchymal-like phenotype in epithelial MCF-7 cells. Arch. Med. Res. 2010, 41, 170–181. [Google Scholar] [CrossRef]
- Perez-Yepez, E.A.; Ayala-Sumuano, J.-T.; Lezama, R.; Meza, I. A novel β-catenin signaling pathway activated by IL-1β leads to the onset of epithelial-mesenchymal transition in breast cancer cells. Cancer Lett. 2014, 354, 164–171. [Google Scholar] [CrossRef]
- Ito-Kureha, T.; Koshikawa, N.; Yamamoto, M.; Semba, K.; Yamaguchi, N.; Yamamoto, T.; Seiki, M.; Inoue, J.-I. Tropomodulin 1 expression driven by NF-κB enhances breast cancer growth. Cancer Res. 2015, 75, 62–72. [Google Scholar] [CrossRef]
- Dixon, J.M. Endocrine Resistance in Breast Cancer. New J. Sci. 2014, 2014, 390618. [Google Scholar] [CrossRef]
- Barrios, C.; Forbes, J.F.; Jonat, W.; Conte, P.; Gradishar, W.; Buzdar, A.; Gelmon, K.; Gnant, M.; Bonneterre, J.; Toi, M.; et al. The sequential use of endocrine treatment for advanced breast cancer: Where are we? Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012, 23, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.I.; West, N.R.; Murphy, L.C.; Watson, P.H. Intratumoural inflammation and endocrine resistance in breast cancer. Endocr. Relat. Cancer 2015, 22, R51–R67. [Google Scholar] [CrossRef] [PubMed]
- Gloyeske, N.C.; Dabbs, D.J.; Bhargava, R. Low ER + Breast Cancer: Is This a Distinct Group? Am. J. Clin. Pathol. 2014, 141, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.M.; Clark, G.M.; Osborne, C.K.; Allred, D.C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol. 1999, 17, 1474–1481. [Google Scholar] [CrossRef]
- Kuukasjärvi, T.; Kononen, J.; Helin, H.; Holli, K.; Isola, J. Loss of estrogen receptor in recurrent breast cancer is associated with poor response to endocrine therapy. J. Clin. Oncol. 1996, 14, 2584–2589. [Google Scholar] [CrossRef]
- McGuire, W.L. Steroid receptors in human breast cancer. Cancer Res. 1978, 38, 4289–4291. [Google Scholar]
- Prabhu, J.S.; Korlimarla, A.; Desai, K.; Alexander, A.; Raghavan, R.; Anupama, C.; Dendukuri, N.; Manjunath, S.; Correa, M.; Raman, N.; et al. A Majority of Low (1–10%) ER Positive Breast Cancers Behave Like Hormone Receptor Negative Tumors. J. Cancer 2014, 5, 156–165. [Google Scholar] [CrossRef]
- Arnedos, M.; Bihan, C.; Delaloge, S.; Andre, F. Triple-negative breast cancer: Are we making headway at least? Ther. Adv. Med. Oncol. 2012, 4, 195–210. [Google Scholar] [CrossRef]
- Jiménez-Garduño, A.M.; Mendoza-Rodríguez, M.G.; Urrutia-Cabrera, D.; Domínguez-Robles, M.C.; Pérez-Yépez, E.A.; Ayala-Sumuano, J.T.; Meza, I. IL-1β induced methylation of the estrogen receptor ERα gene correlates with EMT and chemoresistance in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 490, 780–785. [Google Scholar] [CrossRef]
- Nawas, A.F.; Kanchwala, M.; Thomas-Jardin, S.E.; Dahl, H.; Daescu, K.; Bautista, M.; Anunobi, V.; Wong, A.; Meade, R.; Mistry, R.; et al. IL-1-conferred gene expression pattern in ERα + BCa and AR + PCa cells is intrinsic to ERα-BCa and AR-PCa cells and promotes cell survival. BMC Cancer 2020, 20, 46. [Google Scholar] [CrossRef]
- Valinezhad Sani, F.; Alamolhodaei, N.S.; Rashidpoor, H.; Gharaee, M.E.; Behravan, J.; Mosaffa, F. The effect of IL-1β on MRP2 expression and tamoxifen toxicity in MCF-7 breast cancer cells. Breast Dis. 2021, 40, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Eppenberger-Castori, S.; Marx, C.; Yau, C.; Scott, G.K.; Eppenberger, U.; Benz, C.C. Activation of nuclear factor-kappaB (NFkappaB) identifies a high-risk subset of hormone-dependent breast cancers. Int. J. Biochem. Cell Biol. 2005, 37, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, A.; Rodriguez, G.V.; Dabrosin, C. Fulvestrant-Mediated Attenuation of the Innate Immune Response Decreases ER + Breast Cancer Growth In Vivo More Effectively than Tamoxifen. Cancer Res. 2020, 80, 4487–4499. [Google Scholar] [CrossRef] [PubMed]
- Nehra, R.; Riggins, R.B.; Shajahan, A.N.; Zwart, A.; Crawford, A.C.; Clarke, R. BCL2 and CASP8 regulation by NF-kappaB differentially affect mitochondrial function and cell fate in antiestrogen-sensitive and -resistant breast cancer cells. FASEB J. 2010, 24, 2040–2055. [Google Scholar] [CrossRef] [PubMed]
- Riggins, R.B.; Zwart, A.; Nehra, R.; Clarke, R. The nuclear factor kappa B inhibitor parthenolide restores ICI 182,780 (Faslodex; fulvestrant)-induced apoptosis in antiestrogen-resistant breast cancer cells. Mol. Cancer Ther. 2005, 4, 33–41. [Google Scholar]
- de Graffenried, L.A.; Chandrasekar, B.; Friedrichs, W.E.; Donzis, E.; Silva, J.; Hidalgo, M.; Freeman, J.W.; Weiss, G.R. NF-kappa B inhibition markedly enhances sensitivity of resistant breast cancer tumor cells to tamoxifen. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2004, 15, 885–890. [Google Scholar] [CrossRef]
- Khongthong, P.; Roseweir, A.K.; Edwards, J. The NF-KB pathway and endocrine therapy resistance in breast cancer. Endocr. Relat. Cancer 2019, 26, R369–R380. [Google Scholar] [CrossRef]
- White, C.M.; Martin, B.K.; Lee, L.F.; Haskill, J.S.; Ting, J.P. Effects of paclitaxel on cytokine synthesis by unprimed human monocytes, T lymphocytes, and breast cancer cells. Cancer Immunol. Immunother. 1998, 46, 104–112. [Google Scholar] [CrossRef]
- Vyas, D.; Laput, G.; Vyas, A.K. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. Onco. Targets. Ther. 2014, 7, 1015–1023. [Google Scholar] [CrossRef]
- Montagut, C.; Tusquets, I.; Ferrer, B.; Corominas, J.M.; Bellosillo, B.; Campas, C.; Suarez, M.; Fabregat, X.; Campo, E.; Gascon, P.; et al. Activation of nuclear factor-kappa B is linked to resistance to neoadjuvant chemotherapy in breast cancer patients. Endocr. Relat. Cancer 2006, 13, 607–616. [Google Scholar] [CrossRef]
- Buchholz, T.A.; Garg, A.K.; Chakravarti, N.; Aggarwal, B.B.; Esteva, F.J.; Kuerer, H.M.; Singletary, S.E.; Hortobagyi, G.N.; Pusztai, L.; Cristofanilli, M.; et al. The nuclear transcription factor kappaB/bcl-2 pathway correlates with pathologic complete response to doxorubicin-based neoadjuvant chemotherapy in human breast cancer. Clin. Cancer Res. 2005, 11, 8398–8402. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Rodríguez, M.; Arévalo Romero, H.; Fuentes-Pananá, E.M.; Ayala-Sumuano, J.-T.; Meza, I. IL-1β induces up-regulation of BIRC3, a gene involved in chemoresistance to doxorubicin in breast cancer cells. Cancer Lett. 2017, 390, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, S.; Batoo, S.; Okuno, S.; Glück, S. Immunotherapy in breast cancer. J. Carcinog. 2019, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016, 6, 36107. [Google Scholar] [CrossRef]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef]
- Wellenstein, M.D.; Coffelt, S.B.; Duits, D.E.M.; van Miltenburg, M.H.; Slagter, M.; de Rink, I.; Henneman, L.; Kas, S.M.; Prekovic, S.; Hau, C.-S.; et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 2019, 572, 538–542. [Google Scholar] [CrossRef]
- Wang, W.; Nag, S.A.; Zhang, R. Targeting the NFκB signaling pathways for breast cancer prevention and therapy. Curr. Med. Chem. 2015, 22, 264–289. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.; Young, R.R.; Levin, M.K.; Baisch, J.; Timis, R.; Muniz, L.S.; Turner, J.; Pascual, V.; Palucka, K. Safety and immunologic activity of anakinra in HER2-negative metastatic breast cancer (MBC). J. Clin. Oncol. 2016, 34, e14565. [Google Scholar] [CrossRef]
- Lust, J.A.; Lacy, M.Q.; Zeldenrust, S.R.; Witzig, T.E.; Moon-Tasson, L.L.; Dinarello, C.A.; Donovan, K.A. Reduction in C-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am. J. Hematol. 2016, 91, 571–574. [Google Scholar] [CrossRef]
- Isambert, N.; Hervieu, A.; Hennequin, A.; Borg, C.; Rebe, C.; Derangere, V.; Richard, C.; Blanc, J.; Bertaut, A.; Ghiringhelli, F. 5-fluorouracil plus bevacizumab plus anakinra for patients with metastatic colorectal cancer refractory to standard therapies (IRAFU): An investigator-initiated, open-label, single-arm, multicentre, phase 2 study. J. Clin. Oncol. 2018, 36, e15540. [Google Scholar] [CrossRef]
- Carabasi, M.H.; McKean, M.; Stein, M.N.; Schweizer, M.T.; Luke, J.J.; Narayan, V.; Pachynski, R.K.; Parikh, R.A.; Zhang, J.; Fountaine, T.J.; et al. PSMA targeted armored chimeric antigen receptor (CAR) T-cells in patients with advanced mCRPC: A phase I experience. J. Clin.Oncol. 2021, 39, e2534. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Trial-# | Phase | Cancer Type | Disease Characteristics | Treatment | Start Date | Completion Date/Status | Published Results | Reference |
|---|---|---|---|---|---|---|---|---|
| NCT00635154 | II | Multiple Myeloma and Plasma Cell Neoplasm | Indolent or Smoldering Multiple Myeloma (asymptomatic) | Anakinra in combination with dexamethasone | 2002 | 2010 | Improved progression free survival | [99] |
| NCT00072111 | I | Unspecified solid tumors | Progressive metastatic cancer non-responsive to chemotherapy with tumor expression of IL-1 | Anakinra tolerability | 2003 | 2006 | ||
| NCT01802970 | I | Breast Cancer | Locally unresectable, invasive, or metastatic | Anakinra in combination with nab-paclitaxel, capecitabine, eribulin, or vinorelbine | 2012 | 2017 | Pilot study results, n = 11 patients; 2 = tumor volume reduction, 4 = stable disease, 2 = stopped anakinra for injection site reaction, 3 = progressive disease. Reduction in systemic IL-1 transcriptional signature. | [11] |
| NCT01624766 | I | Advanced/metastatic cancers | Non-responsive to standard therapy | Everolimus in combination with anakinra or denosumab | 2012 | 2021 | ||
| NCT02021422 | I | Pancreatic cancer | Inoperable, metastatic | Anakinra in combination with oxaliplatin, Irinotecan, or fluorouracil | 2013 | 2017 | ||
| NCT02090101 | II | Colorectal Cancer | Metastatic, non-responsive to chemotherapy | Anakinra in combination with LV5FU2 and bevacizumab | 2014 | 2017 | Combination therapy was tolerated and increased overall survival | [100] |
| NCT02550327 | I | Pancreatic Cancer | Suspected prior to diagnosis or histologically diagnosed pancreatic cancer | Anakinra in combination with three-drug regimen of nab-paclitaxel, gemcitabine, and cisplatin | 2016 | 2021 | ||
| NCT02492750 | I | Plasma Cell Myeloma | Indolent or Smoldering Plasma Cell Myeloma (asymptomatic) | Anakinra in combination with lenalidomide and dexamethasone | 2016 | 2019 | ||
| NCT03233776 | II | Multiple Myeloma | Diagnosed with multiple myeloma, scheduled to receive an autologous stem cell transplantation fter myeloablative therapy with high-dose melphalan | Anakinra in combination with autologous stem cell transplantation and melphalan | 2017 | 2020 | ||
| NCT03430011 | II | Multiple Myeloma | Relapsed and/or refractory disease, non-responsive to autologous stem cell transplant, immunomodulatory agents, proteosome inhibitors, and anti-CD38 | Anakinra in combination with JCARH125 (CAR-T that targets B-cell maturation antigen) | 2018 | estimated, 2023 | ||
| NCT04099901 | II | Multiple Myeloma | Diagnosed with multiple myeloma, scheduled to receive an autologous stem cell transplantation fter myeloablative therapy with high-dose melphalan | Anakinra in combination with autologous stem cell transplantation and melphalan [placebo added] | 2019 | estimated, 2022 | ||
| NCT04227275 | I | Metastatic Castration Resistance Prostate Cancer | At least 2 prior lines of systemic therapy, including second generation androgen receptor inhibitor and/or CYP17α inhibitor | Anakinra in combination with cyclophosphamide and fludarabine lymphodepletion and CART-PSMA-TGFβRDN | 2019 | estimated, 2036 | Initial observations indicate immune toxicity management strategy needed; prophylactic anakinra instituted. | [101] |
| NCT04150913 | II | B-cell Lymphoma | Relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy | Anakinra In combination with axicabtagene ciloleucel | 2020 | estimated, 2024; recruiting | ||
| NCT04432506 | II | B-cell Lymphoma | Relapsed or refractory B-cell lymphoma, at least 2 prior lines of systemic therapy | Anakinra in combination with axicabtagene ciloleucel, cyclophosphamide, and fludarabine | 2020 | estimated, 2025; recruiting | ||
| NCT04691765 | I | Chronic Lymphocytic Leukemia | Diagnosis of Chronic Lymphocytic Leukemia (CLL) meeting published diagnostic criteria, not currently treated with other agents for CLL. | Anakinra | 2021 | estimated, 2022; not yet recruiting | ||
| NCT04942626 | I | Rectal Cancer | Localized | Anakinra in combination with capecitabine and radiation | 2021 | estimated, 2026; not yet recruiting | ||
| NCT04926467 | II | Pancreatic Cancer | Resectable, locally advanced or potentially resectable pancreatic adenocarcinoma | Anakinra in combination with pre-operative nab-paclitaxel, gemcitabine and cisplatin and post-operative 5-fluorouracil, oxaliplatin, and irinotecan | 2021 | estimated, 2026; not yet recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diep, S.; Maddukuri, M.; Yamauchi, S.; Geshow, G.; Delk, N.A. Interleukin-1 and Nuclear Factor Kappa B Signaling Promote Breast Cancer Progression and Treatment Resistance. Cells 2022, 11, 1673. https://doi.org/10.3390/cells11101673
Diep S, Maddukuri M, Yamauchi S, Geshow G, Delk NA. Interleukin-1 and Nuclear Factor Kappa B Signaling Promote Breast Cancer Progression and Treatment Resistance. Cells. 2022; 11(10):1673. https://doi.org/10.3390/cells11101673
Chicago/Turabian StyleDiep, Sydney, Mahita Maddukuri, Stephanie Yamauchi, Ganamee Geshow, and Nikki A. Delk. 2022. "Interleukin-1 and Nuclear Factor Kappa B Signaling Promote Breast Cancer Progression and Treatment Resistance" Cells 11, no. 10: 1673. https://doi.org/10.3390/cells11101673
APA StyleDiep, S., Maddukuri, M., Yamauchi, S., Geshow, G., & Delk, N. A. (2022). Interleukin-1 and Nuclear Factor Kappa B Signaling Promote Breast Cancer Progression and Treatment Resistance. Cells, 11(10), 1673. https://doi.org/10.3390/cells11101673