
Probing the Drug Dynamics of Chemotherapeutics Using Metasurface-Enhanced Infrared Reflection Spectroscopy of Live Cells

 , , , and
, , , and
Abstract
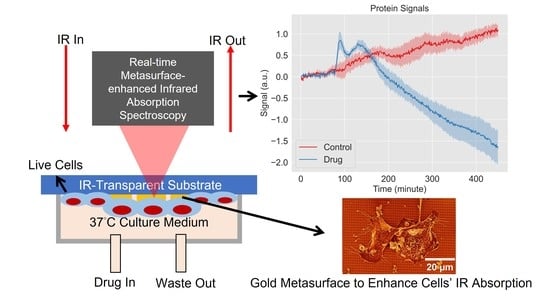
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
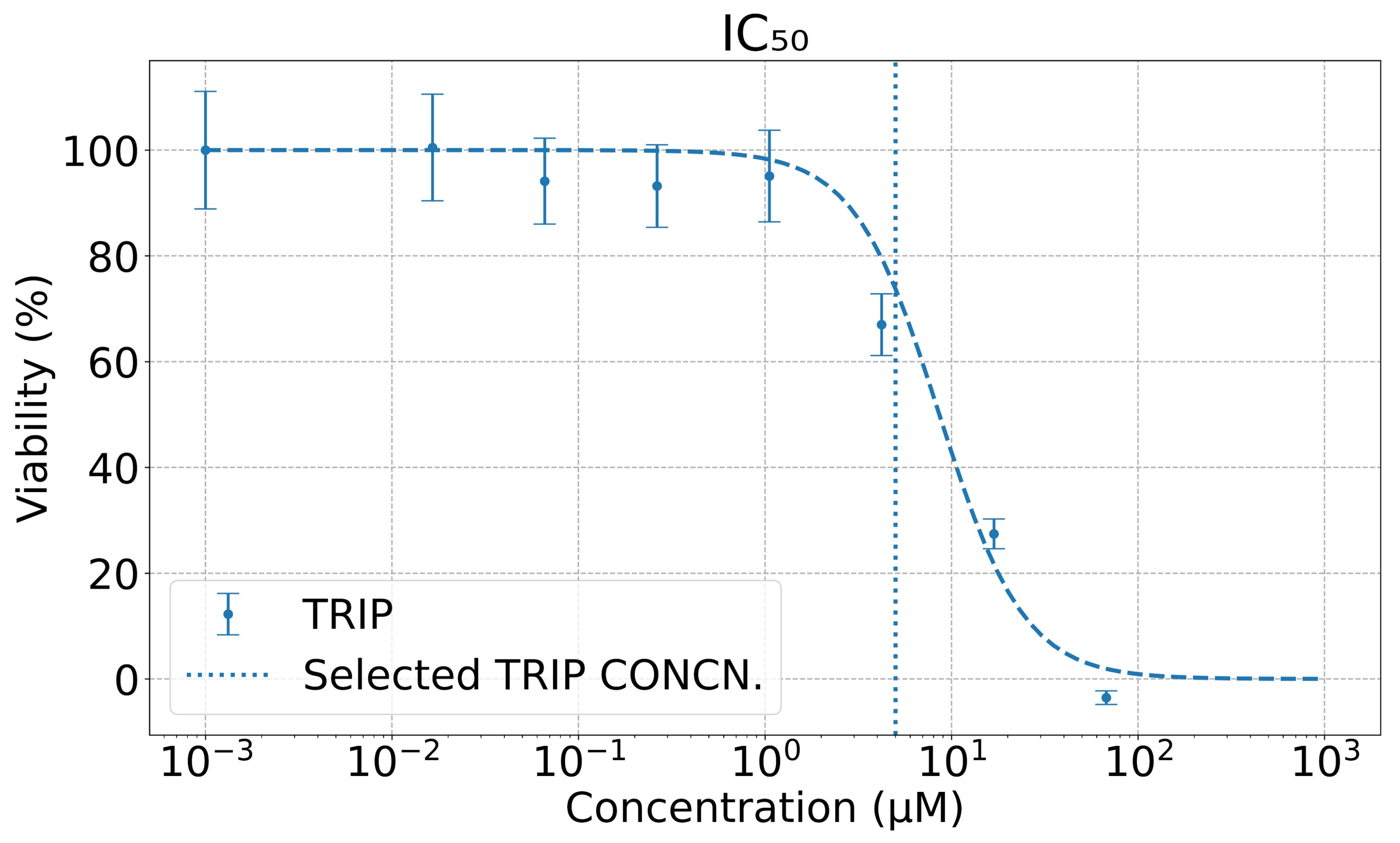
2.2. Viability Assay
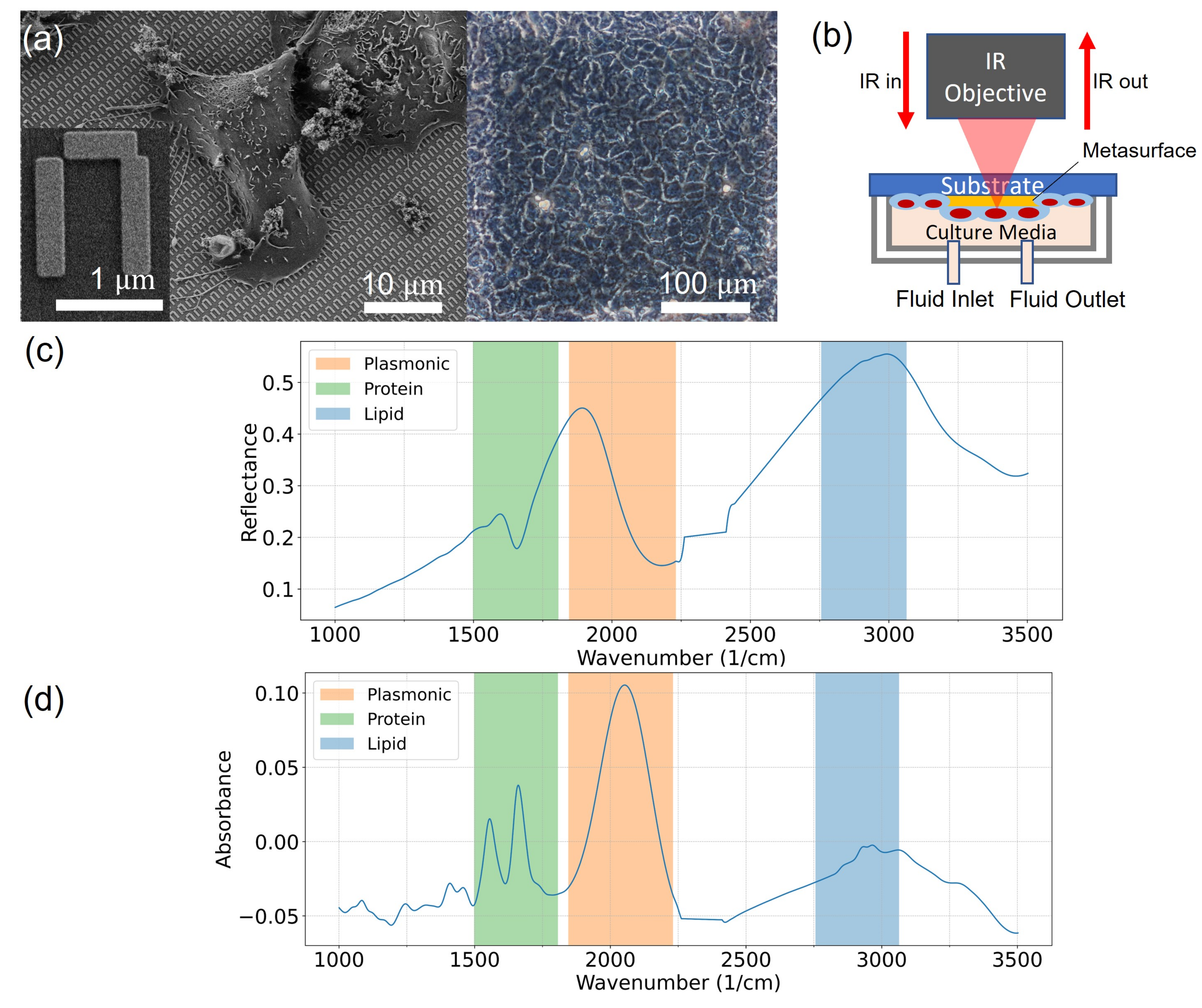
2.3. Metasurface Fabrication
2.4. FTIR Spectroscopy
2.5. Spectral Data Analysis
3. Results
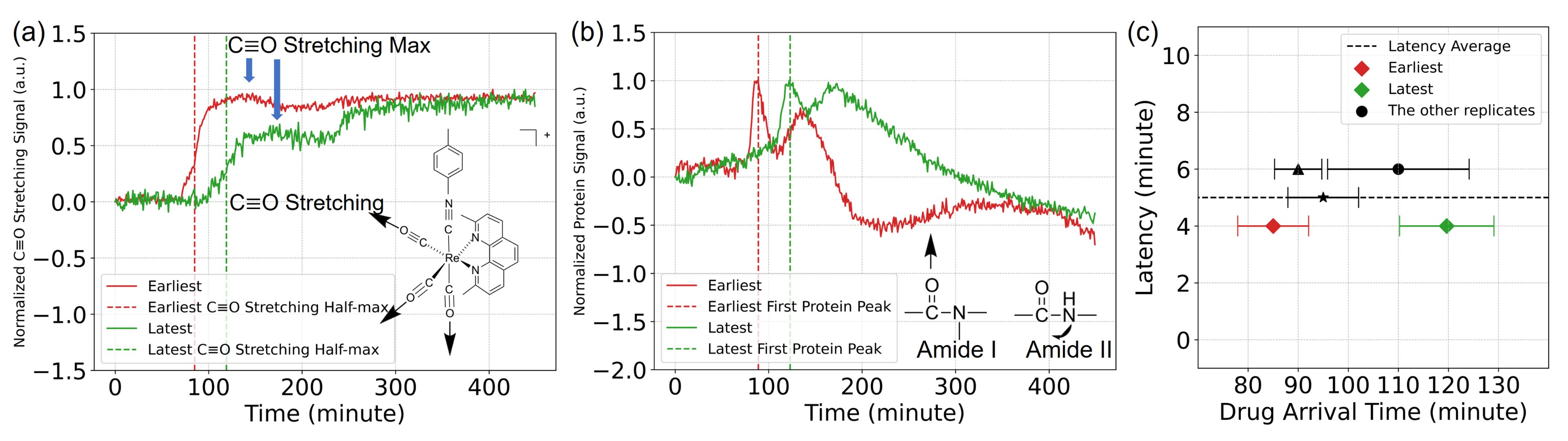
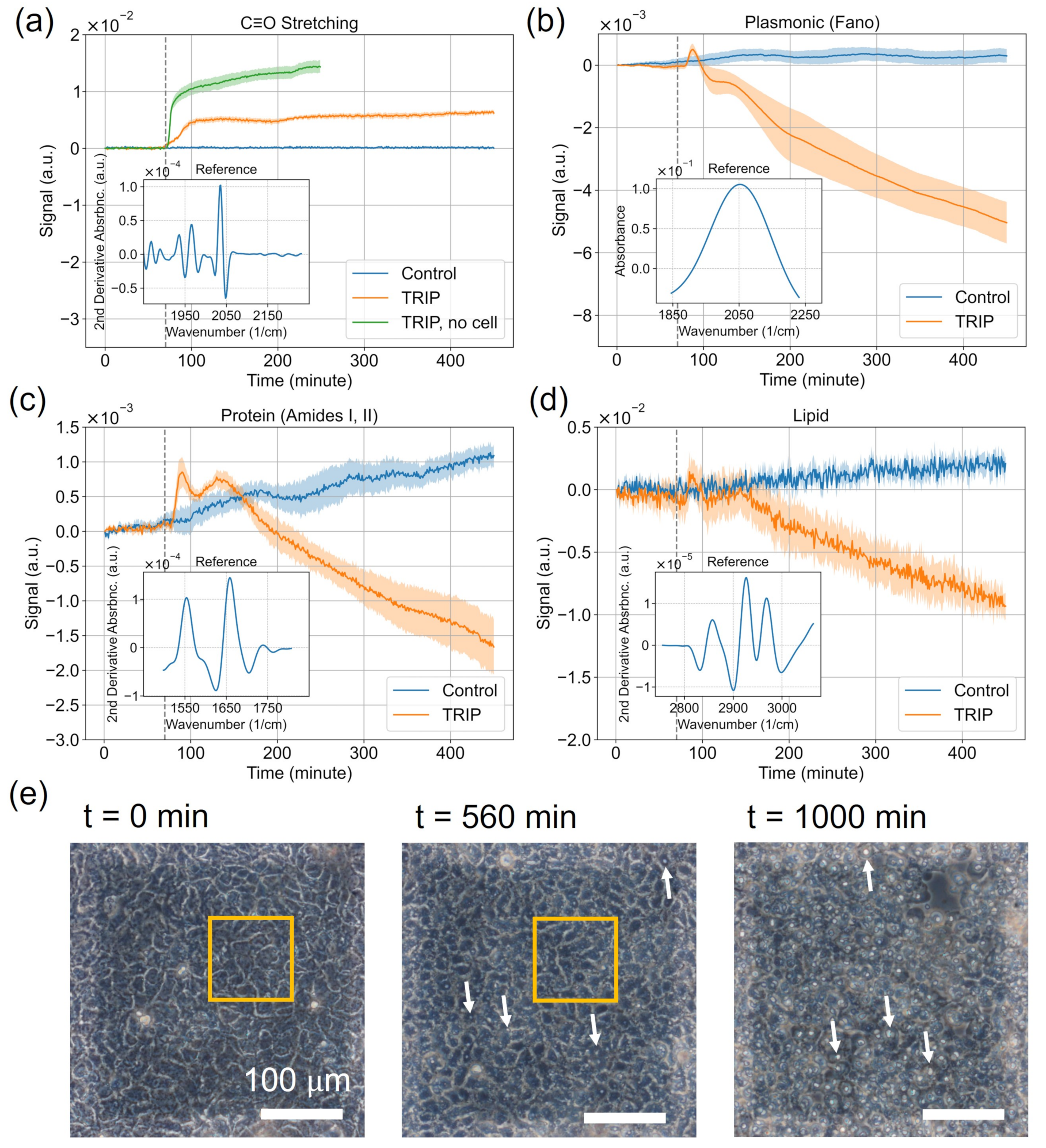
3.1. Delivery of the TRIP Anti-Cancer Complex to Cells Can Be Monitored in Time with MEIRS
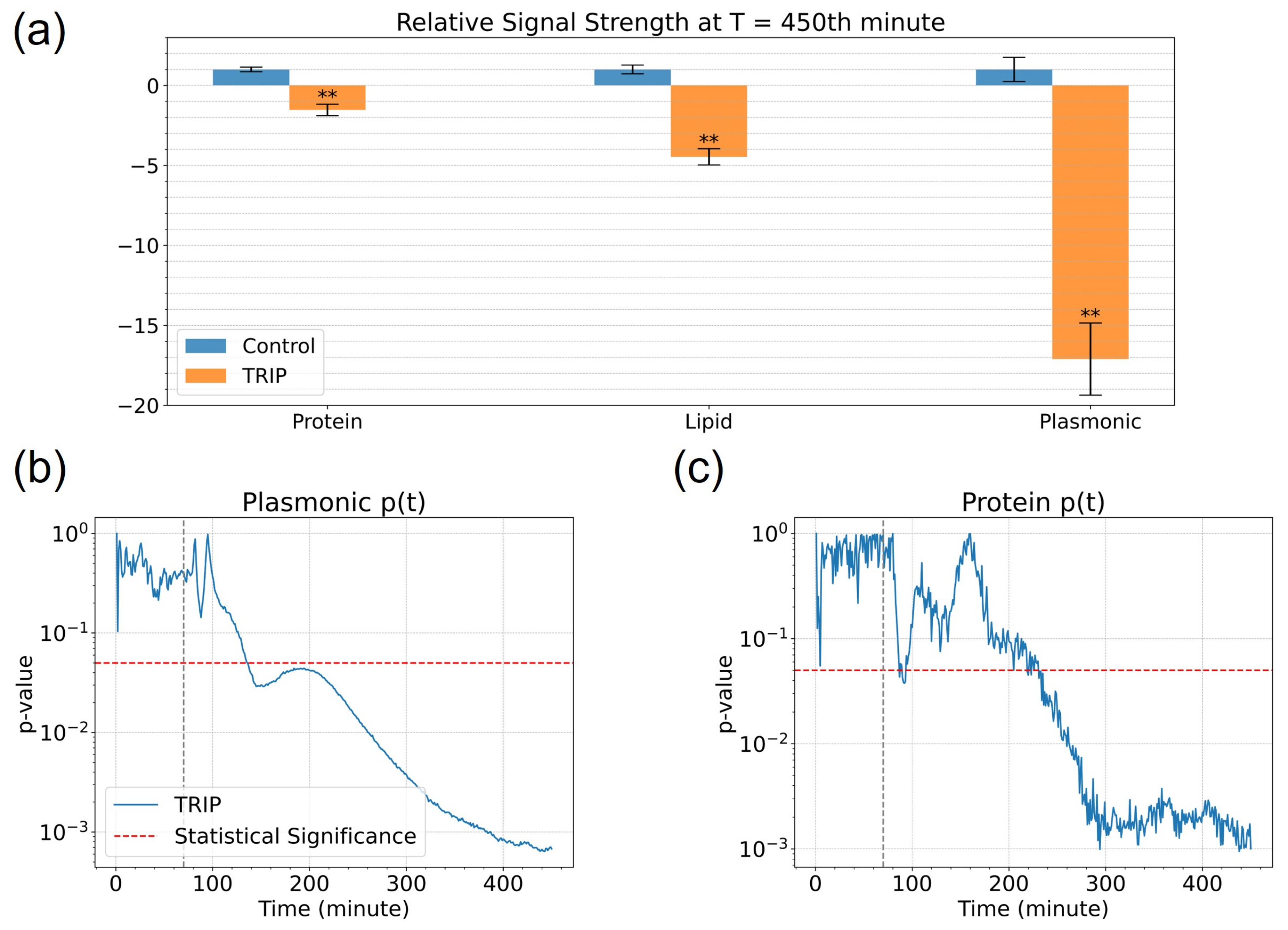
3.2. Analysis of Spectroscopic Changes of Drugs-Influenced Cells in Bio-Relevant Spectral Regions
4. Discussion
4.1. Interpretation of the Spectral Signals
4.2. Rapid Measurement of Drug Action Using MEIRS
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| FTIR | Fourier-transform infrared spectroscopy |
| IR | infrared |
| MoA(s) | mode(s) of action |
| ATR | attenuated total reflection |
| SNR | signal-to-noise ratio |
| QCL | quantum cascade laser |
| MEIRS | metasurface-enhanced infrared reflection spectroscopy |
| SEIRA | surface-enhanced infrared absorption |
| TRIP | tricarbonyl rhenium isonitrile polypyridyl |
| ER | endoplasmic reticulum |
| PDMS | polydimethylsiloxane |
| PMMA | poly(methyl methacrylate) |
| SEM | scanning electron microscope |
| DMEM | Dulbecco’s Modified Eagle Medium |
| FBS | fetal bovine serum |
| MCT | mercury cadmium telluride |
| CRR | cell response reference |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| DMSO | dimethyl sulfoxide |
| IC | half maximal inhibitory concentration |
| DMR | dynamic mass distribution |
| RTCA | real-time cell assay |
| SPR | surface plasmon resonance |
| p-DMR | positive dynamic mass distribution |
| RWG | resonant waveguide grating |
| UPR | unfolded protein response |
| EGF | epidermal growth factor |
| GPCR | G-protein coupled receptor |
| PAR2 | protease-activated receptor 2 |
References
- Baker, M.J.; Trevisan, J.; Bassan, P.; Bhargava, R.; Butler, H.J.; Dorling, K.M.; Fielden, P.R.; Fogarty, S.W.; Fullwood, N.J.; Heys, K.A.; et al. Using Fourier transform IR spectroscopy to analyze biological materials. Nat. Protoc. 2014, 9, 1771–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, M.C.; Nascimento, Y.M.; Araújo, J.M.; Lima, K.M. ATR-FTIR spectroscopy coupled with multivariate analysis techniques for the identification of DENV-3 in different concentrations in blood and serum: A new approach. RSC Adv. 2017, 7, 25640–25649. [Google Scholar] [CrossRef] [Green Version]
- Titus, J.; Ghimire, H.; Viennois, E.; Merlin, D.; Unil Perera, A.G. Protein secondary structure analysis of dried blood serum using infrared spectroscopy to identify markers for colitis screening. J. Biophotonics 2018, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Butler, H.J.; Brennan, P.M.; Cameron, J.M.; Finlayson, D.; Hegarty, M.G.; Jenkinson, M.D.; Palmer, D.S.; Smith, B.R.; Baker, M.J. Development of high-throughput ATR-FTIR technology for rapid triage of brain cancer. Nat. Commun. 2019, 10, 4501. [Google Scholar] [CrossRef] [Green Version]
- Petibois, C.; Déléris, G. Chemical mapping of tumor progression by FT-IR imaging: Towards molecular histopathology. Trends Biotechnol. 2006, 24, 455–462. [Google Scholar] [CrossRef]
- Martin, F.L.; Kelly, J.G.; Llabjani, V.; Martin-Hirsch, P.L.; Patel, I.I.; Trevisan, J.; Fullwood, N.J.; Walsh, M.J. Distinguishing cell types or populations based on the computational analysis of their infrared spectra. Nat. Protoc. 2010, 5, 1748–1760. [Google Scholar] [CrossRef]
- Mostaço-Guidolin, L.B.; Bachmann, L. Application of FTIR spectroscopy for identification of blood and leukemia biomarkers: A review over the past 15 years. Appl. Spectrosc. Rev. 2011, 46, 388–404. [Google Scholar] [CrossRef]
- Mittal, S.; Yeh, K.; Suzanne Leslie, L.; Kenkel, S.; Kajdacsy-Balla, A.; Bhargava, R.; Leslie, L.S.; Kenkel, S.; Kajdacsy-Balla, A.; Bhargava, R. Simultaneous cancer and tumor microenvironment subtyping using confocal infrared microscopy for all-digital molecular histopathology. Proc. Natl. Acad. Sci. USA 2018, 115, E5651–E5660. [Google Scholar] [CrossRef] [Green Version]
- Ly, E.; Piot, O.; Wolthuis, R.; Durlach, A.; Bernard, P.; Manfait, M. Combination of FTIR spectral imaging and chemometrics for tumour detection from paraffin-embedded biopsies. Analyst 2008, 133, 197–205. [Google Scholar] [CrossRef]
- Theophilou, G.; Lima, K.M.; Martin-Hirsch, P.L.; Stringfellow, H.F.; Martin, F.L. ATR-FTIR spectroscopy coupled with chemometric analysis discriminates normal, borderline and malignant ovarian tissue: Classifying subtypes of human cancer. Analyst 2016, 141, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Schnell, M.; Mittal, S.; Falahkheirkhah, K.; Mittal, A.; Yeh, K.; Kenkel, S.; Kajdacsy-Balla, A.; Carney, P.S.; Bhargava, R. All-digital histopathology by infrared-optical hybrid microscopy. Proc. Natl. Acad. Sci. USA 2020, 117, 3388–3396. [Google Scholar] [CrossRef] [PubMed]
- Lotfollahi, M.; Berisha, S.; Daeinejad, D.; Mayerich, D. Digital staining of high-definition Fourier transform infrared (FT-IR) images using deep learning. Appl. Spectrosc. 2019, 73, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Titus, J.; Filfili, C.; Hilliard, J.K.; Ward, J.A.; Unil Perera, A.G. Early detection of cell activation events by means of attenuated total reflection Fourier transform infrared spectroscopy. Appl. Phys. Lett. 2014, 104. [Google Scholar] [CrossRef]
- Sharaha, U.; Rodriguez-Diaz, E.; Riesenberg, K.; Bigio, I.J.; Huleihel, M.; Salman, A. Using infrared spectroscopy and multivariate analysis to detect antibiotics’ resistant escherichia coli bacteria. Anal. Chem. 2017, 89, 8782–8790. [Google Scholar] [CrossRef] [PubMed]
- Derenne, A.; Gasper, R.; Goormaghtigh, E. The FTIR spectrum of prostate cancer cells allows the classification of anticancer drugs according to their mode of action. Analyst 2011, 136, 1134. [Google Scholar] [CrossRef] [PubMed]
- Mignolet, A.; Derenne, A.; Smolina, M.; Wood, B.R.; Goormaghtigh, E. FTIR spectral signature of anticancer drugs. Can drug mode of action be identified? Biochim. Biophys. Acta Proteins Proteom. 2016, 1864, 85–101. [Google Scholar] [CrossRef]
- Ribeiro da Cunha, B.; Fonseca, L.P.; Calado, C.R.C. Simultaneous elucidation of antibiotic mechanism of action and potency with high-throughput Fourier-transform infrared (FTIR) spectroscopy and machine learning. Appl. Microbiol. Biotechnol. 2021, 105, 1269–1286. [Google Scholar] [CrossRef]
- Chan, K.L.A.; Kazarian, S.G.; Andrew Chan, K.L.; Kazarian, S.G. Attenuated total reflection Fourier-transform infrared (ATR-FTIR) imaging of tissues and live cells. Chem. Soc. Rev. 2016, 45, 1850–1864. [Google Scholar] [CrossRef]
- Mitri, E.; Birarda, G.; Vaccari, L.; Kenig, S.; Tormen, M.; Grenci, G. SU-8 bonding protocol for the fabrication of microfluidic devices dedicated to FTIR microspectroscopy of live cells. Lab Chip 2014, 14, 210–218. [Google Scholar] [CrossRef]
- Lehmkuhl, B.; Noblitt, S.D.; Krummel, A.T.; Henry, C.S. Fabrication of IR-Transparent microfluidic devices by anisotropic etching of channels in CaF2. Lab Chip 2015, 15, 4364–4368. [Google Scholar] [CrossRef]
- Fale, P.L.; Altharawi, A.; Chan, K.L.A. In situ Fourier transform infrared analysis of live cells’ response to doxorubicin. Biochim. Biophys. Acta-Mol. Cell Res. 2015, 1853, 2640–2648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meneghel, J.; Passot, S.; Jamme, F.; Lefrançois, S.; Lieben, P.; Dumas, P.; Fonseca, F. FTIR micro-spectroscopy using synchrotron-based and thermal source-based radiation for probing live bacteria. Anal. Bioanal. Chem. 2020, 412, 7049–7061. [Google Scholar] [CrossRef] [PubMed]
- Orondics, F.B.; Ossent, M.J.; Andt, C.S.; Avoute, L.L.; Aponov, D.G.; Ideur, A.H.; Umas, P.D.; Évrier, S.F. Supercontinuum-based Fourier transform infrared spectromicroscopy. Optica 2018, 5, 378–381. [Google Scholar] [CrossRef]
- Yeh, K.; Lee, D.; Bhargava, R. Multicolor discrete frequency infrared spectroscopic imaging. Anal. Chem. 2019, 91, 2177–2185. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Liu, X.; Shi, L.; Stinson, H.T.; Rowlette, J.; Kahl, L.J.; Evans, C.R.; Zheng, C.; Dietrich, L.E.; Min, W. Mid-infrared metabolic imaging with vibrational probes. Nat. Methods 2020, 17, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Kelp, G.; Arju, N.; Lee, A.; Esquivel, E.; Delgado, R.; Yu, Y.; Dutta-Gupta, S.; Sokolov, K.; Shvets, G. Application of metasurface-enhanced infra-red spectroscopy to distinguish between normal and cancerous cell types. Analyst 2019, 144, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Kelp, G.; Li, J.; Lu, J.; DiNapoli, N.; Delgado, R.; Liu, C.; Fan, D.; Dutta-Gupta, S.; Shvets, G. Infrared spectroscopy of live cells from a flowing solution using electrically-biased plasmonic metasurfaces. Lab Chip 2020, 20, 2136–2153. [Google Scholar] [CrossRef]
- Huang, S.H.; Li, J.; Fan, Z.; Delgado, R.; Shvets, G. Monitoring the effects of chemical stimuli on live cells with metasurface-enhanced infrared reflection spectroscopy. Lab Chip 2021, 21, 3991–4004. [Google Scholar] [CrossRef]
- Wu, C.; Khanikaev, A.B.; Adato, R.; Arju, N.; Yanik, A.A.; Altug, H.; Shvets, G. Fano-resonant asymmetric metamaterials for ultrasensitive spectroscopy and identification of molecular monolayers. Nat. Mater. 2012, 11, 69–75. [Google Scholar] [CrossRef]
- Huck, C.; Neubrech, F.; Vogt, J.; Toma, A.; Gerbert, D.; Katzmann, J.; Härtling, T.; Pucci, A. Surface-enhanced infrared spectroscopy using nanometer-sized gaps. ACS Nano 2014, 8, 4908–4914. [Google Scholar] [CrossRef]
- Neubrech, F.; Huck, C.; Weber, K.; Pucci, A.; Giessen, H. Surface-enhanced infrared spectroscopy using resonant nanoantennas. Chem. Rev. 2017, 117, 5110–5145. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, D.; Tittl, A.; Ait-Bouziad, N.; John-Herpin, A.; Limaj, O.; Kelly, C.; Yoo, D.; Wittenberg, N.J.; Oh, S.H.; Lashuel, H.A.; et al. Resolving molecule-specific information in dynamic lipid membrane processes with multi-resonant infrared metasurfaces. Nat. Commun. 2018, 9, 2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Hu, F.; Shen, Y.; Chen, Z.; Yu, Y.; Lin, C.C.; Wang, M.C.; Min, W. Live-cell imaging of alkyne-tagged small biomolecules by stimulated Raman scattering. Nat. Methods 2014, 11, 410–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, A.P.; Marker, S.C.; Swanda, R.V.; Woods, J.J.; Qian, S.B.; Wilson, J.J. A rhenium isonitrile complex induces unfolded protein response-mediated apoptosis in cancer cells. Chem. Eur. J. 2019, 25, 9206–9210. [Google Scholar] [CrossRef]
- Marker, S.C.; King, A.P.; Granja, S.; Vaughn, B.; Woods, J.J.; Boros, E.; Wilson, J.J. Exploring the in vivo and in vitro anticancer activity of rhenium isonitrile complexes. Inorg. Chem. 2020, 59, 10285–10303. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Schultz, Z.D.; Levin, I.W. Vibrational spectroscopy of biomembranes. Annu. Rev. Anal. Chem. 2011, 4, 343–366. [Google Scholar] [CrossRef]
- Fang, Y.; Ferrie, A.M.; Fontaine, N.H.; Mauro, J.; Balakrishnan, J. Resonant waveguide grating biosensor for living cell sensing. Biophys. J. 2006, 91, 1925–1940. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Ferrie, A.M. Optical biosensor differentiates signaling of endogenous PAR1 and PAR2 in A431 cells. BMC Cell Biol. 2007, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Fang, Y. Duplexed label-free G protein-coupled receptor assays for high-throughput screening. J. Biomol. Screen. 2008, 13, 975–985. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Wang, C.; Fang, Y. Label-free cell phenotypic assessment of the molecular mechanism of action of epidermal growth factor receptor inhibitors. RSC Adv. 2013, 3, 10370–10378. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.W.; Peters, M.F. Label-free whole-cell assays: Expanding the scope of GPCR screening. Drug Discov. Today 2010, 15, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Lieb, S.; Michaelis, S.; Plank, N.; Bernhardt, G.; Buschauer, A.; Wegener, J. Label-free analysis of GPCR-stimulation: The critical impact of cell adhesion. Pharmacol. Res. 2016, 108, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Giebel, K.; Bechinger, C.; Herminghaus, S.; Riedel, M.; Leiderer, P.; Weiland, U.; Bastmeyer, M. Imaging of Cell/Substrate contacts of living cells with surface plasmon resonance microscopy. Biophys. J. 1999, 76, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Yanase, Y.; Hiragun, T.; Ishii, K.; Kawaguchi, T.; Yanase, T.; Kawai, M.; Sakamoto, K.; Hide, M. Surface plasmon resonance for cell-based clinical diagnosis. Sensors 2014, 14, 4948–4959. [Google Scholar] [CrossRef] [PubMed]
- Hiragun, T.; Yanase, Y.; Kose, K.; Kawaguchi, T.; Uchida, K.; Tanaka, S.; Hide, M. Surface plasmon resonance-biosensor detects the diversity of responses against epidermal growth factor in various carcinoma cell lines. Biosens. Bioelectron. 2012, 32, 202–207. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yang, S.; Kong, J.; Dong, A.; Yu, S. Obtaining information about protein secondary structures in aqueous solution using Fourier transform IR spectroscopy. Nat. Protoc. 2015, 10, 382–396. [Google Scholar] [CrossRef]
- Lipp, P.; Reither, G. Protein kinase c: The “Masters” of calcium and lipid. Cold Spring Harb. Perspect. Biol. 2011, 3, a004556. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Origin | Protein [26] | Lipid [37] | Plasmonic [28] | TRIP [35] |
|---|---|---|---|---|
| (Refractive Index) | ||||
| Spectral Window | ||||
| for | 1498–1807 | 2756–3064 | 1845–2231 | 1845–2135 |
| Reference Spectrum | (bioorthogonal) | |||
| (cm) | ||||
| Amide II ≈ 1550: | -CH- antisymm. stretching: | |||
| (NH) + v(CN) | 2925 | |||
| Vibrational Modes | Amide I ≈ 1660: | -CH- symm. stretching: | Fano resonance: | C≡O stretching: |
| (cm) | v(C=O) + (NH), | 2862 | 2080 | 1928, 1967, 2031 |
| merged with | CH antisymm. stretching: | |||
| HO absorption | 2970 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, P.-T.; Huang, S.H.; Huang, Z.; Wilson, J.J.; Shvets, G. Probing the Drug Dynamics of Chemotherapeutics Using Metasurface-Enhanced Infrared Reflection Spectroscopy of Live Cells. Cells 2022, 11, 1600. https://doi.org/10.3390/cells11101600
Shen P-T, Huang SH, Huang Z, Wilson JJ, Shvets G. Probing the Drug Dynamics of Chemotherapeutics Using Metasurface-Enhanced Infrared Reflection Spectroscopy of Live Cells. Cells. 2022; 11(10):1600. https://doi.org/10.3390/cells11101600
Chicago/Turabian StyleShen, Po-Ting, Steven H. Huang, Zhouyang Huang, Justin J. Wilson, and Gennady Shvets. 2022. "Probing the Drug Dynamics of Chemotherapeutics Using Metasurface-Enhanced Infrared Reflection Spectroscopy of Live Cells" Cells 11, no. 10: 1600. https://doi.org/10.3390/cells11101600
APA StyleShen, P.-T., Huang, S. H., Huang, Z., Wilson, J. J., & Shvets, G. (2022). Probing the Drug Dynamics of Chemotherapeutics Using Metasurface-Enhanced Infrared Reflection Spectroscopy of Live Cells. Cells, 11(10), 1600. https://doi.org/10.3390/cells11101600