
Identification of miRNA–mRNA Regulatory Modules Involved in Lipid Metabolism and Seed Development in a Woody Oil Tree (Camellia oleifera)


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site and Sampling
2.2. Small RNA Library Construction and Sequencing
2.3. Identification and Analysis of miRNA Sequencing Data
2.4. Analysis of Differentially Expressed miRNAs
2.5. Prediction and Identification of miRNA–mRNA Regulatory Modules
2.6. Identification of the Expression of miRNAs and Their Target Genes by qRT-PCR
2.7. Dual-Luciferase Reporter Assay
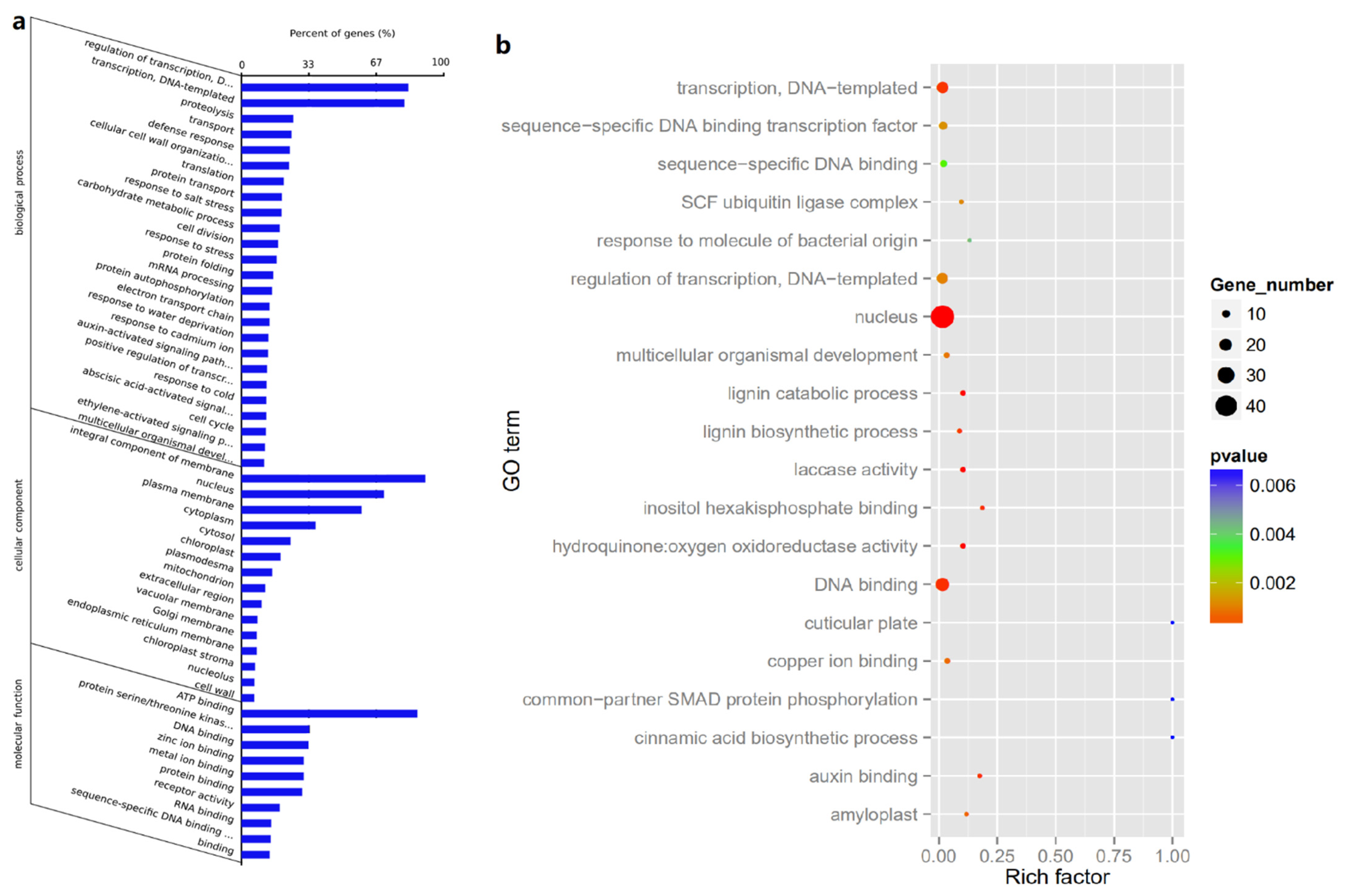
3. Results
3.1. Brief Outline for Sequencing Data, from Raw Data to Cleaned Sequences
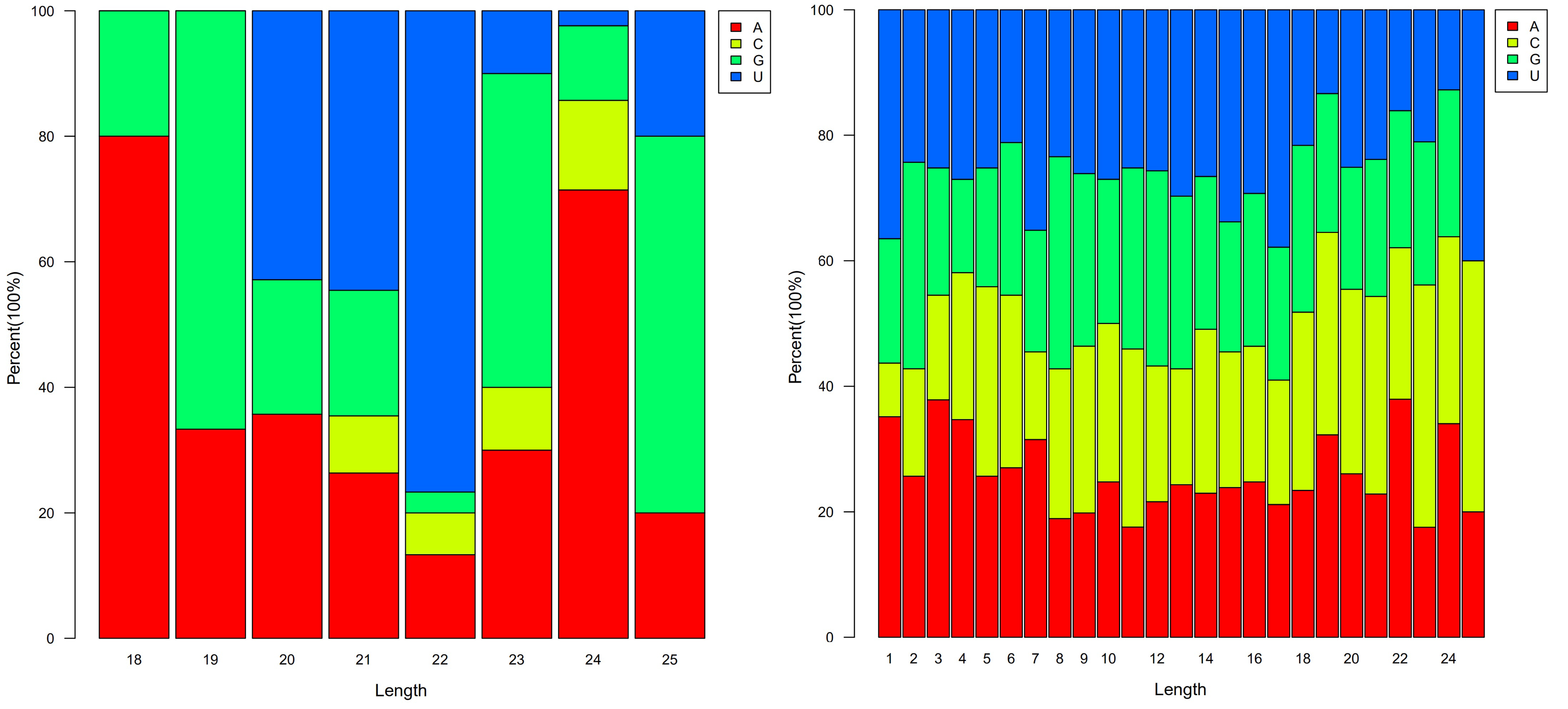
3.2. Identification of Known and Novel miRNAs in Developing Seeds of Tea Oil Camelia
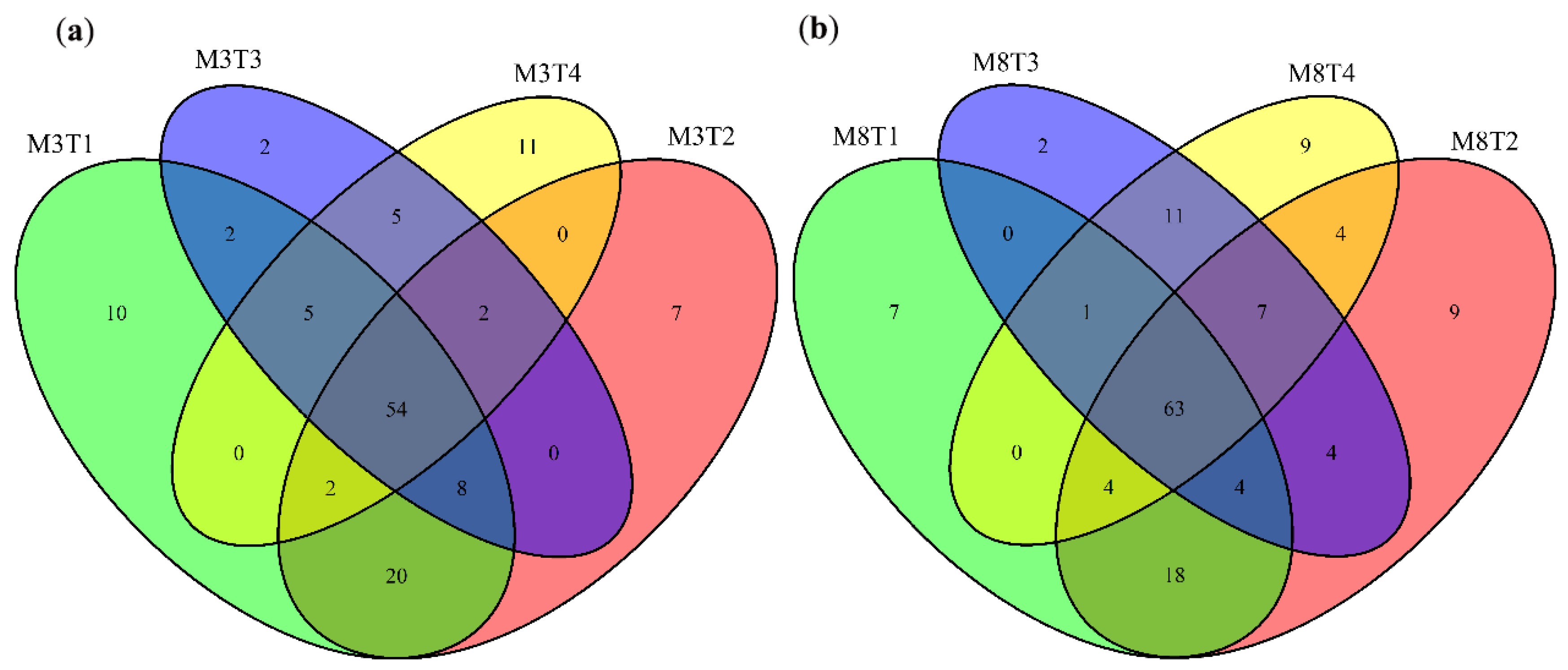
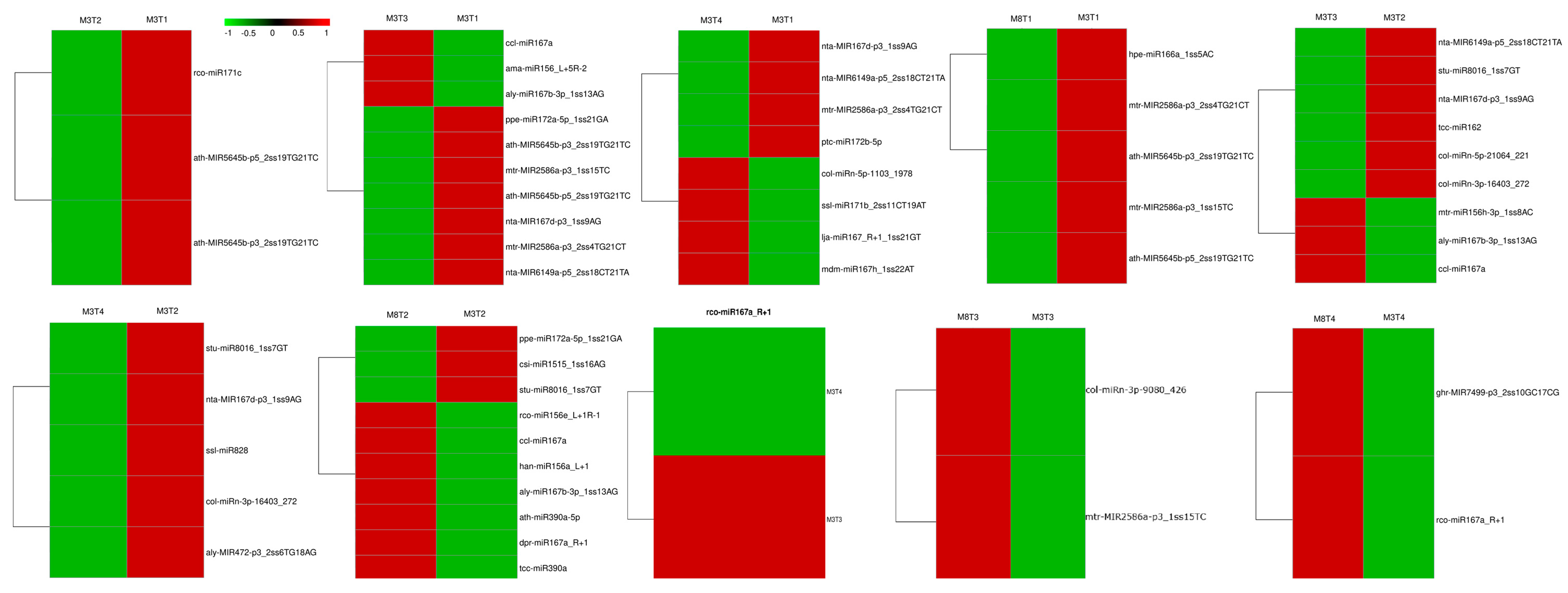
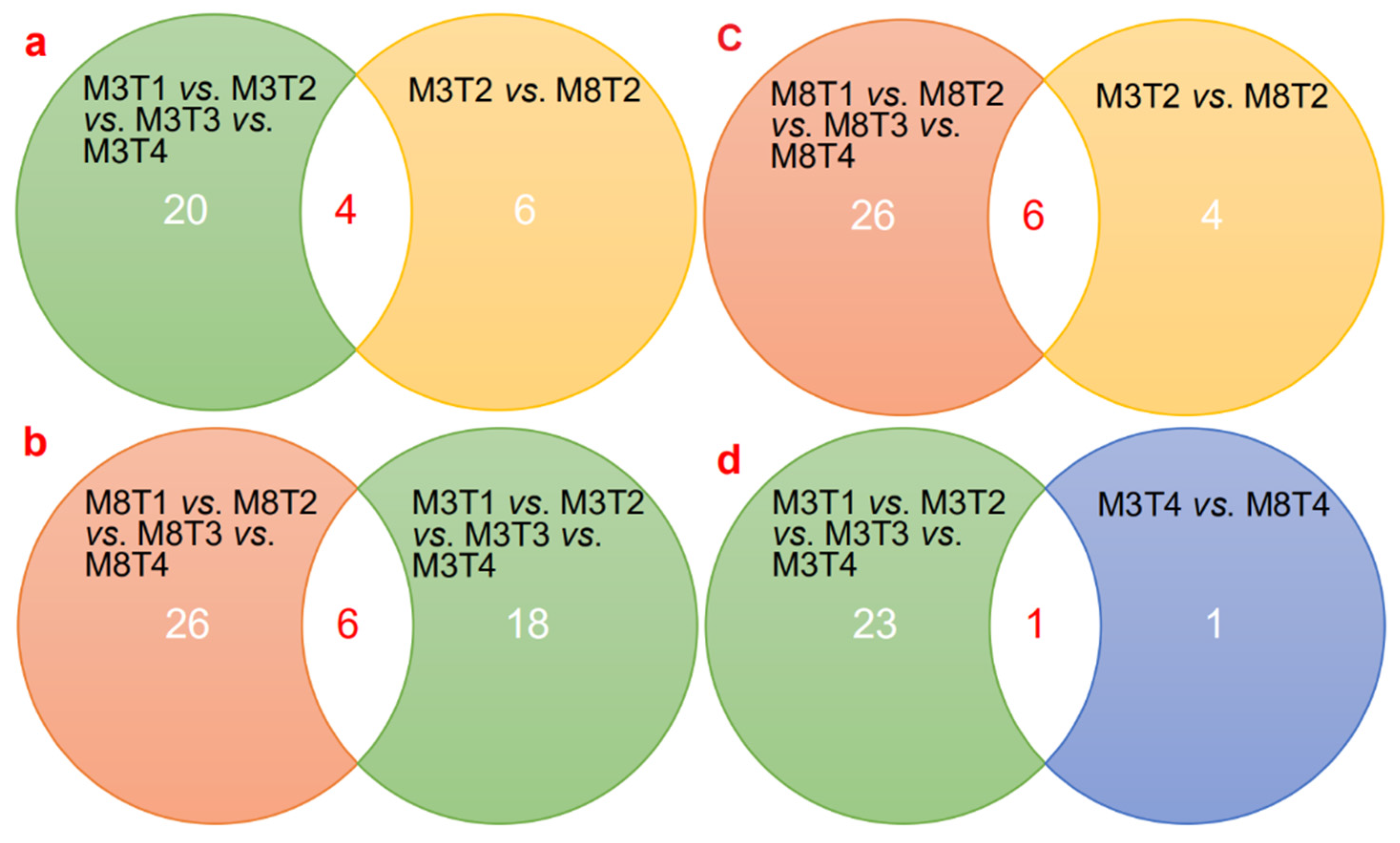
3.3. Identification of Differentially Expressed miRNAs
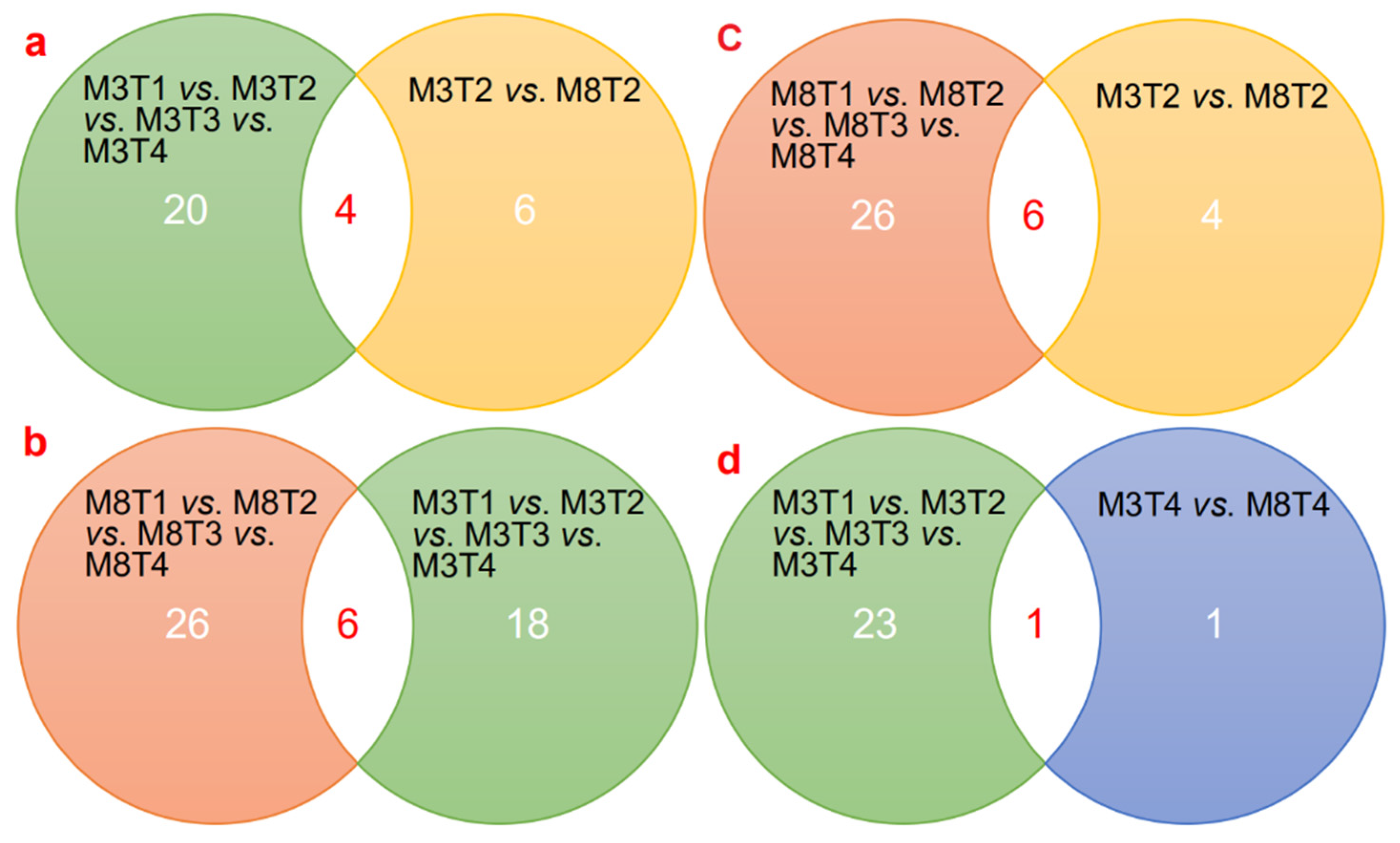
3.4. Prediction and Identification of miRNA–mRNA Regulatory Modules
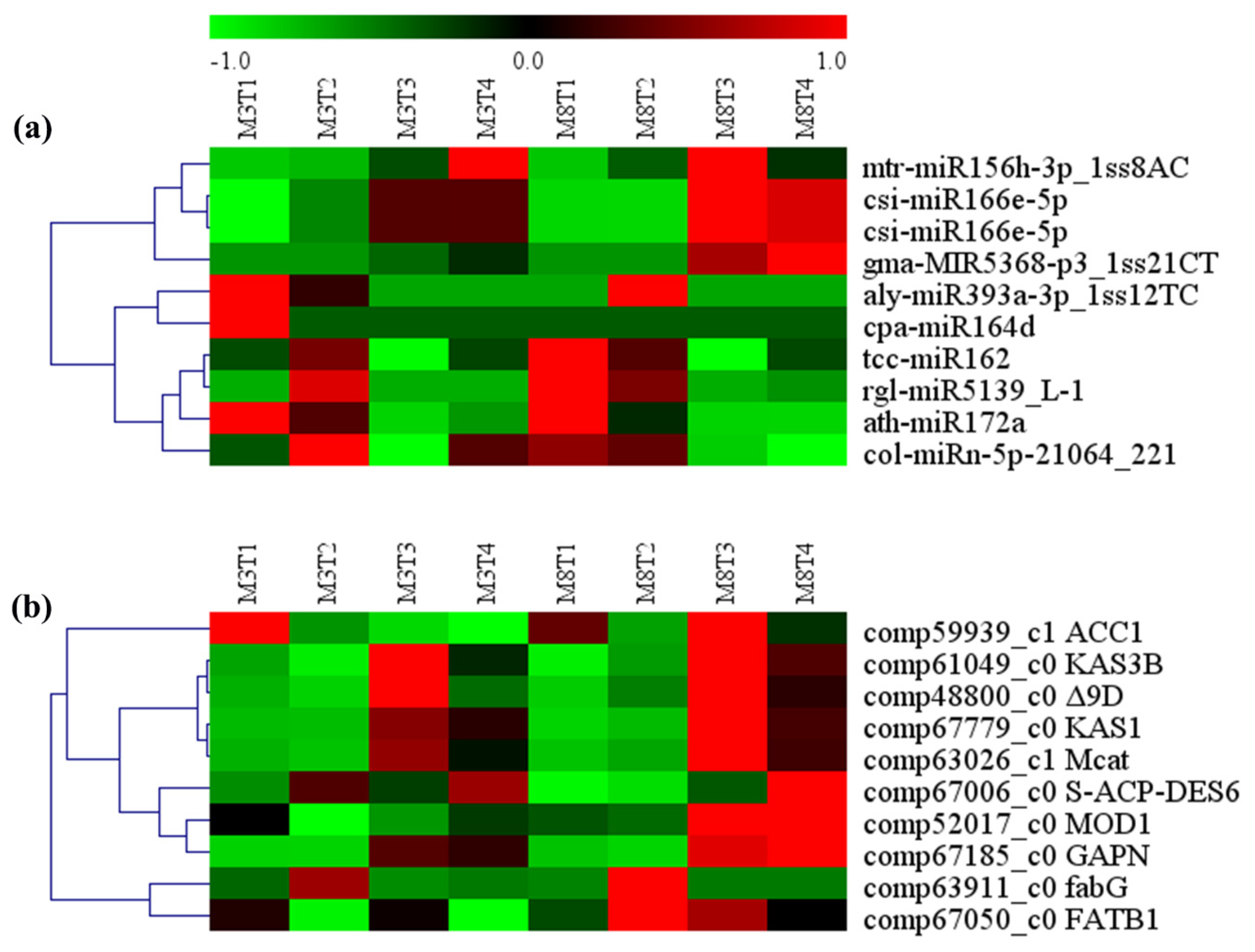
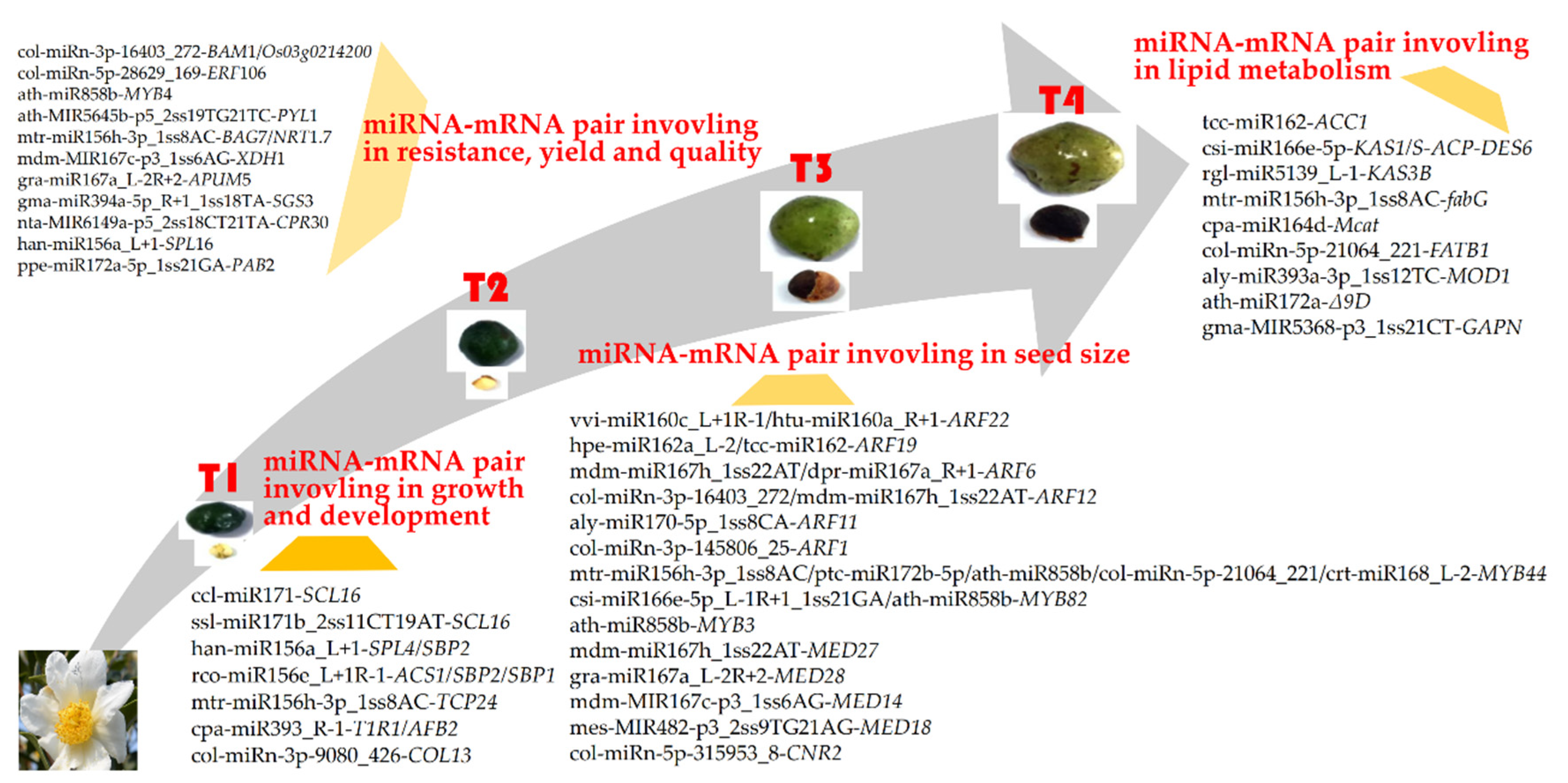
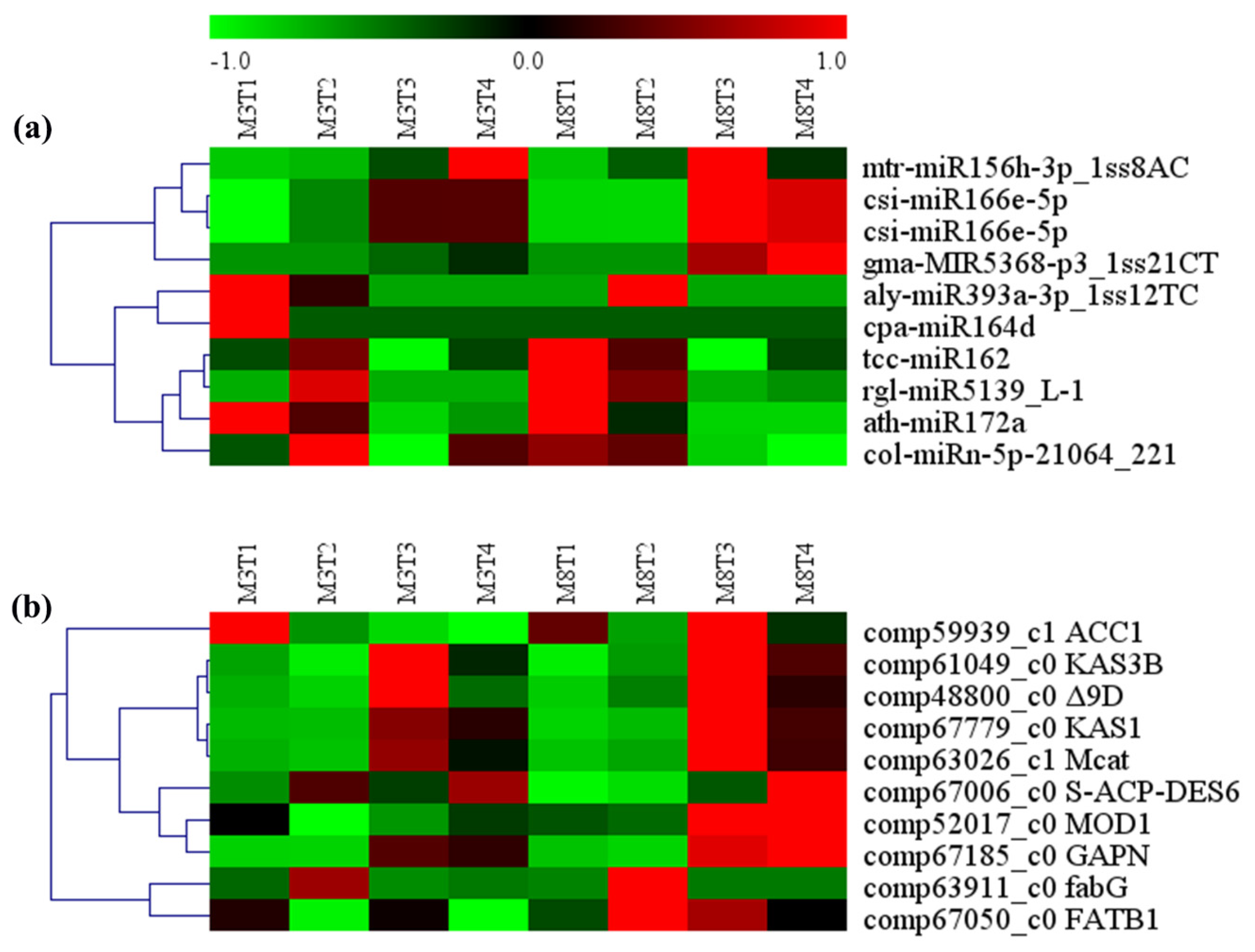
3.5. MiRNA–mRNA Regulation Modules Involved in Lipid Metabolism
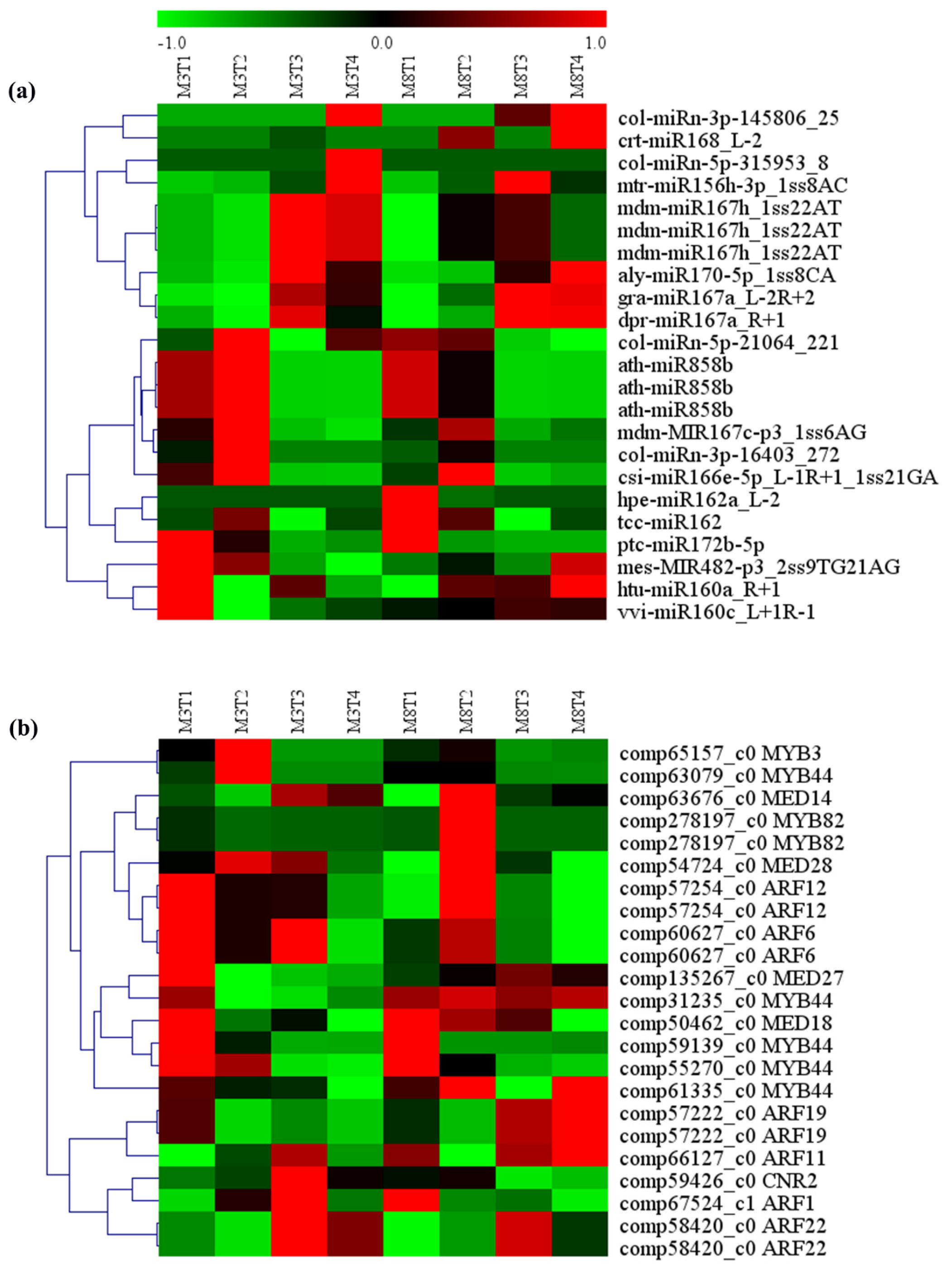
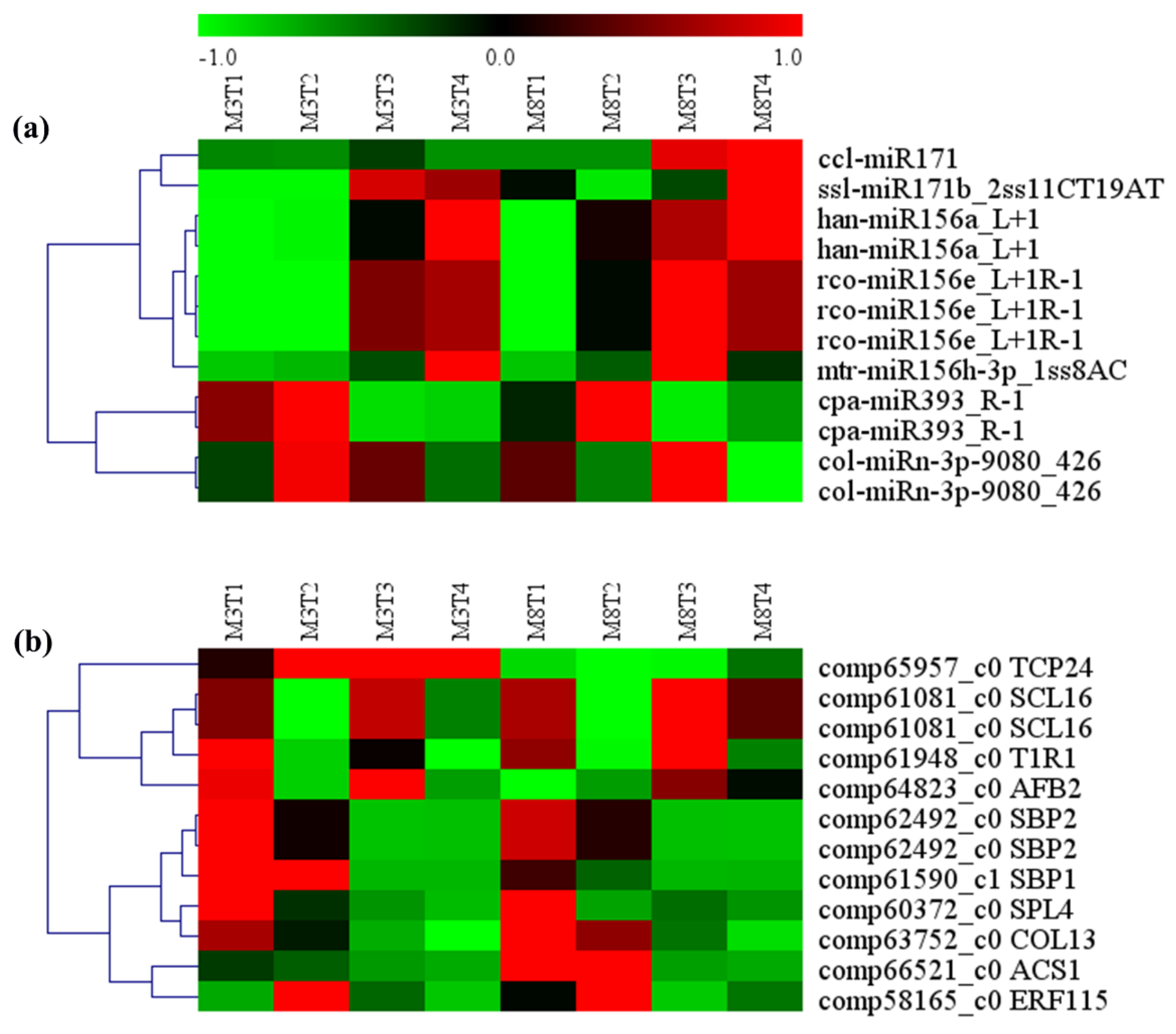
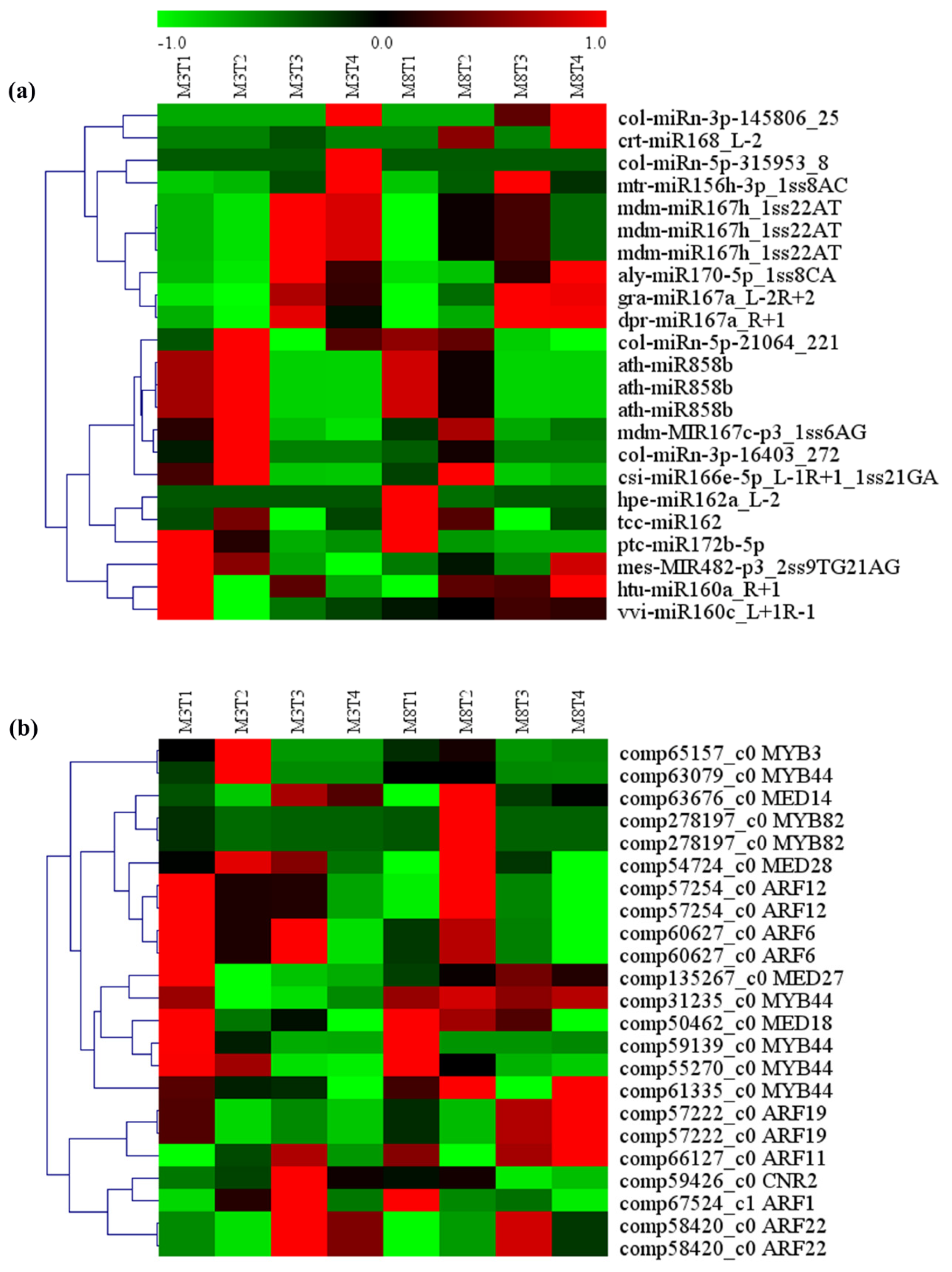
3.6. MiRNA–mRNA Regulation Modules Involved in Seed Size
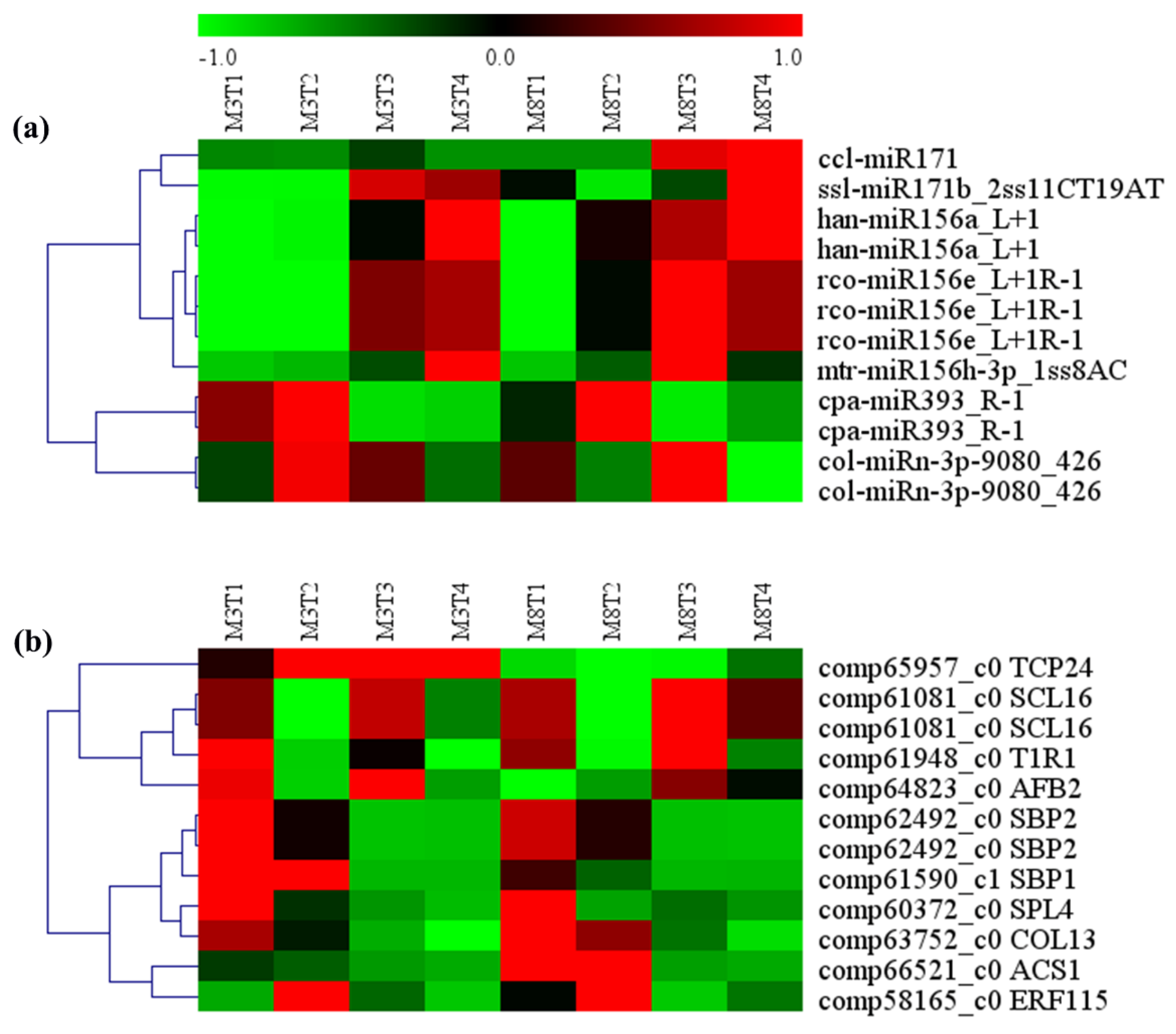
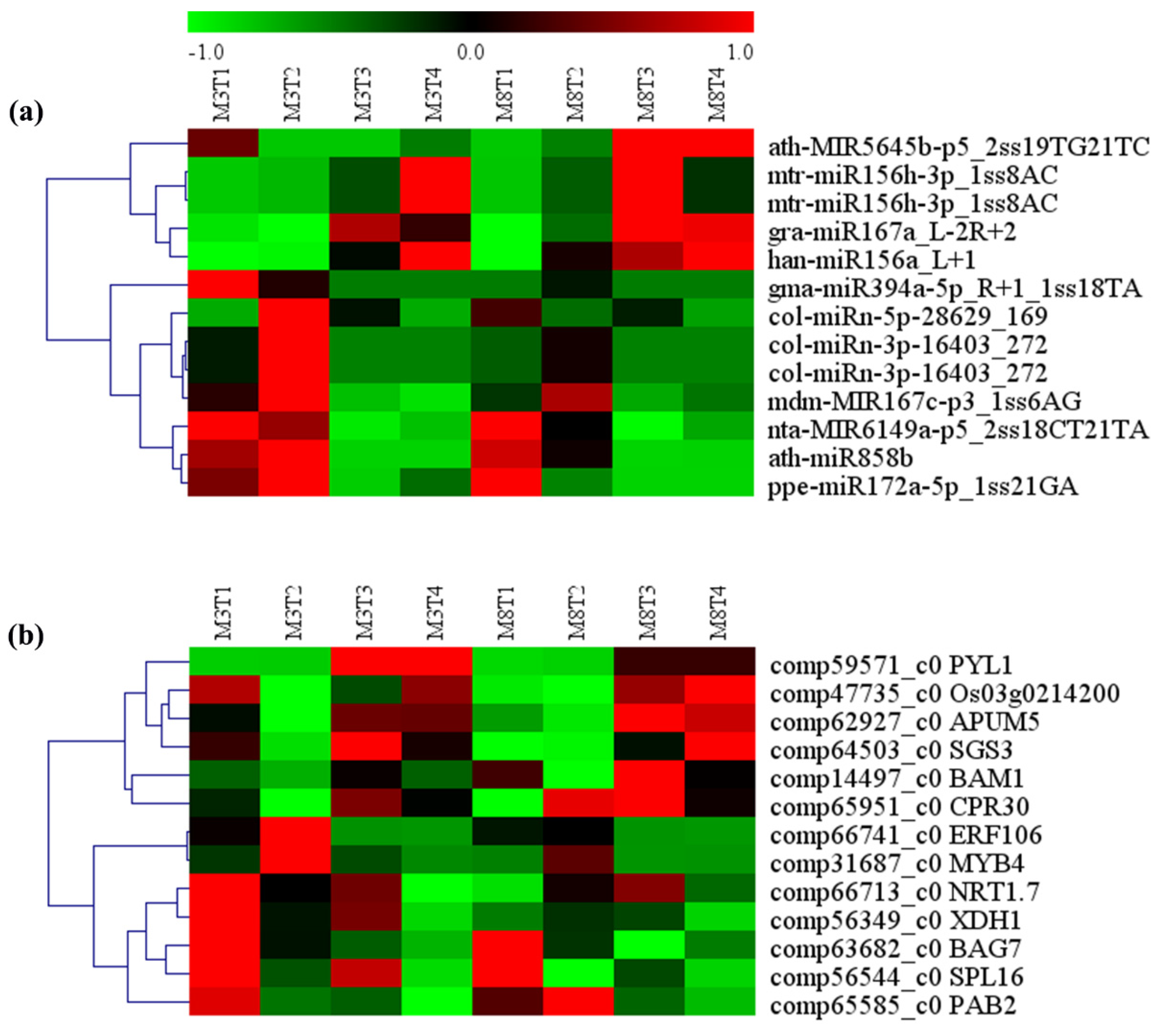
3.7. MiRNA–mRNA Regulatory Module Involved in Growth and Development
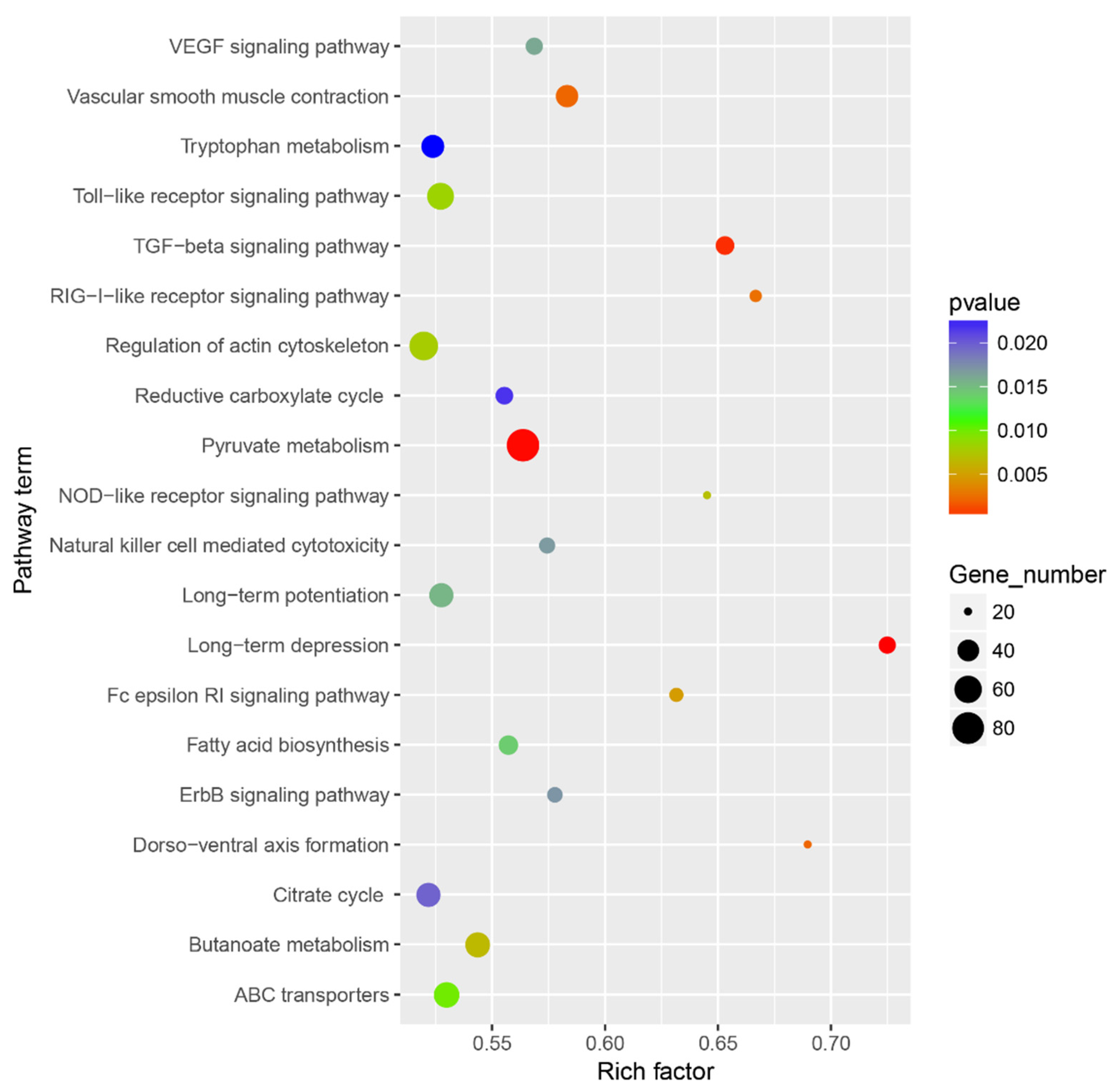
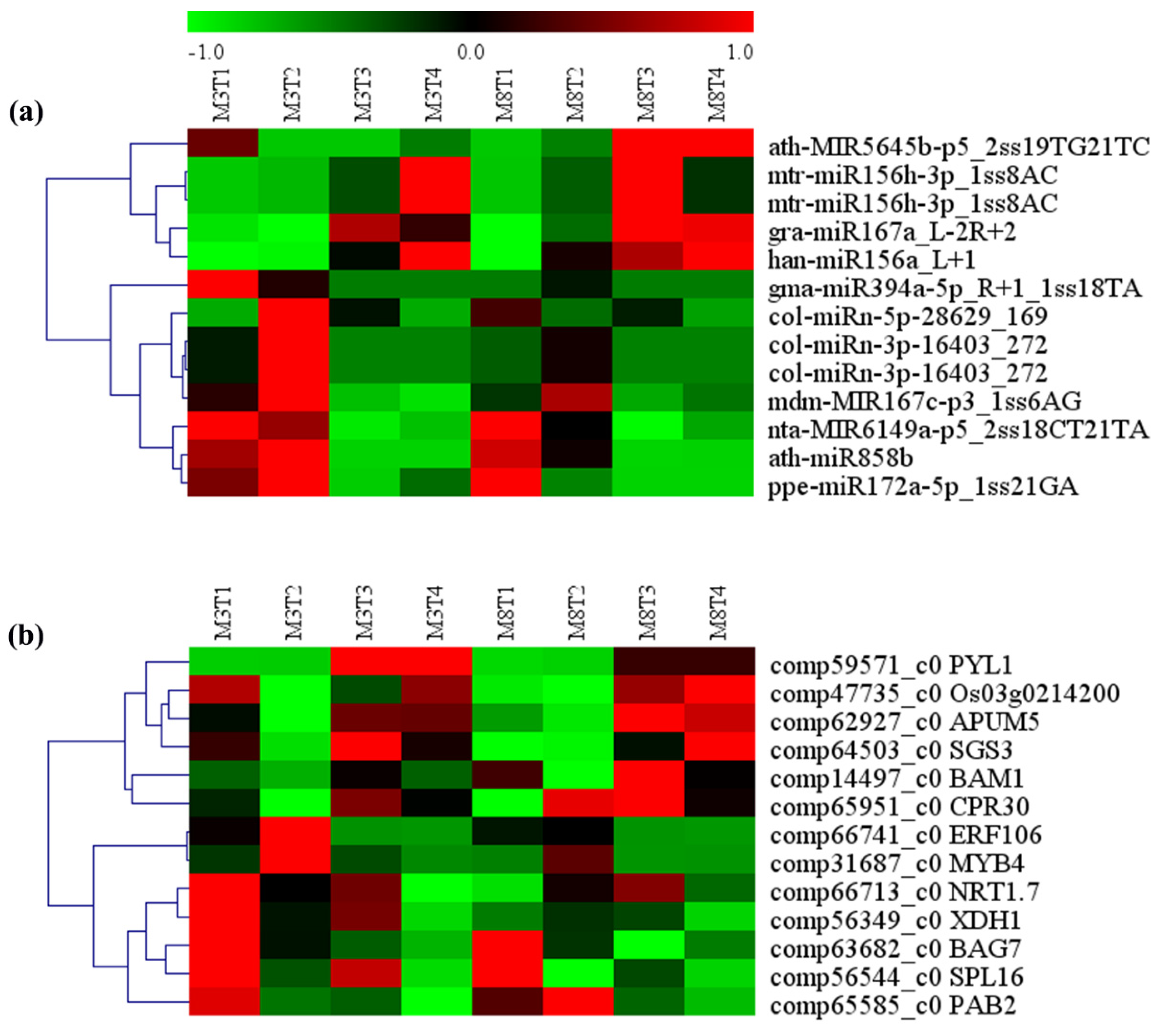
3.8. MiRNA–mRNA Regulatory Modules Involved in Resistance, Yield, and Quality
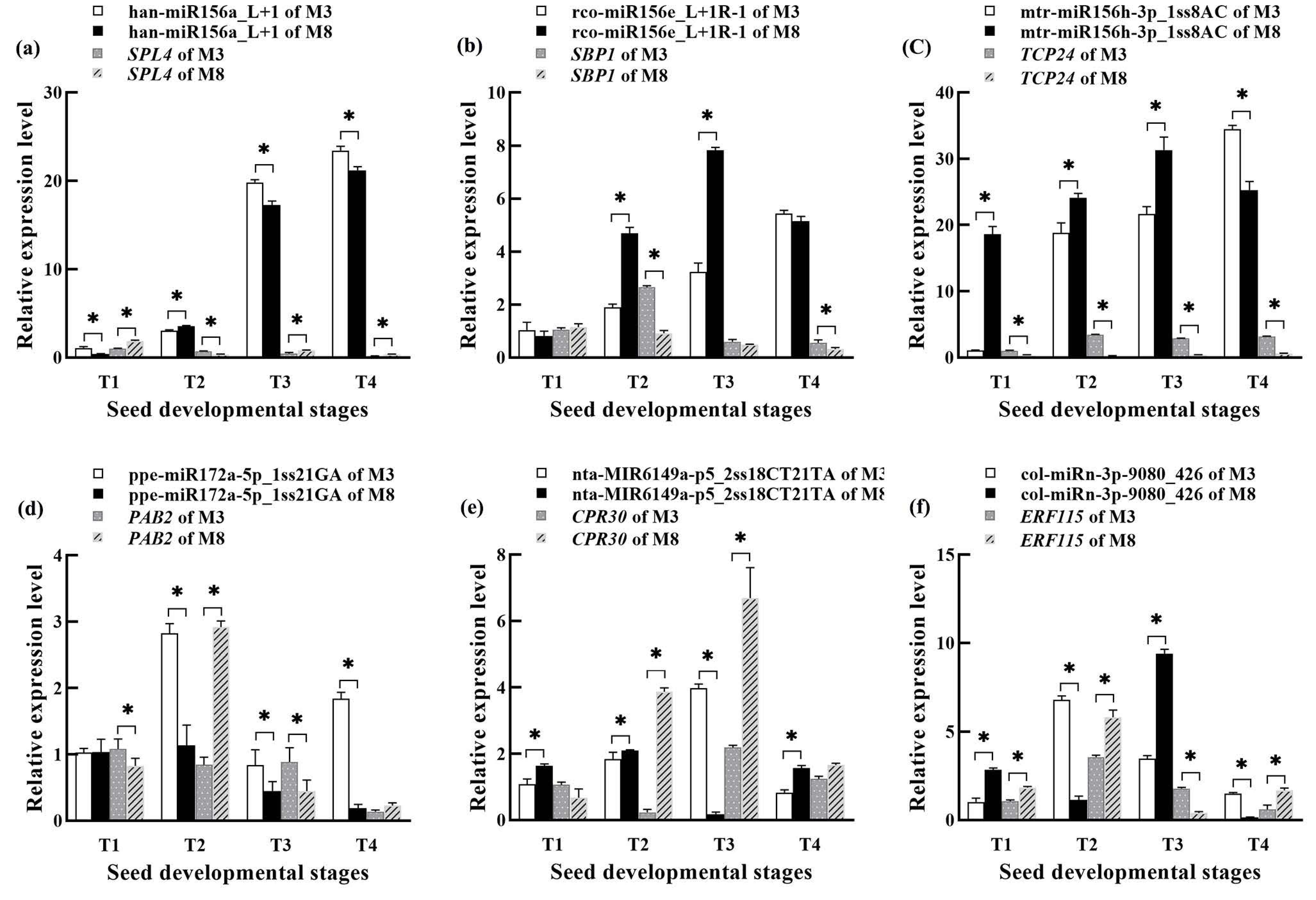
3.9. Validation of Expression Levels of miRNAs and Its Target mRNAs by qRT-PCR
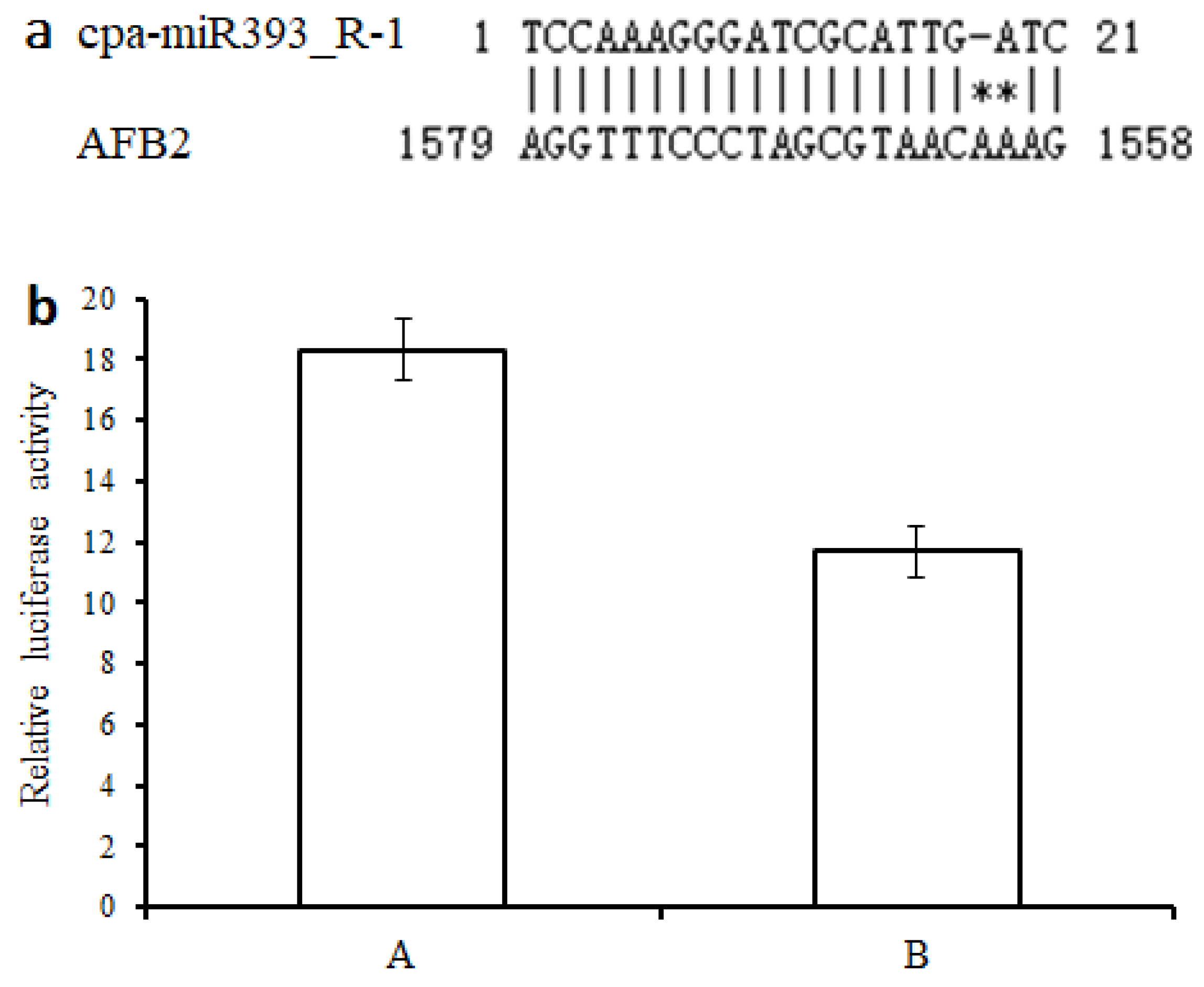
3.10. Validation of Targeting Relationship of the cpa-miR393_R-1-AFB2 Regulatory Module by Luciferase Assays
4. Discussion
4.1. Identification of miRNAs in Developing Seeds of Tea Oil Camellia
4.2. MiRNA–mRNA Modules Involved in Lipid Metabolism in Tea Oil Camellia
4.3. MiRNA–mRNA Regulatory Modules Involved in Seed Size in Tea Oil Camellia
4.4. MiRNA-Target Genes Involved in Growth, Development, and Resistance in Tea Oil Camellia
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ohlrogge, J.B. Design of new plant products: Engineering of fatty acid metabolism. Plant Physiol. 1999, 104, 821–826. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhou, G.Y.; Zhang, H.Y.; Liu, J.A. Research progress on the health function of tea oil. J. Med. Plants Res. 2011, 5, 485–489. [Google Scholar]
- Lee, C.P.; Yen, G.C. Antioxidant activity and bioactive compounds of tea seed (Camellia oleifera Abel.) oil. J. Agric. Food Chem. 2006, 54, 779–784. [Google Scholar] [CrossRef]
- Yu, Y.; Ren, S.; Tan, K. Study on climatic regionalization and layer and belt distribution of oiltea Camellia quality in China. J. Nat. Res. 1999, 14, 123–127. [Google Scholar]
- Chen, J.; Yang, X.; Huang, X.; Duan, S.; Long, C.; Chen, J.; Rong, J. Leaf transcriptome analysis of a subtropical evergreen broadleaf plant, wild oil-tea camellia (Camellia oleifera), revealing candidate genes for cold acclimation. BMC Genom. 2017, 18, 211. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, X.; Liu, J.; Kiba, T.; Woo, J.; Ojo, T.; Hafner, M.; Tuschl, T.; Chua, N.H.; Wang, X.J. Deep sequencing of small RNAs specifically associated with Arabidopsis AGO1 and AGO4 uncovers new AGO functions. Plant J. 2011, 67, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Chen, D.; Wu, X.M.; Huang, D.; Chen, L.L.; Li, L.; Deng, X.X.; Xu, Q. Genome-wide comparison of microRNAs and their targeted transcripts among leaf, flower and fruit of sweet orange. BMC Genom. 2014, 15, 695. [Google Scholar] [CrossRef] [Green Version]
- Yin, D.D.; Li, S.S.; Shu, Q.Y.; Gu, Z.Y.; Wang, L.S. Identification of microRNAs and long non-coding RNAs involved in fatty acid biosynthesis in tree peony seeds. Gene 2018, 666, 72–82. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, G.; Liu, X.; Yu, Z.; Peng, S. Integrated analysis of seed microrna and mrna transcriptome reveals important functional genes and microrna-targets in the process of walnut (Juglans regia) seed oil accumulation. Int. J. Mol. Sci. 2020, 21, 9093. [Google Scholar] [CrossRef]
- Wang, L.; Ruan, C.J.; Bao, A.; Li, H. Small RNA proling for identification of microRNAs involved in regulation of seed development and lipid biosynthesis in yellowhorn. BMC Plant Biol. 2021, 21, 464. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Zhu, Q.H.; Shen, W.; Jiao, X.; Zhao, X.; Wang, M.B.; Liu, L.; Singh, S.P.; Liu, Q. Comparative profiling of microRNA expression in developing seeds of high linoleic and high oleic safflower (Carthamus tinctorius L.) plants. Front. Plant Sci. 2013, 4, 489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, J.; Wang, J.; An, J.; Liu, L.; Lin, Z.; Wang, R.; Wang, L.; Ma, C.; Shi, L.; Lin, S. Integrated mRNA and miRNA transcriptome reveal a cross-talk between developing response and hormone signaling for the seed kernels of Siberian apricot. Sci. Rep. 2016, 6, 35675. [Google Scholar] [CrossRef] [Green Version]
- Mutum, R.D.; Kumar, S.; Balyan, S.; Kansal, S.; Mathur, S.; Raghuvanshi, S. Identification of novel miRNAs from drought tolerant rice variety Nagina 22. Sci. Rep. 2016, 6, 30786. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, S.; Steer, C.J. MicroRNAs as gatekeepers of apoptosis. J. Cell. Physiol. 2010, 223, 289–298. [Google Scholar] [CrossRef]
- Cuperus, J.T.; Fahlgren, N.; Carrington, J.C. Evolution and functional diversification of miRNA genes. Plant Cell 2011, 23, 431–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths-Jones, S.; Saini, H.K.; Dongen, S.V.; Enright, A.J. miRbase: Tools for microRNA genomics. Nucl. Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.X.; Liu, Y.F.; Hu, X.Y.; Zhang, W.K.; Ma, B.; Chen, S.Y.; Zhang, J.S. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 2011, 11, 5. [Google Scholar] [CrossRef] [Green Version]
- Reyes, J.L.; Chua, N.H. ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 2007, 49, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Körbes, A.P.; Machado, R.D.; Guzman, F.; Almerão, M.P.; de Oliveira, L.F.; Loss-Morais, G.; Turchetto-Zolet, A.C.; Cagliari, A.; dos Santos Maraschin, F.; Margis-Pinheiro, M.; et al. Identifying conserved and novel microRNAs in developing seeds of Brassica napus using deep sequencing. PLoS ONE 2012, 7, e50663. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Sun, H.; Qiao, M.; Zhao, Y.; Du, Y. Differentially expressed microRNA cohorts in seed development may contribute to poor grain filling of inferior spikelets in rice. BMC Plant Biol. 2014, 14, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Q.; Xue, Z.; Dong, C.; Wang, Y.; Chu, L.; Xu, Y. Identification and characterization of microRNAs from tree peony (Paeonia ostii) and their response to copper stress. PLoS ONE 2015, 10, e0117584. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, L.; Liu, X.; Cui, D.; Chen, T.; Zhang, H. Deep sequencing of maize small RNAs reveals a diverse set of microRNA in dry and imbibed seeds. PLoS ONE 2013, 8, e55107. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Ruan, C.; Guan, Y. Identification of microRNAs involved in lipid biosynthesis and seed size in developing sea buckthorn seeds using high-throughput sequencing. Sci. Rep. 2018, 8, 4022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belide, S.; Petrie, J.R.; Shrestha, P.; Singh, S.P. Modification of seed oil composition in Arabidopsis by artificial microRNA-mediated gene silencing. Front. Plant Sci. 2012, 3, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, G.N.; Mu, X.; Grabowski, P.; Schmutz, J.; Lu, C. Enhancing microrna167a expression in seed decreases the α-linolenic acid content and increases seed size in Camelina sativa. Plant J. 2019, 98, 346–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, H.; Yang, X.; Zhang, F.; Zheng, X.; Qu, C.; Mu, J.; Fu, F.; Li, J.; Guan, R.; Zhang, H.; et al. Enhanced seed oil production in Canola by conditional expression of Brassica napus LEAFY COTYLEDON1 and LEC1-LIKE in developing seeds. Plant Physiol. 2011, 156, 1577–1588. [Google Scholar] [CrossRef] [Green Version]
- Kwong, R.W.; Bui, A.Q.; Lee, H.; Kwong, L.W.; Fischer, R.L.; Goldberg, R.B.; Harada, J.J. LEAFY COTYLEDON1-LIKE defines a class of regulators essential for embryo development. Plant Cell 2003, 15, 5–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, M.; Bhaskar, P.B.; Sriram, S.; Wang, P.H. Integration of omics approaches to understand oil/protein content during seed development in oilseed crops. Plant Cell Rep. 2016, 36, 637–652. [Google Scholar] [CrossRef]
- Khemka, N.; Rajkumar, M.S.; Garg, R.; Jain, M. Genome-wide profiling of miRNAs during seed development reveals their functional relevance in seed size/weight determination in chickpea. Plant Direct 2021, 5, e00299. [Google Scholar] [CrossRef] [PubMed]
- Parreira, J.R.; Cappuccio, M.; Balestrazzi, A.; Fevereiro, P.; de Sousa Araújo, S. Micrornas expression dynamics reveal post-transcriptional mechanisms regulating seed development in Phaseolus vulgaris L. Hortic. Res. 2021, 8, 18. [Google Scholar] [CrossRef]
- Liu, P.P.; Montgomery, T.A.; Fahlgren, N.; Kasschau, K.D.; Carrington, J.C. Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 2010, 52, 133–146. [Google Scholar] [CrossRef]
- Nodine, M.D.; Bartel, D.P. MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Gene Dev. 2010, 24, 2678–2692. [Google Scholar] [CrossRef] [Green Version]
- Allen, R.S.; Li, J.; Stahle, M.I.; Aurelie, D.; Gubler, F.; Millar, A.A. Genetic analysis reveals functional redundancy and the major target genes of the Arabidopsis miR159 family. Proc. Natl. Acad. Sci. USA 2007, 104, 16371–16376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.B.; Ding, J.; Yu, R.; Li, H.; Ruan, C.J. Identification and expression analysis of critical microrna-transcription factor regulatory modules related to seed development and oil accumulation in developing Hippophae rhamnoides seeds. Ind. Crops Prod. 2019, 137, 33–42. [Google Scholar] [CrossRef]
- Liu, J.; Luo, Q.; Zhang, X.; Zhang, Q.; Cheng, Y. Identification of vital candidate microRNA/mRNA pairs regulating ovule development using high-throughput sequencing in hazel. BMC Dev. Biol. 2020, 20, 13. [Google Scholar] [CrossRef]
- Harikrishna, K.; Othman, R.Y.; Hoon, L.S.; Harikrishna, J.A. Computational prediction of microRNAs from oil palm (Elaeis guineensis Jacq.) expressed sequence tags. Asia-Pac. J. Mol. Biol. Biotechnol. 2007, 15, 107–113. [Google Scholar]
- Donaire, L.L.; Pedrola, L.L.; Rosa, R.D.L.; Liave, C. High-throughput sequencing of RNA silencing-associated small RNAs in olive (Olea europaea L.). PLoS ONE 2011, 6, e27916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Ruan, C.J.; Du, W.; Guan, Y. RNA-seq data reveals a coordinated regulation mechanism of multigenes involved in the high accumulation of palmitoleic acid and oil in sea buckthorn berry pulp. BMC Plant Biol. 2019, 19, 207. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Ruan, C.J.; Ding, J.; Du, W.; Li, J.B.; Yan, R.; Liu, L.R. Coordinated expression of source and sink genes involved in lipid biosynthesis and accumulation during camellia oleifera seed development. Acta Bot. Boreal.-Occident. Sin. 2017, 37, 2483–2488, (In Chinese with an English abstract). [Google Scholar]
- Wu, B.; Ruan, C.J.; Han, P.; Ruan, D.; Xiong, C.W.; Ding, J.; Liu, S.H. Comparative transcriptomic analysis of high- and low-oil Camellia oleifera reveals a coordinated mechanism for the regulation of upstream and downstream multigenes for high oleic acid accumulation. 3 Biotech 2019, 9, 257. [Google Scholar] [CrossRef]
- Han, X.; Yin, H.; Song, X.; Zhang, Y.; Liu, M.; Sang, J.; Jiang, J.; Li, J.; Zhuo, R. Integration of small RNAs, degradome and transcriptome sequencing in hyperaccumulator Sedum alfredii uncovers a complex regulatory network and provides insights into cadmium phytoremediation. Plant Biotechnol. J. 2016, 14, 1470–1483. [Google Scholar] [CrossRef] [Green Version]
- miRBase 20.0. Available online: ftp://mirbase.org/pub/mirbase/CURRENT/ (accessed on 20 July 2015).
- Ambady, S.; Wu, Z.Y.; Dominko, T. Identifification of novel microRNAs in Xenopus laevis Metaphase II arrested eggs. Genesis 2012, 50, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Shahid, M.Q.; Wu, J.W.; Wang, L.; Liu, X.D.; Lu, Y.G. Comparative small RNA analysis of pollen development in autotetraploid and diploid rice. Int. J. Mol. Sci. 2016, 17, 499. [Google Scholar] [CrossRef] [PubMed]
- Cer, R.Z.; Herrera-Galeano, J.E.; Anderson, J.J.; Bishop-Lilly, K.A.; Mokashi, V.P. miRNA Temporal Analyzer (mirnaTA): A bioinformatics tool for identifying differentially expressed microRNAs in temporal studies using normal quantile transformation. GigaScience 2014, 3, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahlgren, N.; Carrington, J.C. miRNA target prediction in plants. Methods Mol. Biol. 2010, 592, 51–57. [Google Scholar] [PubMed]
- Schwab, R.; Palatnik, J.F.; Riester, M.; Schommer, C.; Schmid, M.; Weigel, D. Specific effects of microRNAs on the plant transcriptome. Dev. Cell 2005, 8, 517–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Vidal, E.A.; Araus, V.; Lu, C.; Parry, G.; Green, P.J.; Coruzzi, G.M.; Gutierrez, R.A. Nitrate-responsive miR393/AFB3 regulatory module controls root system architecture in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2010, 107, 4477–4482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakehal, A.; Chaabouni, S.; Cavel, E.; Hir, R.L.; Bellini, C. A molecular framework for the control of adventitious rooting by the TIR1/AFB2-Aux/IAA-Dependent auxin signaling in Arabidopsis. Mol. Plant 2019, 12, 1499–1514. [Google Scholar] [CrossRef]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Agatheeswaran, S.; Pattnayak, N.C.; Chakraborty, S. Identification and functional characterization of the miRNA-gene regulatory network in chronic myeloid leukemia lineage negative cells. Sci. Rep. 2016, 6, 32493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, B.; Jain, P.; Das, S.; Ghosal, S.; Hazra, B.; Trivedi, A.C.; Basu, A.; Chakrabarti, J.; Vrati, S.; Banerjee, J. Dynamic changes in global microRNAome and transcriptome reveal complex miRNA-mRNA regulated host response to Japanese encephalitis Virus in microglial cells. Sci. Rep. 2016, 6, 20263. [Google Scholar] [CrossRef] [Green Version]
- Qian, P.; Jiang, T.; Wang, X.; Song, F.; Shen, X. Bmo-miR-275 down-regulates expression of Bombyx mori sericin gene 2 in vitro. PLoS ONE 2018, 13, e0190464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, 155–159. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, E.; Wuyts, J.; Rouzé, P.; van de Peer, Y. Detection of 91 potential conserved plant microRNAs in Arabidopsis thaliana and Oryza sativa identifies important target genes. Proc. Natl. Acad. Sci. USA 2004, 101, 11511–11516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, C.C.; Sieber, P.; Wellmer, F.; Meyerowitz, E.M. The early extra petals1 mutant uncovers a role for microRNA miR164c in regulating petal number in Arabidopsis. Curr. Biol. 2005, 15, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.J. New human and mouse microRNA genes found by homology search. FEBS J. 2010, 272, 59–73. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, X.; Cannon, C.H.; Cobb, G.P.; Anderson, T.A. Conservation and divergence of plant microRNA genes. Plant J. 2010, 46, 243–259. [Google Scholar] [CrossRef]
- Zhao, Y.T.; Wang, M.; Fu, S.X.; Yang, W.C.; Qi, C.K.; Wang, X.J. Small RNA profiling in two Brassica napus cultivars identifies microRNAs with oil production- and development-correlated expression and new small RNA classes. Plant Physiol. 2012, 158, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.Y.; Cronan, J.E. Isotation and characterization of β-ketoacyl-acyl carrier protein reductase (fabG) mutants of Escherichia coli and Salmonella enterica serovar Tyhimurium. J. Bacteriol. 2004, 186, 1869–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Koh, C.; Feurtado, J.A.; Tsang, E.W.; Cutler, A.J. MicroRNAs and their putative targets in Brassica napus seed maturation. BMC Genom. 2013, 14, 140. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tan, X.; Jiang, J.; Zhang, L. Molecular cloning and expression of two genes encoding accase subunits of Camellia oleifera (Theaceae). Pak. J. Bot. 2018, 50, 103–110. [Google Scholar]
- Andre, C.; Haslam, R.P.; Shanklin, J. Feedback regulation of plastidic acetyl-CoA carboxylase by 18:1-acyl carrier protein in Brassica napus. Proc. Natl. Acad. Sci. USA 2012, 109, 10107–10112. [Google Scholar] [CrossRef] [Green Version]
- Nakkaew, A.; Chotigeat, W.; Eksomtramage, T.; Phongdara, A. Cloning and expression of a plastid-encoded subunit, beta-carboxyltransferase gene (accD) and a nuclear-encoded subunit, biotin carboxylase of acetyl-CoA carboxylase from oil palm (Elaeis guineensis Jacq.). Plant Sci. 2008, 175, 497–504. [Google Scholar] [CrossRef]
- Lee, B.; Shin, S.E.; Choi, G.G.; Jeong, B.; Park, M.S.; Yang, J.W. Effects of Overexpression of GAPDH on Growth and Lipid Accumulation in Chlamydomonas reinhardtii. 2013. Available online: https://koasas.kaist.ac.kr/handle/10203/188091 (accessed on 23 December 2021).
- Nofiani, R.; Philmus, B.; Nindita, Y.; Mahmud, T. 3-ketoacyl-acp synthase (kas) iii homologues and their roles in natural product biosynthesis. MedChemComm 2019, 10, 1517–1530. [Google Scholar] [CrossRef]
- Sang, Y.S.; Sang, H.K.; Velusamy, V.; Lee, Y.M. Comparative gene expression analysis in a highly anthocyanin pigmented mutant of colorless chrysanthemum. Mol. Biol. Rep. 2013, 40, 5177–5189. [Google Scholar] [CrossRef]
- Heath, R.J.; Su, N.; Murphy, C.K.; Rock, C.O. The Enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J. Biol. Chem. 2000, 275, 40128–40133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuno, R.; von Wettstein-Knowles, P.; Wada, H. Identification and molecular characterization of the beta-Ketoacyl-[Acyl Carrier Protein] synthase component of the Arabidopsis mitochondrial fatty acid synthase. J. Biol. Chem. 2004, 279, 8242–8251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.Q.; Tan, X.F.; Hu, F.M. The cDNA cloning and characteristic of stearoyl-ACP desaturase gene of Camellia oleifera. Acta Hortic. 2008, 769, 55–61. [Google Scholar] [CrossRef]
- Kachroo, A.; Shanklin, J.; Whittle, E.; Lapchyk, L.; Hildebrand, D.; Kachroo, P. The Arabidopsis stearoyl-acyl carrier protein-desaturase family and the contribution of leaf isoforms to oleic acid synthesis. Plant Mol. Biol. 2007, 63, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Dörmann, P.; Voelker, T.A.; Ohlrogge, J.B. Accumulation of palmitate in Arabidopsis mediated by the acyl-acyl carrier protein thioesterase FATB1. Plant Physiol. 2000, 123, 637–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Li, Y. Control of final organ size by mediator complex subunit 25 in Arabidopsis thaliana. Development 2011, 22, 4545–4554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Simmons, C.R. Cell number counts—The fw2.2 and CNR genes and implications for controlling plant fruit and organ size. Plant Sci. 2011, 181, 1–7. [Google Scholar] [CrossRef]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdalla, A.H.S. Characterization of a MYB Gene from Tomato (Solanum lycopersicum) Involved in Fruit Development. Master’s Thesis, Universitat Politècnica de València, Valencia, Spain, 2013. [Google Scholar]
- Li, D.; Jin, C.; Duan, S.; Zhu, Y.; Qi, S.; Liu, K.; Gao, C.; Ma, H.; Zhang, M.; Liao, Y.; et al. MYB89 transcription factor represses seed oil accumulation. Plant Physiol. 2017, 173, 1211–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, W.; Mao, Y.; Yang, J.; He, Y. TCP24 modulates secondary cell wall thickening and anther endothecium development. Front. Plant Sci. 2015, 6, 436. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.Y.; Li, C.Q.; Li, J.G.J. Isolation of acs1 gene and the relationship between its expression and fruitlet abscission in litchi. J. Fruit Sci. 2017, 34, 817–827. [Google Scholar]
- Yu, Z.; Ma, X.; Zeng, B.; Wang, J.; Aymt, A. Research progress on sbp1 gene of plant gametophytic self-incompatibility. Mol. Plant Breed. 2019, 17, 5285–5290, (In Chinese with an English abstract). [Google Scholar]
- Freitas, R.L.; Carvalho, C.M.; Fietto, L.G.; Loureiro, M.E.; Almeida, A.M.; Fontes, E. Distinct repressing modules on the distal region of the sbp2 promoter contribute to its vascular tissue-specific expression in different vegetative organs. Plant Mol. Biol. 2007, 65, 603–614. [Google Scholar] [CrossRef]
- Sunkar, R.; Zhu, J.-K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, S.U.; Kim, M.J.; Paek, K.H. Arabidopsis Pumilio protein APUM5 suppresses Cucumber mosaic virus infection via direct binding of viral RNAs. Proc. Natl. Acad. Sci. USA 2013, 110, 779–784. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.M.; Xie, L.L.; Ouyang, X.; Zhang, Y.L.; Dai, Z.Z.; Liu, F. The Structure and Functional Characteristics of Plant BAG Protein Family. Hunan Agric. Sci. 2016, 9, 115–120, (in Chinese with an English abstract). [Google Scholar]
- Gou, M.; Nan, S.; Zheng, J.; Huai, J.; Wang, G. An f-box gene, cpr30, functions as a negative regulator of the defense response in Arabidopsis. Plant J. 2010, 60, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Srivastava, Y.; Sangwan, R.S.; Sangwan, N.S. Mining and functional analysis of ap2/erf gene in. Sci. Rep. 2020, 10, 4877. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J. The Mechanism of MYB4 Gene Involved in the Response of Drought Stress in Plants. Master’s Thesis, Hefei University of Technology, Hefei, China, 2019. [Google Scholar]
- Pan, Z.Y. Mapping of QTLs Controlling Seed Low Temperature Germinability and Fine Mapping and Transcriptome Analysis of qLTGR3-1 in Rice. Master’s Thesis, Huazhong Agricultural University, Wuhan, China, 2018. [Google Scholar]
- Peragine, A.; Yoshikawa, M.; Wu, G.; Albrecht, H.L.; Poethig, R.S. SGS3 and SGS2/SDE1/RDR6 are required for juvenile development and the production of trans-acting siRNAs in Arabidopsis. Genes Dev. 2004, 18, 2368–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Li, S.; Liu, Q. The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat. Genet. 2014, 47, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, W.; Yang, Y. The Arabidopsis NLP7 gene regulates nitrate signaling via NRT1.1–dependent pathway in the presence of ammonium. Sci. Rep. 2018, 8, 1487. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA ID | Gene ID | Gene Name | Gene Annotation | KEGG Pathway |
|---|---|---|---|---|
| tcc-miR162 | comp59939_c1 | ACC1 | acetyl-CoA carboxylase/biotin carboxylase | Pyruvate metabolism |
| csi-miR166e-5p | comp67779_c0 | KAS1 | 3-oxoacyl-[acyl-carrier-protein] synthase I | |
| csi-miR166e-5p | comp67006_c0 | S-ACP-DES6 | acyl-[acyl-carrier-protein]-desaturase | |
| rgl-miR5139_L-1 | comp61049_c0 | KAS3B | 3-oxoacyl-[acyl-carrier-protein] synthase III | |
| mtr-miR156h-3p_1ss8AC | comp63911_c0 | fabG | 3-oxoacyl-[acyl-carrier protein] reductase | |
| cpa-miR164d | comp63026_c1 | Mcat | [acyl-carrier-protein] S-malonyltransferase | Fatty acid biosynthesis metabolic |
| col-miRn-5p-21064_221 | comp67050_c0 | FATB1 | fatty acyl-ACP thioesterase B | |
| aly-miR393a-3p_1ss12TC | comp52017_c0 | MOD1 | enoyl-[acyl-carrier protein] reductase I | |
| ath-miR172a | comp48800_c0 | Δ9D | acyl-[acyl-carrier-protein] desaturase | |
| gma-MIR5368-p3_1ss21CT | comp67185_c0 | GAPN | glyceraldehyde-3-phosphate dehydrogenase (NADP) | Glycolysis/gluconeogenesis |
| miRNA ID | Gene ID | Gene Name | Gene Annotation |
|---|---|---|---|
| vvi-miR160c_L+1R-1 | comp58420_c0 | ARF22 | auxin response factor 22 |
| htu-miR160a_R+1 | comp58420_c0 | ARF22 | auxin response factor 22 |
| hpe-miR162a_L-2 | comp57222_c0 | ARF19 | auxin response factor 19 |
| tcc-miR162 | comp57222_c0 | ARF19 | auxin response factor 19 |
| mdm-miR167h_1ss22AT | comp60627_c0 | ARF6 | auxin response factor 6 |
| dpr-miR167a_R+1 | comp60627_c0 | ARF6 | auxin response factor 6 |
| col-miRn-3p-16403_272 | comp57254_c0 | ARF12 | auxin response factor 12 |
| mdm-miR167h_1ss22AT | comp57254_c0 | ARF12 | auxin response factor 12 |
| aly-miR170-5p_1ss8CA | comp66127_c0 | ARF11 | auxin response factor 11 |
| col-miRn-3p-145806_25 | comp67524_c1 | ARF1 | auxin response factor 1 |
| mtr-miR156h-3p_1ss8AC | comp55270_c0 | MYB44 | Transcription factor MYB44 |
| ptc-miR172b-5p | comp61335_c0 | MYB44 | Transcription factor MYB44 |
| ath-miR858b | comp31235_c0 | MYB44 | Transcription factor MYB44 |
| col-miRn-5p-21064_221 | comp63079_c0 | MYB44 | Transcription factor MYB44 |
| crt-miR168_L-2 | comp59139_c0 | MYB44 | Transcription factor MYB44 |
| csi-miR166e-5p_L-1R+1_1ss21GA | comp278197_c0 | MYB82 | Transcription factor MYB82 |
| ath-miR858b | comp278197_c0 | MYB82 | Transcription factor MYB82 |
| ath-miR858b | comp65157_c0 | MYB3 | Transcription factor MYB3 |
| mdm-miR167h_1ss22AT | comp135267_c0 | MED27 | Mediator of RNA polymerase II transcription subunit 27 |
| gra-miR167a_L-2R+2 | comp54724_c0 | MED28 | Mediator of RNA polymerase II transcription subunit 28 |
| mdm-MIR167c-p3_1ss6AG | comp63676_c0 | MED14 | Mediator of RNA polymerase II transcription subunit 14 |
| mes-MIR482-p3_2ss9TG21AG | comp50462_c0 | MED18 | Mediator of RNA polymerase II transcription subunit 18 |
| col-miRn-5p-315953_8 | comp59426_c0 | CNR2 | Cell number regulator 2 |
| miRNA ID | Gene ID | Gene Name | Gene Annotation |
|---|---|---|---|
| ccl-miR171 | comp61081_c0 | SCL16 | Scarecrow-like protein 6 |
| ssl-miR171b_2ss11CT19AT | comp61081_c0 | SCL16 | Scarecrow-like protein 6 |
| han-miR156a_L+1 | comp60372_c0 | SPL4 | Squamosa promoter-binding-like protein 4 |
| han-miR156a_L+1 | comp62492_c0 | SBP2 | Squamosa promoter-binding protein 2 |
| rco-miR156e_L+1R-1 | comp66521_c0 | ACS1 | 1-aminocyclopropane-1-carboxylate synthase |
| rco-miR156e_L+1R-1 | comp62492_c0 | SBP2 | Squamosa promoter-binding protein 2 |
| rco-miR156e_L+1R-1 | comp61590_c1 | SBP1 | Squamosa promoter-binding protein 1 |
| mtr-miR156h-3p_1ss8AC | comp65957_c0 | TCP24 | Transcription factor TCP24 |
| cpa-miR393_R-1 | comp61948_c0 | T1R1 | transport inhibitor response 1 |
| cpa-miR393_R-1 | comp64823_c0 | AFB2 | Protein AUXIN SIGNALING F-BOX 2 |
| col-miRn-3p-9080_426 | comp63752_c0 | COL13 | Zinc finger protein CONSTANS-LIKE 13 |
| col-miRn-3p-9080_426 | comp58165_c0 | ERF115 | EREBP-like factor |
| miRNA ID | Gene ID | Gene Name | Gene Annotation |
|---|---|---|---|
| col-miRn-3p-16403_272 | comp14497_c0 | BAM1 | leucine-rich repeat receptor-like serine/threonine-protein kinase, BAM1 |
| col-miRn-3p-16403_272 | comp47735_c0 | Os03g0214200 | Ninja-family protein, Os03g0214200 |
| col-miRn-5p-28629_169 | comp66741_c0 | ERF106 | EREBP-like factor |
| ath-miR858b | comp31687_c0 | MYB4 | Myb-related protein, Myb4 |
| ath-MIR5645b-p5_2ss19TG21TC | comp59571_c0 | PYL1 | Abscisic acid receptor, PYL1 |
| mtr-miR156h-3p_1ss8AC | comp63682_c0 | BAG7 | BAG family molecular chaperone regulator 7 |
| mtr-miR156h-3p_1ss8AC | comp66713_c0 | NRT1.7 | proton-dependent oligopeptide transporter, POT family |
| mdm-MIR167c-p3_1ss6AG | comp56349_c0 | XDH1 | xanthine dehydrogenase/oxidas |
| gra-miR167a_L-2R+2 | comp62927_c0 | APUM5 | Pumilio homolog 5 |
| gma-miR394a-5p_R+1_1ss18TA | comp64503_c0 | SGS3 | Protein SUPPRESSOR OF GENE SILENCING 3 |
| nta-MIR6149a-p5_2ss18CT21TA | comp65951_c0 | CPR30 | F-box protein CPR30 |
| han-miR156a_L+1 | comp56544_c0 | SPL16 | Squamosa promoter-binding-like protein 16 |
| ppe-miR172a-5p_1ss21GA | comp65585_c0 | PAB2 | 20S proteasome subunit alpha 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, B.; Ruan, C.; Shah, A.H.; Li, D.; Li, H.; Ding, J.; Li, J.; Du, W. Identification of miRNA–mRNA Regulatory Modules Involved in Lipid Metabolism and Seed Development in a Woody Oil Tree (Camellia oleifera). Cells 2022, 11, 71. https://doi.org/10.3390/cells11010071
Wu B, Ruan C, Shah AH, Li D, Li H, Ding J, Li J, Du W. Identification of miRNA–mRNA Regulatory Modules Involved in Lipid Metabolism and Seed Development in a Woody Oil Tree (Camellia oleifera). Cells. 2022; 11(1):71. https://doi.org/10.3390/cells11010071
Chicago/Turabian StyleWu, Bo, Chengjiang Ruan, Asad Hussain Shah, Denghui Li, He Li, Jian Ding, Jingbin Li, and Wei Du. 2022. "Identification of miRNA–mRNA Regulatory Modules Involved in Lipid Metabolism and Seed Development in a Woody Oil Tree (Camellia oleifera)" Cells 11, no. 1: 71. https://doi.org/10.3390/cells11010071
APA StyleWu, B., Ruan, C., Shah, A. H., Li, D., Li, H., Ding, J., Li, J., & Du, W. (2022). Identification of miRNA–mRNA Regulatory Modules Involved in Lipid Metabolism and Seed Development in a Woody Oil Tree (Camellia oleifera). Cells, 11(1), 71. https://doi.org/10.3390/cells11010071