
The Human Ntn-Hydrolase Superfamily: Structure, Functions and Perspectives


Abstract
1. Introduction
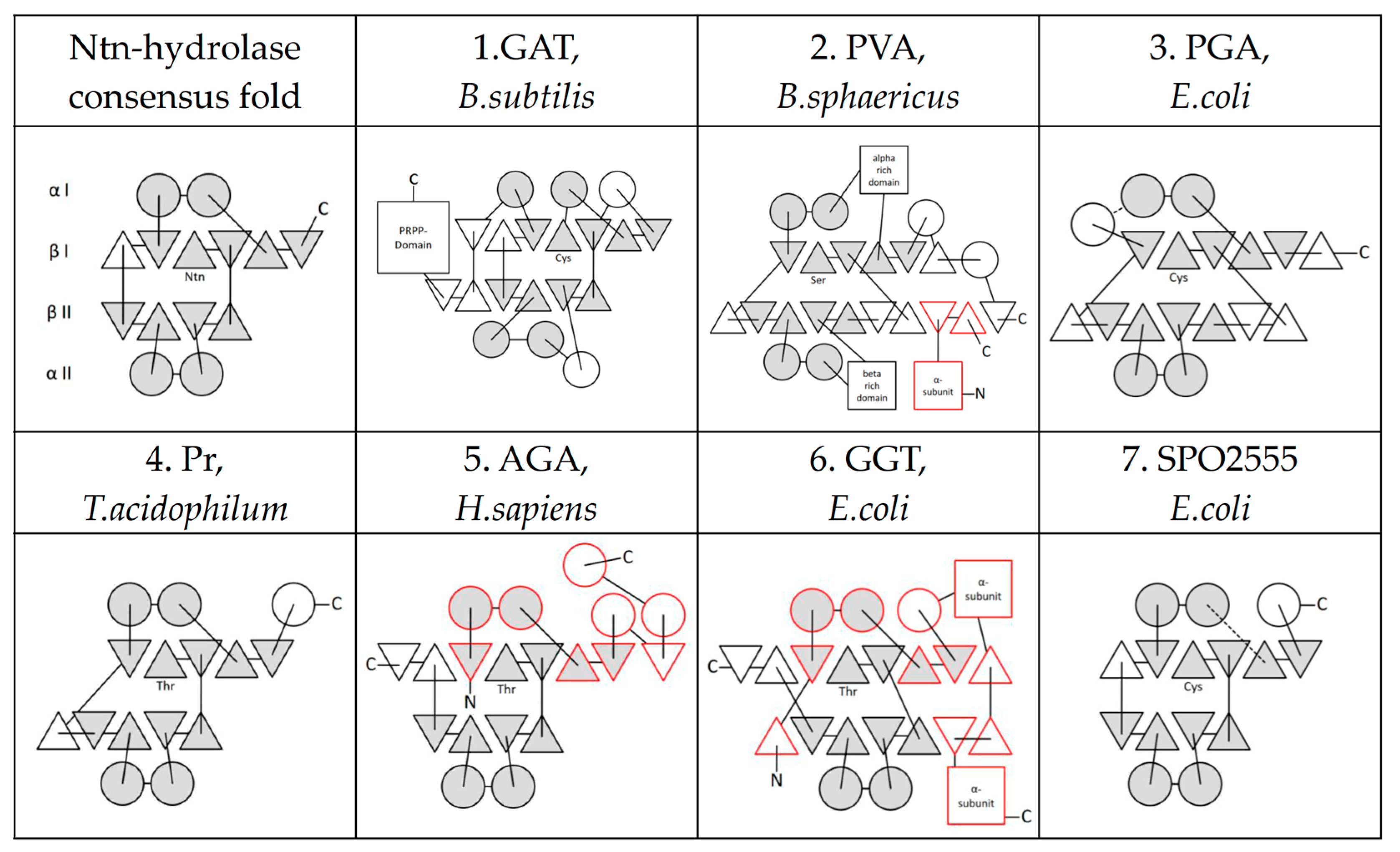
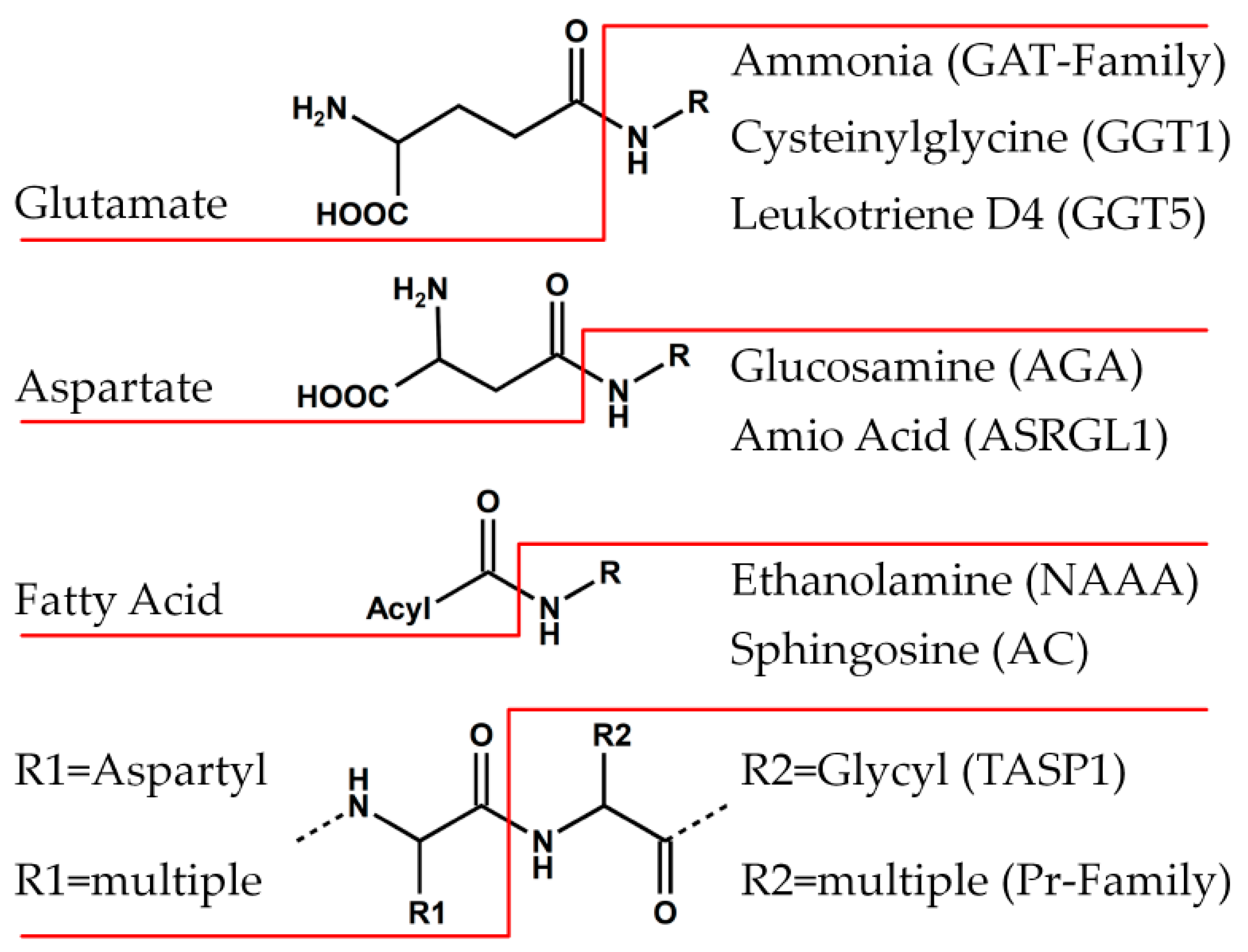
2. The Ntn-Hydrolase Superfamily
3. Ntn-Hydrolase Consensus Fold
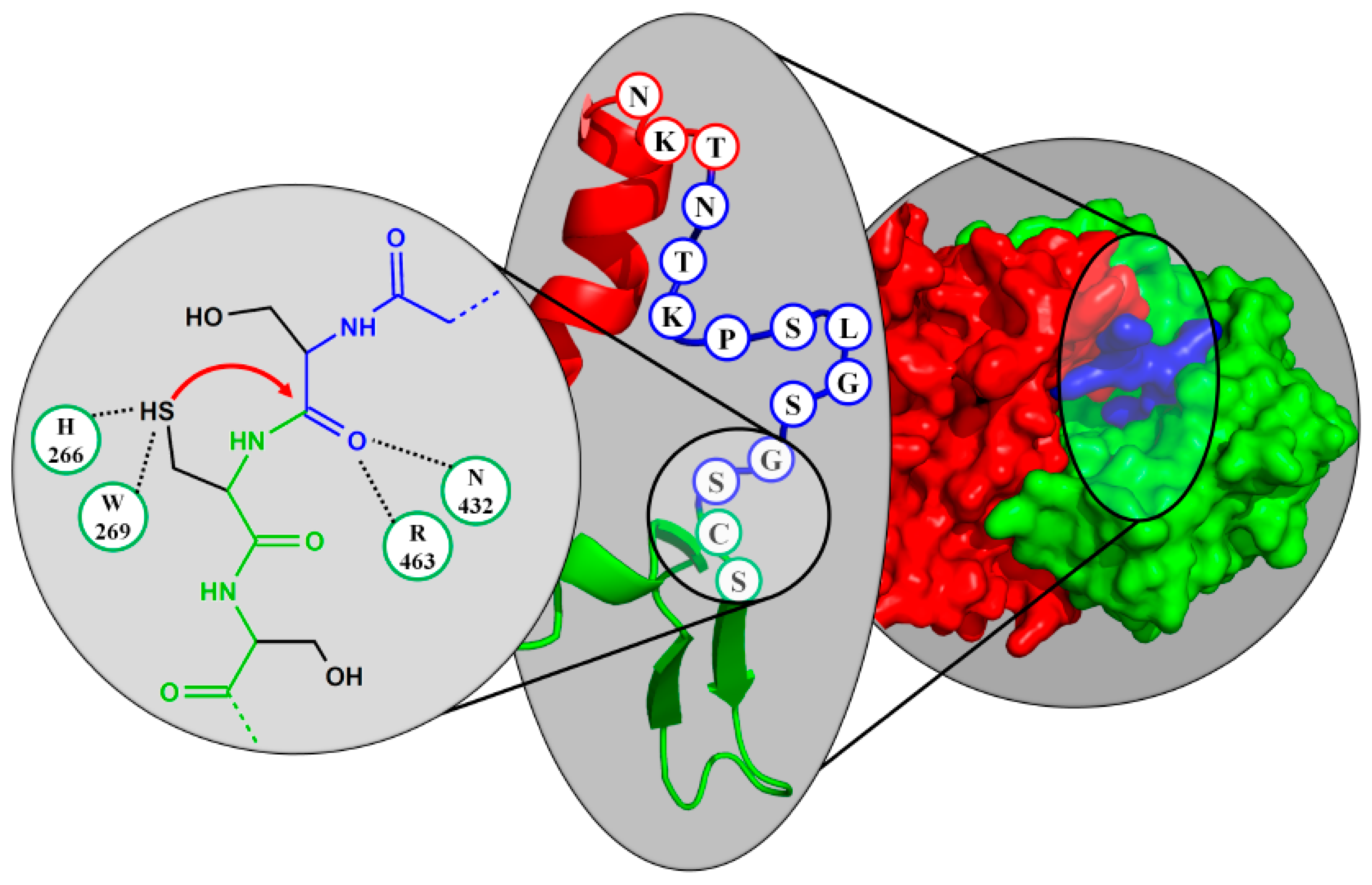
4. Active Site Residues
5. Further Superfamilies with Ntn-Hydrolase-like Folds
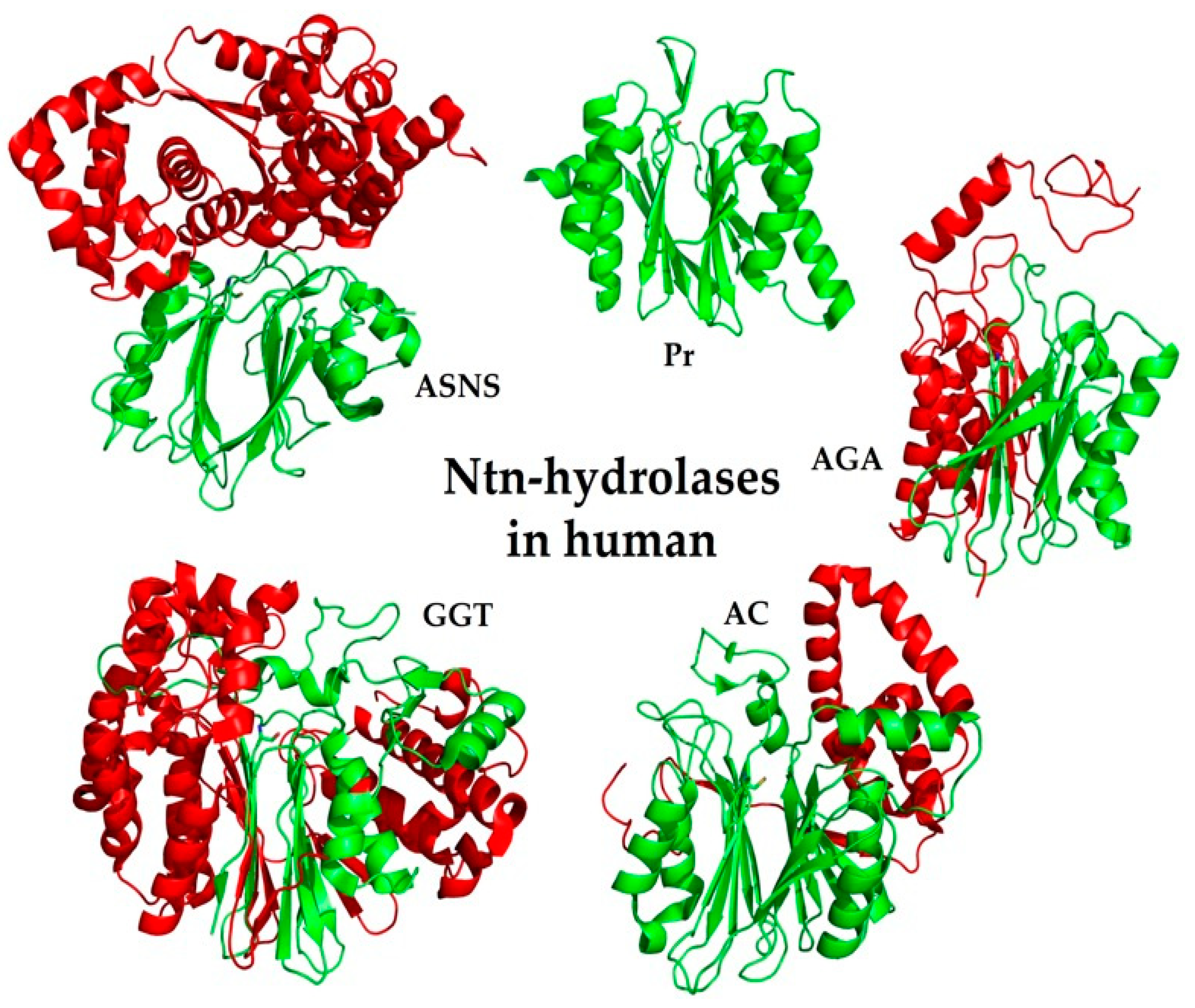
6. Ntn-Hydrolases in Human
6.1. Family of Class II Glutamine Amidotransferases (GAT)
6.2. PVA-Subfamily of Lysosomal Hydrolases
6.3. Family of Proteasome Subunits (Pr)
6.4. Family of Asparaginases (AGA)
6.5. Family of γ-Glutamyl Transpeptidases (GGT)
7. Concluding Remarks and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Mahesh, S.; Tang, K.-C.; Raj, M. Amide Bond Activation of Biological Molecules. Molecules 2018, 23, 2615. [Google Scholar] [CrossRef] [PubMed]
- Goswami, A.; Van Lanen, S.G. Enzymatic strategies and biocatalysts for amide bond formation: Tricks of the trade outside of the ribosome. Mol. BioSyst. 2015, 11, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Brannigan, J.A.; Dodson, G.; Duggleby, H.J.; Moody, P.; Smith, J.L.; Tomchick, D.; Murzin, A.G. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature 1995, 378, 416–419. [Google Scholar] [CrossRef]
- Smith, J.L.; Zaluzec, E.J.; Wery, J.-P.; Niu, L.; Switzer, R.L.; Zalkin, H.; Satow, Y. Structure of the Allosteric Regulatory Enzyme of Purine Biosynthesis. Science 1994, 264, 1427–1433. [Google Scholar] [CrossRef]
- Duggleby, H.J.; Tolley, S.P.; Hill, C.P.; Dodson, E.J.; Dodson, G.; Moody, P.C. Penicillin acylase has a single-amino-acid catalytic centre. Nature 1995, 373, 264–268. [Google Scholar] [CrossRef]
- Löwe, J.; Stock, D.; Jap, B.; Zwickl, P.; Baumeister, W.; Huber, R. Crystal Structure of the 20 S Proteasome from the Archaeon T. acidophilum at 3.4 Å Resolution. Science 1995, 268, 533–539. [Google Scholar] [CrossRef]
- Oinonen, C.; Tikkanen, R.; Rouvinen, J.; Peltonen, L. Three-dimensional structure of human lysosomal aspartylglucosaminidase. Nat. Struct. Biol. 1995, 2, 1102–1108. [Google Scholar] [CrossRef]
- Suresh, C.G.; Pundle, A.V.; Sivaraman, H.; Rao, K.N.; Brannigan, J.A.; McVey, C.E.; Verma, C.S.; Dauter, Z.; Dodson, E.J.; Dodson, G.G. Penicillin V acylase crystal structure reveals new Ntn-hydrolase family members. Nat. Struct. Biol. 1999, 6, 414–416. [Google Scholar] [CrossRef]
- Okada, T.; Suzuki, H.; Wada, K.; Kumagai, H.; Fukuyama, K. Crystal structures of γ-glutamyltranspeptidase from Escherichia coli, a key enzyme in glutathione metabolism, and its reaction intermediate. Proc. Natl. Acad. Sci. USA 2006, 103, 6471–6476. [Google Scholar] [CrossRef]
- Chandonia, J.-M.; Fox, N.K.; Brenner, S.E. SCOPe: Classification of large macromolecular structures in the structural classification of proteins—extended database. Nucleic Acids Res. 2019, 47, D475–D481. [Google Scholar] [CrossRef]
- Kim, Y.; Yoon, K.-H.; Khang, Y.; Turley, S.; Hol, W.G. The 2.0 Å Crystal Structure of Cephalosporin Acylase. Structure 2000, 8, 1059–1068. [Google Scholar] [CrossRef]
- Shtraizent, N.; Eliyahu, E.; Park, J.-H.; He, X.; Shalgi, R.; Schuchman, E.H. Autoproteolytic Cleavage and Activation of Human Acid Ceramidase. J. Biol. Chem. 2008, 283, 11253–11259. [Google Scholar] [CrossRef] [PubMed]
- Sleat, D.E.; Lackland, H.; Wang, Y.; Sohar, I.; Xiao, G.; Li, H.; Lobel, P. The human brain mannose 6-phosphate glycoproteome: A complex mixture composed of multiple isoforms of many soluble lysosomal proteins. Proteomics 2005, 5, 1520–1532. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, K.; Mutenda, K.E.; Balleininger, M.; Eckermann, E.; von Figura, K.; Schmidt, B.; Lübke, T. Identification of novel lysosomal matrix proteins by proteome analysis. Proteomics 2005, 5, 3966–3978. [Google Scholar] [CrossRef]
- Artymiuk, P.J. A sting in the (N-terminal) tail. Nat. Struct. Biol. 1995, 2, 1035–1037. [Google Scholar] [CrossRef]
- Oinonen, C.; Rouvinen, J. Structural comparison of Ntn-hydrolases. Protein Sci. 2000, 9, 2329–2337. [Google Scholar] [CrossRef]
- Lakomek, K.; Dickmanns, A.; Kettwig, M.; Urlaub, H.; Ficner, R.; Lübke, T. Initial insight into the function of the lysosomal 66.3 kDa protein from mouse by means of X-ray crystallography. BMC Struct. Biol. 2009, 9, 56. [Google Scholar] [CrossRef]
- Riikonen, A.; Tikkanen, R.; Jalanko, A.; Peltonen, L. Immediate Interaction between the Nascent Subunits and Two Conserved Amino Acids Trp34 and Thr206 Are Needed for the Catalytic Activity of Aspartylglucosaminidase. J. Biol. Chem. 1995, 270, 4903–4907. [Google Scholar] [CrossRef]
- Schalk, A.M.; Lavie, A. Structural and Kinetic Characterization of Guinea Pig l-Asparaginase Type III. Biochemistry 2014, 53, 2318–2328. [Google Scholar] [CrossRef]
- Braulke, T.; Bonifacino, J.S. Sorting of lysosomal proteins. Biochim. Biophys. Acta 2009, 1793, 605–614. [Google Scholar] [CrossRef]
- Hewitt, L.; Kasche, V.; Lummer, K.; Lewis, R.J.; Murshudov, G.N.; Verma, C.S.; Dodson, G.G.; Wilson, K.S. Structure of a slow processing precursor penicillin acylase from Escherichia coli reveals the linker peptide blocking the active-site cleft. J. Mol. Biol. 2000, 302, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Deng, Z.; Zhao, G.; Huang, X. The N-terminal Nucleophile Serine of Cephalosporin Acylase Executes the Second Autoproteolytic Cleavage and Acylpeptide Hydrolysis. J. Biol. Chem. 2011, 286, 24476–24486. [Google Scholar] [CrossRef] [PubMed]
- Saarela, J.; Laine, M.; Tikkanen, R.; Oinonen, C.; Jalanko, A.; Rouvinen, J.; Peltonen, L.; Tikkanen, R. Activation and Oligomerization of Aspartylglucosaminidase. J. Biol. Chem. 1998, 273, 25320–25328. [Google Scholar] [CrossRef]
- Ikonen, E.; Julkunen, I.; Tollersrud, O.K.; Kalkkinen, N.; Peltonen, L. Lysosomal aspartylglucosaminidase is processed to the active subunit complex in the endoplasmic reticulum. EMBO J. 1993, 12, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Ekici, Ö.D.; Paetzel, M.; Dalbey, R.E. Unconventional serine proteases: Variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 2008, 17, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Zhiryakova, D.; Ivanov, I.; Ilieva, S.; Guncheva, M.; Galunsky, B.; Stambolieva, N. Do N-terminal nucleophile hydrolases indeed have a single amino acid catalytic center? FEBS J. 2009, 276, 2589–2598. [Google Scholar] [CrossRef]
- Peräkylä, M.; Rouvinen, J. Ab Initio Quantum Mechanical Model Calculations on the Catalytic Mechanism of Aspartylglucosaminidase (AGA): A Serine Protease-Like Mechanism with an N-terminal Threonine and Substrate-Assisted Catalysis. Chem. A Eur. J. 1996, 2, 1548–1551. [Google Scholar] [CrossRef]
- Tikkanen, R.; Riikonen, A.; Oinonen, C.; Rouvinen, R.; Peltonen, L. Functional analyses of active site residues of human lysosomal aspartylglucosaminidase: Implications for catalytic mechanism and autocatalytic activation. EMBO J. 1996, 15, 2954–2960. [Google Scholar] [CrossRef]
- Lodola, A.; Branduardi, D.; De Vivo, M.; Capoferri, L.; Mor, M.; Piomelli, D.; Cavalli, A. A Catalytic Mechanism for Cysteine N-Terminal Nucleophile Hydrolases, as Revealed by Free Energy Simulations. PLoS ONE 2012, 7, e32397. [Google Scholar] [CrossRef]
- Gopal-Srivastava, R.; Hylemon, P.B. Purification and characterization of bile salt hydrolase from Clostridium perfringens. J. Lipid Res. 1988, 29, 1079–1085. [Google Scholar] [CrossRef]
- Buller, A.R.; Townsend, C.A. Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad. Proc. Natl. Acad. Sci. USA 2013, 110, E653–E661. [Google Scholar] [CrossRef] [PubMed]
- Buller, A.R.; Freeman, M.F.; Wright, N.T.; Schildbach, J.F.; Townsend, C.A. Insights into cis-autoproteolysis reveal a reactive state formed through conformational rearrangement. Proc. Natl. Acad. Sci. USA 2012, 109, 2308–2313. [Google Scholar] [CrossRef] [PubMed]
- Huber, E.M.; Heinemeyer, W.; Li, X.; Arendt, C.S.; Hochstrasser, M.; Groll, M. A unified mechanism for proteolysis and autocatalytic activation in the 20S proteasome. Nat. Commun. 2016, 7, 10900. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Songyang, Z.; Goldberg, A.L. Why Does Threonine, and Not Serine, Function as the Active Site Nucleophile in Proteasomes? J. Biol. Chem. 2000, 275, 14831–14837. [Google Scholar] [CrossRef]
- Liu, Y.; Guan, C.; Aronson, N.N., Jr. Site-directed Mutagenesis of Essential Residues Involved in the Mechanism of Bacterial Glycosylasparaginase. J. Biol. Chem. 1998, 273, 9688–9694. [Google Scholar] [CrossRef]
- Fanuel, L.; Goffin, C.; Cheggour, A.; Devreese, B.; Van Driessche, G.; Joris, B.; Van Beeumen, J.; Frere, J.M. The DmpA aminopeptidase from Ochrobactrum anthropi LMG7991 is the prototype of a new terminal nucleophile hydrolase family. Biochem. J. 1999, 341 Pt 1, 147–155. [Google Scholar] [CrossRef]
- Bompard-Gilles, C.; Villeret, V.; Davies, G.; Fanuel, L.; Joris, B.; Frère, J.-M.; Van Beeumen, J. A new variant of the Ntn hydrolase fold revealed by the crystal structure of l-aminopeptidase d-Ala-esterase/amidase from Ochrobactrum anthropi. Structure 2000, 8, 153–162. [Google Scholar] [CrossRef][Green Version]
- Cheng, H.; Grishin, N.V. DOM-fold: A structure with crossing loops found in DmpA, ornithine acetyltransferase, and molybdenum cofactor-binding domain. Protein Sci. 2005, 14, 1902–1910. [Google Scholar] [CrossRef]
- Saridakis, V.; Christendat, D.; Thygesen, A.; Arrowsmith, C.; Edwards, A.M.; Pai, E. Crystal structure ofMethanobacterium thermoautotrophicum conserved protein MTH1020 reveals an NTN-hydrolase fold. Proteins Struct. Funct. Bioinform. 2002, 48, 141–143. [Google Scholar] [CrossRef][Green Version]
- Kang, Y.-N.; Tran, A.; White, R.H.; Ealick, S.E. A Novel Function for the N-Terminal Nucleophile Hydrolase Fold Demonstrated by the Structure of an Archaeal Inosine Monophosphate Cyclohydrolase. Biochemistry 2007, 46, 5050–5062. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, D503–D509. [Google Scholar] [CrossRef] [PubMed]
- Zalkin, H. The Amidotransferases. Adv. Enzymol. Relat. Areas Mol. Biol. 1993, 66, 203–309. [Google Scholar] [CrossRef] [PubMed]
- Isupov, M.N.; Obmolova, G.; Butterworth, S.; Badet-Denisot, M.-A.; Badet, B.; Polikarpov, I.; Littlechild, J.A.; Teplyakov, A. Substrate binding is required for assembly of the active conformation of the catalytic site in Ntn amidotransferases: Evidence from the 1.8 å crystal structure of the glutaminase domain of glucosamine 6-phosphate synthase. Structure 1996, 4, 801–810. [Google Scholar] [CrossRef]
- Teplyakov, A.; Obmolova, G.; Badet, B.; Badet-Denisot, M.-A. Channeling of ammonia in glucosamine-6-phosphate synthase. J. Mol. Biol. 2001, 313, 1093–1102. [Google Scholar] [CrossRef]
- Richards, N.G.J.; Schuster, S.M. Mechanistic Issues in Asparagine Synthetase Catalysis. Adv. Enzymol. Relat. Areas. Mol. Biol. 1998, 72, 145–198. [Google Scholar] [CrossRef]
- Ruzzo, E.K.; Capo-Chichi, J.-M.; Ben-Zeev, B.; Chitayat, D.; Mao, H.; Pappas, A.L.; Hitomi, Y.; Lu, Y.-F.; Yao, X.; Hamdan, F.F.; et al. Deficiency of Asparagine Synthetase Causes Congenital Microcephaly and a Progressive Form of Encephalopathy. Neuron 2013, 80, 429–441. [Google Scholar] [CrossRef]
- Lomelino, C.L.; Andring, J.T.; McKenna, R.; Kilberg, M.S. Asparagine synthetase: Function, structure, and role in disease. J. Biol. Chem. 2017, 292, 19952–19958. [Google Scholar] [CrossRef]
- Aslanian, A.M.; Kilberg, M.S. Multiple adaptive mechanisms affect asparagine synthetase substrate availability in asparaginase-resistant MOLT-4 human leukaemia cells. Biochem. J. 2001, 358, 59–67. [Google Scholar] [CrossRef]
- Kim, J.H.; Krahn, J.M.; Tomchick, D.R.; Smith, J.L.; Zalkin, H. Structure and Function of the Glutamine Phosphoribosylpyrophosphate Amidotransferase Glutamine Site and Communication with the Phosphoribosylpyrophosphate Site. J. Biol. Chem. 1996, 271, 15549–15557. [Google Scholar] [CrossRef]
- Smith, J.L. Glutamine PRPP amidotransferase: Snapshots of an enzyme in action. Curr. Opin. Struct. Biol. 1998, 8, 686–694. [Google Scholar] [CrossRef]
- Zhao, H.; French, J.B.; Fang, Y.; Benkovic, S.J. The purinosome, a multi-protein complex involved in the de novo biosynthesis of purines in humans. Chem. Commun. 2013, 49, 4444–4452. [Google Scholar] [CrossRef] [PubMed]
- Baresova, V.; Krijt, M.; Skopova, V.; Součková, O.; Kmoch, S.; Zikanova, M. CRISPR-Cas9 induced mutations along de novo purine synthesis in HeLa cells result in accumulation of individual enzyme substrates and affect purinosome formation. Mol. Genet. Metab. 2016, 119, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Pelet, A.; Skopova, V.; Steuerwald, U.; Baresova, V.; Zarhrate, M.; Plaza, J.-M.; Hnizda, A.; Krijt, M.; Souckova, O.; Wibrand, F.; et al. PAICS deficiency, a new defect of de novo purine synthesis resulting in multiple congenital anomalies and fatal outcome. Hum. Mol. Genet. 2019, 28, 3805–3814. [Google Scholar] [CrossRef]
- Yamazaki, K.; Mizui, Y.; Oki, T.; Okada, M.; Tanaka, I. Cloning and characterization of mouse glutamine:fructose-6-phosphate amidotransferase 2 gene promoter. Gene 2000, 261, 329–336. [Google Scholar] [CrossRef]
- Oki, T.; Yamazaki, K.; Kuromitsu, J.; Okada, M.; Tanaka, I. cDNA Cloning and Mapping of a Novel Subtype of Glutamine:fructose-6-phosphate Amidotransferase (GFAT2) in Human and Mouse. Genomics 1999, 57, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Broschat, K.O.; Gorka, C.; Page, J.D.; Martin-Berger, C.L.; Davies, M.S.; Huang, H.-C.; Gulve, E.A.; Salsgiver, W.J.; Kasten, T.P. Kinetic Characterization of Human Glutamine-fructose-6-phosphate Amidotransferase I: Potent feedback inhibition by glucosamine 6-phosphate. J. Biol. Chem. 2002, 277, 14764–14770. [Google Scholar] [CrossRef] [PubMed]
- Moloughney, J.G.; Kim, P.K.; Vega-Cotto, N.M.; Wu, C.-C.; Zhang, S.; Adlam, M.; Lynch, T.; Chou, P.-C.; Rabinowitz, J.D.; Werlen, G.; et al. mTORC2 Responds to Glutamine Catabolite Levels to Modulate the Hexosamine Biosynthesis Enzyme GFAT1. Mol. Cell 2016, 63, 811–826. [Google Scholar] [CrossRef]
- Senderek, J.; Müller, J.S.; Dusl, M.; Strom, T.M.; Guergueltcheva, V.; Diepolder, I.; Laval, S.H.; Maxwell, S.; Cossins, J.; Krause, S.; et al. Hexosamine Biosynthetic Pathway Mutations Cause Neuromuscular Transmission Defect. Am. J. Hum. Genet. 2011, 88, 162–172. [Google Scholar] [CrossRef]
- Elbein, S.C.; Zheng, H.; Jia, Y.; Chu, W.; Cooper, J.J.; Hale, T.; Zhang, Z. Molecular screening of the human glutamine–fructose-6-phosphate amidotransferase 1 (GFPT1) gene and association studies with diabetes and diabetic nephropathy. Mol. Genet. Metab. 2004, 82, 321–328. [Google Scholar] [CrossRef]
- Curnow, A.W.; Hong, K.-W.; Yuan, R.; Kim, S.-I.; Martins, O.; Winkler, W.; Henkin, T.M.; Söll, D. Glu-tRNA Gln amidotransferase: A novel heterotrimeric enzyme required for correct decoding of glutamine codons during translation. Proc. Natl. Acad. Sci. USA 1997, 94, 11819–11826. [Google Scholar] [CrossRef]
- Wu, J.; Bu, W.; Sheppard, K.; Kitabatake, M.; Kwon, S.-T.; Söll, D.; Smith, J.L. Insights into tRNA-Dependent Amidotransferase Evolution and Catalysis from the Structure of the Aquifex aeolicus Enzyme. J. Mol. Biol. 2009, 391, 703–716. [Google Scholar] [CrossRef] [PubMed]
- LaRonde-LeBlanc, N.; Resto, M.; Gerratana, B. Regulation of active site coupling in glutamine-dependent NAD+ synthetase. Nat. Struct. Mol. Biol. 2009, 16, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Pace, H.C.; Brenner, C. The nitrilase superfamily: Classification, structure and function. Genome Biol. 2001, 2, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Moser, A.B.; Moser, H.W. Role of lysosomal acid ceramidase in the metabolism of ceramide in human skin fibroblasts. Arch. Biochem. Biophys. 1981, 208, 444–455. [Google Scholar] [CrossRef]
- Yu, F.P.S.; Amintas, S.; Levade, T.; Medin, J.A. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J. Rare Dis. 2018, 13, 1–19. [Google Scholar] [CrossRef]
- Coant, N.; Sakamoto, W.; Mao, C.; Hannun, Y.A. Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv. Biol. Regul. 2016, 63, 122–131. [Google Scholar] [CrossRef]
- Ferraz, M.J.; Marques, A.R.A.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef]
- Gebai, A.; Gorelik, A.; Li, Z.; Illes, K.; Nagar, B. Structural basis for the activation of acid ceramidase. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Schulze, H.; Schepers, U.; Sandhoff, K. Overexpression and mass spectrometry analysis of mature human acid ceramidase. Biol. Chem. 2007, 388, 1333–1343. [Google Scholar] [CrossRef]
- Ferlinz, K.; Kopal, G.; Bernardo, K.; Linke, T.; Bär, J.; Breiden, B.; Neumann, U.; Lang, F.; Schuchman, E.H.; Sandhoff, K. Human Acid Ceramidase: Processing, glycosylation, and lysosomal targeting. J. Biol. Chem. 2001, 276, 35352–35360. [Google Scholar] [CrossRef]
- Azuma, N.; Obrien, J.; Moser, H.; Kishimoto, Y. Stimulation of Acid Ceramidase Activity by Saposin D. Arch. Biochem. Biophys. 1994, 311, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Henseler, M.; Klein, C.; Suzuki, K.; Harzer, K.; Sandhoff, K. Sphingolipid Activator Protein D (sap-D) Stimulates the Lysosomal Degradation of Ceramide in Vivo. Biochem. Biophys. Res. Commun. 1994, 200, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Vasiliauskaité-Brooks, I.; Healey, R.D.; Rochaix, P.; Saint-Paul, J.; Sounier, R.; Grison, C.; Waltrich-Augusto, T.; Fortier, M.; Hoh, F.; Saied, E.M.; et al. Structure of a human intramembrane ceramidase explains enzymatic dysfunction found in leukodystrophy. Nat. Commun. 2018, 9, 5437. [Google Scholar] [CrossRef] [PubMed]
- Airola, M.V.; Allen, W.J.; Pulkoski-Gross, M.J.; Obeid, L.M.; Rizzo, R.C.; Hannun, Y.A. Structural Basis for Ceramide Recognition and Hydrolysis by Human Neutral Ceramidase. Structure 2015, 23, 1482–1491. [Google Scholar] [CrossRef]
- Ueda, N.; Tsuboi, K.; Uyama, T. N-acylethanolamine metabolism with special reference to N-acylethanolamine-hydrolyzing acid amidase (NAAA). Prog. Lipid Res. 2010, 49, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Sasso, O.; Summa, M.; Armirotti, A.; Pontis, S.; De Mei, C.; Piomelli, D. The N-Acylethanolamine Acid Amidase Inhibitor ARN077 Suppresses Inflammation and Pruritus in a Mouse Model of Allergic Dermatitis. J. Investig. Dermatol. 2018, 138, 562–569. [Google Scholar] [CrossRef]
- Tai, T.; Tsuboi, K.; Uyama, T.; Masuda, K.; Cravatt, B.F.; Houchi, H.; Ueda, N. Endogenous Molecules Stimulating N-Acylethanolamine-Hydrolyzing Acid Amidase (NAAA). ACS Chem. Neurosci. 2012, 3, 379–385. [Google Scholar] [CrossRef]
- Zhao, L.-Y.; Tsuboi, K.; Okamoto, Y.; Nagahata, S.; Ueda, N. Proteolytic activation and glycosylation of N-acylethanolamine-hydrolyzing acid amidase, a lysosomal enzyme involved in the endocannabinoid metabolism. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2007, 1771, 1397–1405. [Google Scholar] [CrossRef]
- Gorelik, A.; Gebai, A.; Illes, K.; Piomelli, D.; Nagar, B. Molecular mechanism of activation of the immunoregulatory amidase NAAA. Proc. Natl. Acad. Sci. USA 2018, 115, E10032–E10040. [Google Scholar] [CrossRef]
- Repo, H.; Kuokkanen, E.; Oksanen, E.; Goldman, A.; Heikinheimo, P. Is the bovine lysosomal phospholipase B-like protein an amidase? Proteins Struct. Funct. Bioinform. 2014, 82, 300–311. [Google Scholar] [CrossRef]
- Peck, R.F.; Shiflett, A.M.; Schwartz, K.J.; McCann, A.; Hajduk, S.L.; Bangs, J.D. The LAMP-like protein p67 plays an essential role in the lysosome of African trypanosomes. Mol. Microbiol. 2008, 68, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhao, L.; Larsson, A.; Venge, P. The identification of a phospholipase B precursor in human neutrophils. FEBS J. 2009, 276, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, H.; Wong, W.-C.; Sem, X.; Han, H.; Ong, S.-M.; Tan, Y.-C.; Yeap, W.-H.; Gan, C.-S.; Ng, K.-Q.; et al. Identification of Novel Functional Differences in Monocyte Subsets Using Proteomic and Transcriptomic Methods. J. Proteome Res. 2009, 8, 4028–4038. [Google Scholar] [CrossRef] [PubMed]
- Joucla, G.; Le Sénéchal, C.; Bégorre, M.; Garbay, B.; Santarelli, X.; Cabanne, C. Cation exchange versus multimodal cation exchange resins for antibody capture from CHO supernatants: Identification of contaminating Host Cell Proteins by mass spectrometry. J. Chromatogr. B 2013, 942–943, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Dixit, N.; Salamat-Miller, N.; Salinas, P.A.; Taylor, K.D.; Basu, S.K. Residual Host Cell Protein Promotes Polysorbate 20 Degradation in a Sulfatase Drug Product Leading to Free Fatty Acid Particles. J. Pharm. Sci. 2016, 105, 1657–1666. [Google Scholar] [CrossRef]
- Yuk, I.H.; Nishihara, J.; Walker, D., Jr.; Huang, E.; Gunawan, F.; Subramanian, J.; Pynn, A.F.; Yu, X.C.; Zhu-Shimoni, J.; Vanderlaan, M.; et al. More similar than different: Host cell protein production using three null CHO cell lines. Biotechnol. Bioeng. 2015, 112, 2068–2083. [Google Scholar] [CrossRef]
- Fischer, S.K.; Cheu, M.; Peng, K.; Lowe, J.; Araujo, J.; Murray, E.; McClintock, D.; Matthews, J.; Siguenza, P.; Song, A. Specific Immune Response to Phospholipase B-Like 2 Protein, a Host Cell Impurity in Lebrikizumab Clinical Material. AAPS J. 2017, 19, 254–263. [Google Scholar] [CrossRef]
- Valente, K.N.; Levy, N.E.; Lee, K.H.; Lenhoff, A.M. Applications of proteomic methods for CHO host cell protein characterization in biopharmaceutical manufacturing. Curr. Opin. Biotechnol. 2018, 53, 144–150. [Google Scholar] [CrossRef]
- Pei, J.; Grishin, N.V. Peptidase family U34 belongs to the superfamily of N-terminal nucleophile hydrolases. Protein Sci. 2003, 12, 1131–1135. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Lindhout, F.W.; Cao, Y.; Kevenaar, J.T.; Bodzęta, A.; Stucchi, R.; Boumpoutsari, M.M.; Katrukha, E.A.; Altelaar, M.; MacGillavry, H.; Hoogenraad, C.C. VAP-SCRN1 interaction regulates dynamic endoplasmic reticulum remodeling and presynaptic function. EMBO J. 2019, 38, e101345. [Google Scholar] [CrossRef] [PubMed]
- Way, G.; Morrice, N.; Smythe, C.; O‘Sullivan, A.J. Purification and Identification of Secernin, a Novel Cytosolic Protein that Regulates Exocytosis in Mast Cells. Mol. Biol. Cell 2002, 13, 3344–3354. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.E.; Levine, T.P. VAP, a Versatile Access Point for the Endoplasmic Reticulum: Review and analysis of FFAT-like motifs in the VAPome. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2016, 1861, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Ditzel, L.; Löwe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S proteasome from yeast at 2.4Å resolution. Nature 1997, 386, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Unno, M.; Mizushima, T.; Morimoto, Y.; Tomisugi, Y.; Tanaka, K.; Yasuoka, N.; Tsukihara, T. The Structure of the Mammalian 20S Proteasome at 2.75 Å Resolution. Structure 2002, 10, 609–618. [Google Scholar] [CrossRef]
- Frentzel, S.; Pesold-Hurt, B.; Seelig, A.; Kloetzel, P.-M. 20 S proteasomes are assembled via distinct precursor complexes processing of LMP2 and LMP7 proproteins takes place in 13–16 S preproteasome complexes. J. Mol. Biol. 1994, 236, 975–981. [Google Scholar] [CrossRef]
- Seemüller, E.; Lupas, A.; Stock, D.; Löwe, J.; Huber, R.; Baumeister, W. Proteasome from Thermoplasma acidophilum: A Threonine Protease. Science 1995, 268, 579–582. [Google Scholar] [CrossRef]
- Van den Eshof, B.L.; Medfai, L.; Nolfi, E.; Wawrzyniuk, M.; Sijts, A.J.A.M. The Function of Immunoproteasomes-An Immunologists’ Perspective. Cells. 2021, 10, 3360. [Google Scholar] [CrossRef]
- Sasaki, K.; Takada, K.; Ohte, Y.; Kondo, H.; Sorimachi, H.; Tanaka, K.; Takahama, Y.; Murata, S. Thymoproteasomes produce unique peptide motifs for positive selection of CD8+ T cells. Nat. Commun. 2015, 6, 7484. [Google Scholar] [CrossRef]
- Da Silva, L.S.; Doonan, L.B.; Pessoa, A., Jr.; de Oliveira, M.A.; Long, P.F. Structural and functional diversity of asparaginases: Overview and recommendations for a revised nomenclature. Biotechnol. Appl. Biochem. 2022, 69, 503–513. [Google Scholar] [CrossRef]
- Michalska, K.; Brzezinski, K.; Jaskolski, M. Crystal Structure of Isoaspartyl Aminopeptidase in Complex with l-Aspartate. J. Biol. Chem. 2005, 280, 28484–28491. [Google Scholar] [CrossRef] [PubMed]
- Nomme, J.; Su, Y.; Konrad, M.; Lavie, A. Structures of Apo and Product-Bound Human l-Asparaginase: Insights into the Mechanism of Autoproteolysis and Substrate Hydrolysis. Biochemistry 2012, 51, 6816–6826. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.R.; Stone, E.M.; Chantranupong, L.; Georgiou, G. The Human Asparaginase-like Protein 1 hASRGL1 Is an Ntn Hydrolase with β-Aspartyl Peptidase Activity. Biochemistry 2009, 48, 11026–11031. [Google Scholar] [CrossRef] [PubMed]
- Morais, S.B.; Pirolla, R.A.S.; Frota, N.F.; Lourenzoni, M.R.; Gozzo, F.C.; Souza, T. The role of the quaternary structure in the activation of human L-asparaginase. J. Proteom. 2020, 224, 103818. [Google Scholar] [CrossRef] [PubMed]
- Borek, D.; Jaskólski, M. Crystallization and preliminary crystallographic studies of a new L-asparaginase encoded by the Escherichia coli genome. Acta Crystallogr. Sect. D Biol. Crystallogr. 2000, 56, 1505–1507. [Google Scholar] [CrossRef] [PubMed]
- Borek, D.; Michalska, K.; Brzeziński, K.; Kisiel, A.; Podkowiński, J.; Bonthron, D.; Krowarsch, D.; Otlewski, J.; Jaskolski, M.; Zmienko, A. Expression, purification and catalytic activity of Lupinus luteus asparagine β-amidohydrolase and its Escherichia coli homolog. JBIC J. Biol. Inorg. Chem. 2004, 271, 3215–3226. [Google Scholar] [CrossRef]
- Mononen, I.; Fisher, K.J.; Kaartinen, V.; Aronson, N.N., Jr. Aspartylglycosaminuria: Protein chemistry and molecular biology of the most common lysosomal storage disorder of glycoprotein degradation. FASEB J. 1993, 7, 1247–1256. [Google Scholar] [CrossRef]
- Tikkanen, R.; Enomaa, N.; Riikonen, A.; Ikonen, E.; Peltonen, L. Intracellular Sorting of Aspartylglucosaminidase: The Role ofN-Linked Oligosaccharides and Evidence of Man-6-P-Independent Lysosomal Targeting. DNA Cell Biol. 1995, 14, 305–312. [Google Scholar] [CrossRef]
- Arvio, M.; Mononen, I. Aspartylglycosaminuria: A review. Orphanet J. Rare Dis. 2016, 11, 162. [Google Scholar] [CrossRef]
- Beck, M. Treatment strategies for lysosomal storage disorders. Dev. Med. Child Neurol. 2017, 60, 13–18. [Google Scholar] [CrossRef]
- Banning, A.; Gülec, C.; Rouvinen, J.; Gray, S.J.; Tikkanen, R. Identification of Small Molecule Compounds for Pharmacological Chaperone Therapy of Aspartylglucosaminuria. Sci. Rep. 2016, 6, 37583. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Snanoudj-Verber, S.; Pollard, L.; Hu, Y.; Cathey, S.S.; Tikkanen, R.; Gray, S.J. Pre-clinical Gene Therapy with AAV9/AGA in Aspartylglucosaminuria Mice Provides Evidence for Clinical Translation. Mol. Ther. 2021, 29, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.-D.; Cheng, E.H.-Y.; Korsmeyer, S.J. Taspase1: A Threonine Aspartase Required for Cleavage of MLL and Proper HOX Gene Expression. Cell 2003, 115, 293–303. [Google Scholar] [CrossRef]
- Khan, J.A.; Dunn, B.M.; Tong, L. Crystal Structure of Human Taspase1, a Crucial Protease Regulating the Function of MLL. Structure 2005, 13, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, R.C.; Clarke, S. Succinimide Formation from Aspartyl and Asparaginyl Peptides as a Model for the Spontaneous Degradation of Proteins. J. Biol. Chem. 1989, 264, 6164–6170. [Google Scholar] [CrossRef]
- Suleiman, J.; Riedhammer, K.M.; Jicinsky, T.; Mundt, M.; Werner, L.; Gusic, M.; Burgemeister, A.L.; AlSaif, H.S.; Abdulrahim, M.; Moghrabi, N.N.; et al. Homozygous loss-of-function variants of TASP1, a gene encoding an activator of the histone methyltransferases KMT2A and KMT2D, cause a syndrome of developmental delay, happy demeanor, distinctive facial features, and congenital anomalies. Hum. Mutat. 2019, 40, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- Niizuma, H.; Cheng, E.H.; Hsieh, J.J. Taspase 1: A protease with many biological surprises. Mol. Cell. Oncol. 2015, 2, e999513. [Google Scholar] [CrossRef]
- Takeda, S.; Chen, D.Y.; Westergard, T.D.; Fisher, J.K.; Rubens, J.A.; Sasagawa, S.; Kan, J.T.; Korsmeyer, S.J.; Cheng, E.H.-Y.; Hsieh, J.J.-D. Proteolysis of MLL family proteins is essential for Taspase1-orchestrated cell cycle progression. Genes Dev. 2006, 20, 2397–2409. [Google Scholar] [CrossRef]
- Schalk, A.M.; Nguyen, H.-A.; Rigouin, C.; Lavie, A. Identification and Structural Analysis of an l-Asparaginase Enzyme from Guinea Pig with Putative Tumor Cell Killing Properties. J. Biol. Chem. 2014, 289, 33175–33186. [Google Scholar] [CrossRef]
- Karamitros, C.S.; Konrad, M. Human 60-kDa Lysophospholipase Contains an N-terminal l-Asparaginase Domain That Is Allosterically Regulated by l-Asparagine. J. Biol. Chem. 2014, 289, 12962–12975. [Google Scholar] [CrossRef]
- Ueno, T.; Ohtawa, K.; Mitsui, K.; Kodera, Y.; Hiroto, M.; Matsushima, A.; Inada, Y.; Nishimura, H. Cell cycle arrest and apoptosis of leukemia cells induced by L-asparaginase. Leukemia 1997, 11, 1858–1861. [Google Scholar] [CrossRef] [PubMed]
- Pieters, R.; Hunger, S.P.; Boos, J.; Rizzari, C.; Silverman, L.; Baruchel, A.; Goekbuget, N.; Schrappe, M.; Pui, C.-H. L-asparaginase treatment in acute lymphoblastic leukemia: A focus on Erwinia asparaginase. Cancer 2011, 117, 238–249. [Google Scholar] [CrossRef]
- Nomme, J.; Su, Y.; Lavie, A. Elucidation of the Specific Function of the Conserved Threonine Triad Responsible for Human l-Asparaginase Autocleavage and Substrate Hydrolysis. J. Mol. Biol. 2014, 426, 2471–2485. [Google Scholar] [CrossRef]
- Wickham, S.; West, M.B.; Cook, P.F.; Hanigan, M.H. Gamma-glutamyl compounds: Substrate specificity of gamma-glutamyl transpeptidase enzymes. Anal. Biochem. 2011, 414, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Keillor, J.W.; Castonguay, R.; Lherbet, C. Gamma-Glutamyl Transpeptidase Substrate Specificity and Catalytic Mechanism. Methods Enzymol. 2005, 401, 449–467. [Google Scholar] [CrossRef]
- Carter, B.Z.; Wiseman, A.L.; Orkiszewski, R.; Ballard, K.D.; Ou, C.-N.; Lieberman, M.W. Metabolism of Leukotriene C4 in γ-Glutamyl Transpeptidase-deficient Mice. J. Biol. Chem. 1997, 272, 12305–12310. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Luo, G.; Shi, Z.-Z.; Barrios, R.; Atwood, D.; Liu, W.; Habib, G.M.; Sifers, R.N.; Corry, D.B.; Lieberman, M.W. γ-Glutamyl Leukotrienase, a Novel Endothelial Membrane Protein, Is Specifically Responsible for Leukotriene D4 Formation in Vivo. Am. J. Pathol. 2002, 161, 481–490. [Google Scholar] [CrossRef]
- Iida, M.; Yasuhara, T.; Mochizuki, H.; Takakura, H.; Yanagisawa, T.; Kubo, H. Two Japanese brothers with hereditary γ-glutamyl transpeptidase deficiency. J. Inherit. Metab. Dis. 2005, 28, 49–55. [Google Scholar] [CrossRef]
- Hammond, J.W.; Potter, M.; Wilcken, B.; Truscott, R. Siblings with ?-glutamyltransferase deficiency. J. Inherit. Metab. Dis. 1995, 18, 82–83. [Google Scholar] [CrossRef] [PubMed]
- Mayatepek, E.; Meissner, T.; Gröbe, H. Acute metabolic crisis with extreme deficiency of glutathione in combination with decreased CSF levels of leukotriene C4 in a patient with glutathione synthetase deficiency. J. Inherit. Metab. Dis. 2004, 27, 297–299. [Google Scholar] [CrossRef]
- Heisterkamp, N.; Groffen, J.; Warburton, D.; Sneddon, T.P. The human gamma-glutamyltransferase gene family. Hum. Genet. 2008, 123, 321–332. [Google Scholar] [CrossRef] [PubMed]
- West, M.B.; Chen, Y.; Wickham, S.; Heroux, A.; Cahill, K.; Hanigan, M.H.; Mooers, B.H.M. Novel Insights into Eukaryotic γ-Glutamyltranspeptidase 1 from the Crystal Structure of the Glutamate-bound Human Enzyme. J. Biol. Chem. 2013, 288, 31902–31913. [Google Scholar] [CrossRef] [PubMed]
- Terzyan, S.S.; Nguyen, L.T.; Burgett, A.W.G.; Heroux, A.; Smith, C.A.; You, Y.; Hanigan, M.H. Crystal structures of glutathione- and inhibitor-bound human GGT1: Critical interactions within the cysteinylglycine binding site. J. Biol. Chem. 2021, 296, 100066. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ntn-Hydrolase-Like | Catalytic Residue | Reference Structure |
|---|---|---|
| Ntn-hydrolase Superfamily | ||
| 1. Class II glutamine amidotransferases | C | GAT, B. subtilis (PDB: 1GPH) |
| 2. Penicillin V Acylases | C | PVA, B. sphaericus (PDB: 3PVA) |
| 3. Penicillin G Acylases | S | PGA, E. coli (PDB: 1PNK) |
| 4. Proteasome subunits | T | Pr, T. acidophilum (PDB: 1PMA) |
| 5. (Glycosyl-)asparaginases | T | AGA, H. sapiens (PDB: 1APY) |
| 6. Gamma-glutamyltranspeptidase-like | T | GGT, E. coli (PDB: 2DBU) |
| 7. SPO2555 | T | SPO2555, S. pomerovi (PDB: 2IMH) |
| Archaeal IMP cyclohydrolase | ||
| 1. Archaeal IMP cyclohydrolase PurO | N/A | MTH1020 M. thermoautotrophicum (PDB: 1KUU) |
| beta-Aminopeptidases | ||
| 1. DmpA like/BapA | S | DmpA, O. anthropi (PDB: 1B65) |
| 2. Ornithin Acyltransferases/ArgJ | T | OAT, E. coli (PDB: IVZ6) |
| SCOPe Family | MEROPS Family | Gene | Protein Name | EC | UniProt |
|---|---|---|---|---|---|
| Class II glutamine amido-transferases (GAT) d.153.1.1 | C44 | ASNS | Glutamine-dependent asparagine synthetase (ASNS) | 6.3.5.4 | P08243 |
| ASNSD1 | Asparagine synthetase domain-containing protein 1 (ASNSD1) | 6.3.5.- | Q9NWL6 | ||
| PPAT | Glutamine phosphoribosyl-pyrophosphate amidotransferase (GPAT), Amidophosphoribosyltransferase PPAT, PUR1 | 2.4.2.14 | Q06203 | ||
| GFPT1 | Glutamine fructose-6-phosphate amidotransferase 1 (GFAT1) Gln-Fru6P-Transaminase 1 (GFPT1) | 2.6.1.16 | Q06210 | ||
| GFPT2 | Glutamine fructose-6-phosphate amidotransferase 2 (GFAT2) Gln-Fru6P-Transaminase 2 (GFPT2) | 2.6.1.16 | O94808 | ||
| Penicillin acylase (PVA) d.153.1.3 | C89 | ASAH1 | Acid Ceramidase (AC, aCDase), N-acylsphingosine amidohydrolase 1 (ASAH1) | 3.5.1.23 | Q13510 |
| NAAA | N-acylethanolamine-hydrolyzing acid amidase (NAAA) | 3.5.1.60 | Q02083 | ||
| C95 | PLBD1 | Phospholipase B domain-containing protein 1, FLJ22662 | tbd | Q6P4A8 | |
| PLBD2 | Phospholipase B domain-containing protein 2 (PLBD2), Phospholipase B-like 2 (PLBL2), P76, 66.3-kDa protein | tbd | Q8NHP8 | ||
| C69 | SCRN1 | Secernin-1, U34-dipeptidase homologue | N/A | Q12765 | |
| SCRN2 | Secernin-2, U34-dipeptidase homologue | tbd | Q96FV2 | ||
| SCRN3 | Secernin-3, U34-dipeptidase homologue | tbd | Q0VDG4 | ||
| Proteasome subunits (Pr) d.153.1.4 | T1 | PSMB6 | Proteasome subunit β1, Y | 3.4.25.1 | P28072 |
| PSMB7 | Proteasome subunit β2, Z | 3.4.25.1 | Q99436 | ||
| PSMB5 | Proteasome subunit β5, X | 3.4.25.1 | P28074 | ||
| PSMB9 | Proteasome subunit β1i, LMP2 | 3.4.25.1 | P28065 | ||
| PSMB10 | Proteasome subunit β2i, MECL-1 | 3.4.25.1 | P40306 | ||
| PSMB8 | Proteasome subunit β5i, LMP7 | 3.4.25.1 | P28062 | ||
| PSMB11 | Proteasome subunit β5t | 3.4.25.1 | A5LHX3 | ||
| (Glycosyl) asparaginase (AGA) d.153.1.5 | T2 | AGA | Aspartylglucosaminidase (ASPG) | 3.5.1.26 | P20933 |
| ASRGL1 | Isoaspartyl peptidase, Asparaginase-like protein 1, beta-aspartyl-peptidase, L-Asparaginase, ALP, hASNase3, CRASH, glial asparaginase | 3.4.19.5 | Q7L266 | ||
| TASP1 | Aspartyl endopeptidase, threonine aspartase 1 | 3.4.25.- | Q9H6P5 | ||
| Gamma-glutamyl-transpeptidase-like (GGT) d.153.1.6 | T3 | GGT1 | Glutathione hydrolase 1 proenzyme, γ-Glutamyl transpeptidase 1 | 3.4.19.13 | P19440 |
| GGT2 | γ-Glutamyl transpeptidase 2 | tbd | P36268 | ||
| GGT3 | γ-Glutamyl transpeptidase 3 | tbd | A6NGU5 | ||
| GGT5 | γ-Glutamyl leukotrienase, γ-Glutamyl transpeptidase 5 | 3.4.19.14 | P36269 | ||
| GGT6 | γ-Glutamyl transpeptidase 6 | tbd | Q6P531 | ||
| GGT7 | γ-Glutamyl transpeptidase 7 | tbd | Q9UJ14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linhorst, A.; Lübke, T. The Human Ntn-Hydrolase Superfamily: Structure, Functions and Perspectives. Cells 2022, 11, 1592. https://doi.org/10.3390/cells11101592
Linhorst A, Lübke T. The Human Ntn-Hydrolase Superfamily: Structure, Functions and Perspectives. Cells. 2022; 11(10):1592. https://doi.org/10.3390/cells11101592
Chicago/Turabian StyleLinhorst, Arne, and Torben Lübke. 2022. "The Human Ntn-Hydrolase Superfamily: Structure, Functions and Perspectives" Cells 11, no. 10: 1592. https://doi.org/10.3390/cells11101592
APA StyleLinhorst, A., & Lübke, T. (2022). The Human Ntn-Hydrolase Superfamily: Structure, Functions and Perspectives. Cells, 11(10), 1592. https://doi.org/10.3390/cells11101592