
Up-Regulation of Fibroblast Growth Factor 23 Gene Expression in UMR106 Osteoblast-like Cells with Reduced Viability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Quantitative Real Time PCR
2.3. Viability Assay (MTT Assay)
2.4. Enzyme Linked Immunosorbent Assay (ELISA)
2.5. Western Blot
2.6. Statistics
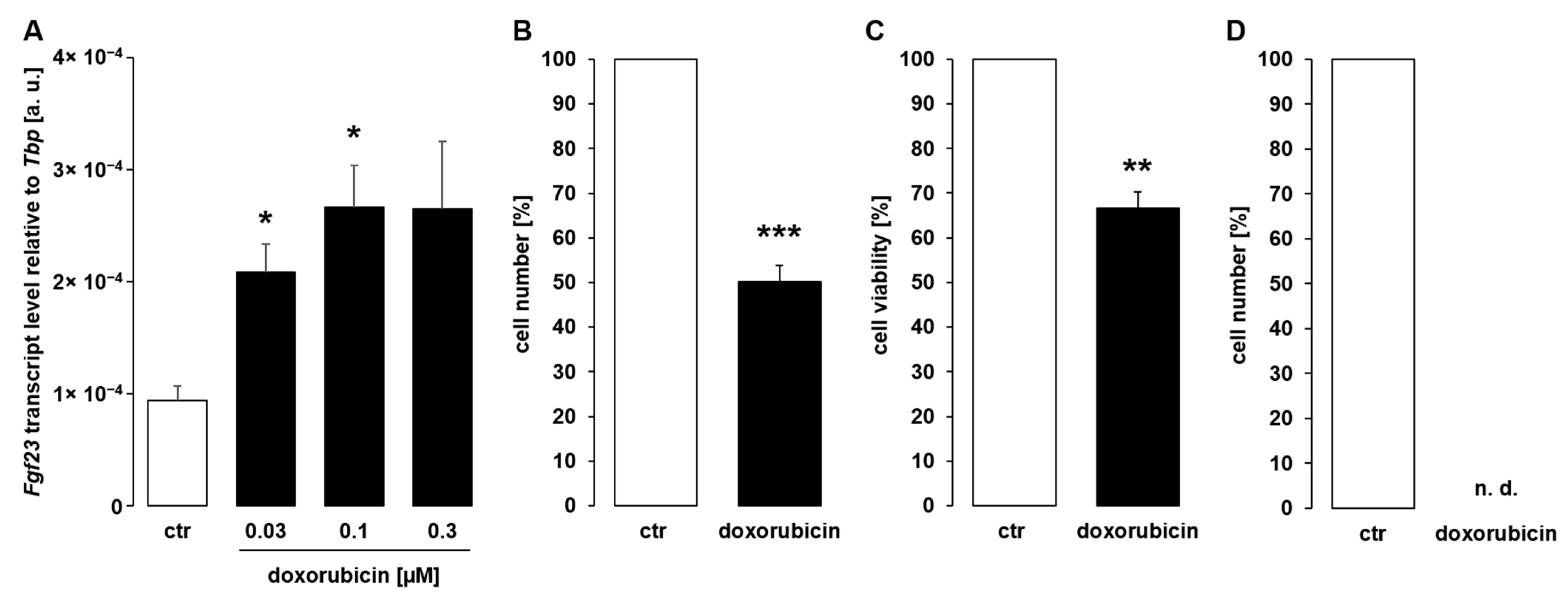
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Han, Y.; You, X.; Xing, W.; Zhang, Z.; Zou, W. Paracrine and endocrine actions of bone—The functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res. 2018, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Leifheit-Nestler, M.; Haffner, D. Paracrine Effects of FGF23 on the Heart. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Moe, O.W. Role of αKlotho and FGF23 in regulation of type II Na-dependent phosphate co-transporters. Pflug. Arch. 2019, 471, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Chanakul, A.; Zhang, M.Y.H.; Louw, A.; Armbrecht, H.J.; Miller, W.L.; Portale, A.A.; Perwad, F. FGF-23 Regulates CYP27B1 Transcription in the Kidney and in Extra-Renal Tissues. PLoS ONE 2013, 8, e72816. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 Is a Potent Regulator of Vitamin D Metabolism and Phosphate Homeostasis. J. Bone Miner. Res. 2004, 19, 429–435. [Google Scholar] [CrossRef]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-o, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shiizaki, K.; Kuro-o, M.; Moe, O.W. Fibroblast Growth Factor 23 and Klotho: Physiology and Pathophysiology of an Endocrine Network of Mineral Metabolism. Annu. Rev. Physiol. 2013, 75, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Mytych, J.; Sołek, P.; Będzińska, A.; Rusinek, K.; Warzybok, A.; Tabęcka-Łonczyńska, A.; Koziorowski, M. Towards Age-Related Anti-Inflammatory Therapy: Klotho Suppresses Activation of ER and Golgi Stress Response in Senescent Monocytes. Cells 2020, 9, 261. [Google Scholar] [CrossRef] [PubMed]
- Rusinek, K.; Sołek, P.; Tabęcka-Łonczyńska, A.; Koziorowski, M.; Mytych, J. Focus on the Role of Klotho Protein in Neuro-Immune Interactions in HT-22 Cells Upon LPS Stimulation. Cells 2020, 9, 1231. [Google Scholar] [CrossRef]
- Imura, A.; Iwano, A.; Tohyama, O.; Tsuji, Y.; Nozaki, K.; Hashimoto, N.; Fujimori, T.; Nabeshima, Y.-I. Secreted Klotho protein in sera and CSF: Implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. 2004, 565, 143–147. [Google Scholar] [CrossRef]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Razzaque, M.S.; Lanske, B. Hypervitaminosis D and premature aging: Lessons learned from Fgf23 and Klotho mutant mice. Trends Mol. Med. 2006, 12, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Wahl, P.; Wolf, M. FGF23 in Chronic Kidney Disease. In Endocrine FGFs and Klothos; Kuro-o, M., Ed.; Springer: New York, NY, USA, 2012; pp. 107–125. ISBN 9781461408864. [Google Scholar]
- Chu, C.; Elitok, S.; Zeng, S.; Xiong, Y.; Hocher, C.-F.; Hasan, A.A.; Krämer, B.K.; Hocher, B. C-terminal and intact FGF23 in kidney transplant recipients and their associations with overall graft survival. BMC Nephrol. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Peng, C.; Huang, W.; Zhang, J.; Xia, M.; Zhang, Y.; Ling, W. Circulating Fibroblast Growth Factor 23 Is Associated with Angiographic Severity and Extent of Coronary Artery Disease. PLoS ONE 2013, 8, e72545. [Google Scholar] [CrossRef]
- Mirza, M.A.I.; Hansen, T.; Johansson, L.; Ahlström, H.; Larsson, A.; Lind, L.; Larsson, T.E. Relationship between circulating FGF23 and total body atherosclerosis in the community. Nephrol. Dial. Transplant. 2009, 24, 3125–3131. [Google Scholar] [CrossRef] [PubMed]
- Di Giuseppe, R.; Kühn, T.; Hirche, F.; Buijsse, B.; Dierkes, J.; Fritsche, A.; Kaaks, R.; Boeing, H.; Stangl, G.I.; Weikert, C. Plasma fibroblast growth factor 23 and risk of cardiovascular disease: Results from the EPIC-Germany case-cohort study. Eur. J. Epidemiol. 2015, 30, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Figurek, A.; Rroji, M.; Spasovski, G. The Complexity of FGF23 Effects on Cardiomyocytes in Normal and Uremic Milieu. Cells 2021, 10, 1266. [Google Scholar] [CrossRef]
- Fitzpatrick, E.A.; Han, X.; Xiao, Z.; Quarles, L.D. Role of Fibroblast Growth Factor-23 in Innate Immune Responses. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T. Fibroblast growth factor 23 and adverse clinical outcomes in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2012, 21, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Vervloet, M.G.; van Ittersum, F.J.; Büttler, R.M.; Heijboer, A.C.; Blankenstein, M.A.; ter Wee, P.M. Effects of dietary phosphate and calcium intake on fibroblast growth factor-23. Clin. J. Am. Soc. Nephrol. 2011, 6, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Meir, T.; Durlacher, K.; Pan, Z.; Amir, G.; Richards, W.G.; Silver, J.; Naveh-Many, T. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014, 86, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, R.; Stockmans, I.; Torrekens, S.; van Looveren, R.; Maes, C.; Carmeliet, P.; Bouillon, R.; Carmeliet, G. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J. Clin. Investig. 2006, 116, 3150–3159. [Google Scholar] [CrossRef]
- Bär, L.; Feger, M.; Fajol, A.; Klotz, L.-O.; Zeng, S.; Lang, F.; Hocher, B.; Föller, M. Insulin suppresses the production of fibroblast growth factor 23 (FGF23). Proc. Natl. Acad. Sci. USA 2018, 115, 5804–5809. [Google Scholar] [CrossRef]
- Daryadel, A.; Bettoni, C.; Haider, T.; Imenez Silva, P.H.; Schnitzbauer, U.; Pastor-Arroyo, E.M.; Wenger, R.H.; Gassmann, M.; Wagner, C.A. Erythropoietin stimulates fibroblast growth factor 23 (FGF23) in mice and men. Pflug. Arch. 2018, 470, 1569–1582. [Google Scholar] [CrossRef]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Durlacher-Betzer, K.; Hassan, A.; Levi, R.; Axelrod, J.; Silver, J.; Naveh-Many, T. Interleukin-6 contributes to the increase in fibroblast growth factor 23 expression in acute and chronic kidney disease. Kidney Int. 2018, 94, 315–325. [Google Scholar] [CrossRef]
- Glosse, P.; Fajol, A.; Hirche, F.; Feger, M.; Voelkl, J.; Lang, F.; Stangl, G.I.; Föller, M. A high-fat diet stimulates fibroblast growth factor 23 formation in mice through TNFα upregulation. Nutr. Diabetes 2018, 8. [Google Scholar] [CrossRef]
- Zhang, B.; Yan, J.; Umbach, A.T.; Fakhri, H.; Fajol, A.; Schmidt, S.; Salker, M.S.; Chen, H.; Alexander, D.; Spichtig, D.; et al. NFκB-sensitive Orai1 expression in the regulation of FGF23 release. J. Mol. Med. 2016, 94, 557–566. [Google Scholar] [CrossRef]
- Bold, R.J.; Termuhlen, P.M.; McConkey, D.J. Apoptosis, cancer and cancer therapy. Surg. Oncol. 1997, 6, 133–142. [Google Scholar] [CrossRef]
- Makin, G.; Hickman, J.A. Apoptosis and cancer chemotherapy. Cell Tissue Res. 2000, 301, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Kemp, C.J.; Henikoff, S. Anthracyclines induce double-strand DNA breaks at active gene promoters. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2015, 773, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Peterson, Q.P.; Goode, D.R.; West, D.C.; Ramsey, K.N.; Lee, J.J.Y.; Hergenrother, P.J. PAC-1 activates procaspase-3 in vitro through relief of zinc-mediated inhibition. J. Mol. Biol. 2009, 388, 144–158. [Google Scholar] [CrossRef]
- Higuchi, A.; Shimmura, S.; Takeuchi, T.; Suematsu, M.; Tsubota, K. Elucidation of apoptosis induced by serum deprivation in cultured conjunctival epithelial cells. Br. J. Ophthalmol. 2006, 90, 760–764. [Google Scholar] [CrossRef]
- Vyas, D.; Laput, G.; Vyas, A.K. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. OncoTargets Ther. 2014, 7, 1015–1023. [Google Scholar] [CrossRef]
- Ludwig, T.; Riethmüller, C.; Gekle, M.; Schwerdt, G.; Oberleithner, H. Nephrotoxicity of platinum complexes is related to basolateral organic cation transport. Kidney Int. 2004, 66, 196–202. [Google Scholar] [CrossRef]
- Volkova, M.; Russell, R. Anthracycline cardiotoxicity: Prevalence, pathogenesis and treatment. Curr. Cardiol. Rev. 2011, 7, 214–220. [Google Scholar] [CrossRef]
- Saini, R.K.; Kaneko, I.; Jurutka, P.W.; Forster, R.; Hsieh, A.; Hsieh, J.-C.; Haussler, M.R.; Whitfield, G.K. 1,25-dihydroxyvitamin D(3) regulation of fibroblast growth factor-23 expression in bone cells: Evidence for primary and secondary mechanisms modulated by leptin and interleukin-6. Calcif. Tissue Int. 2013, 92, 339–353. [Google Scholar] [CrossRef]
- González-Bermúdez, L.; Anglada, T.; Genescà, A.; Martín, M.; Terradas, M. Identification of reference genes for RT-qPCR data normalisation in aging studies. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Abuna, R.P.F.; Oliveira, F.S.; Ramos, J.I.R.; Lopes, H.B.; Freitas, G.P.; Souza, A.T.P.; Beloti, M.M.; Rosa, A.L. Selection of reference genes for quantitative real-time polymerase chain reaction studies in rat osteoblasts. J. Cell. Physiol. 2018, 234, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Bär, L.; Hase, P.; Föller, M. PKC regulates the production of fibroblast growth factor 23 (FGF23). PLoS ONE 2019, 14, e0211309. [Google Scholar] [CrossRef] [PubMed]
- Oflazoglu, U.; Alacacioglu, A.; Varol, U.; Kucukzeybek, Y.; Salman, T.; Onal, H.T.; Yilmaz, H.E.; Yildiz, Y.; Taskaynatan, H.; Saray, S.; et al. The role of inflammation in adjuvant chemotherapy-induced sarcopenia (Izmir Oncology Group (IZOG) study). Support Care Cancer 2020, 28, 3965–3977. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F.; Wacker, M.J. FGF23 production by osteocytes. Pediatr. Nephrol. 2013, 28, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Gao, M.; Wu, L.; Zhao, X.; Mao, H.; Xing, C. The suppressive effect of soluble Klotho on fibroblastic growth factor 23 synthesis in UMR-106 osteoblast-like cells. Cell Biol. Int. 2018, 42, 1270–1274. [Google Scholar] [CrossRef]
- Vidal, A.; Rios, R.; Pineda, C.; Lopez, I.; Muñoz-Castañeda, J.R.; Rodriguez, M.; Aguilera-Tejero, E.; Raya, A.I. Direct regulation of fibroblast growth factor 23 by energy intake through mTOR. Sci. Rep. 2020, 10, 1795. [Google Scholar] [CrossRef]
- Samadfam, R.; Richard, C.; Nguyen-Yamamoto, L.; Bolivar, I.; Goltzman, D. Bone formation regulates circulating concentrations of fibroblast growth factor 23. Endocrinology 2009, 150, 4835–4845. [Google Scholar] [CrossRef][Green Version]
- Takashi, Y.; Kosako, H.; Sawatsubashi, S.; Kinoshita, Y.; Ito, N.; Tsoumpra, M.K.; Nangaku, M.; Abe, M.; Matsuhisa, M.; Kato, S.; et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc. Natl. Acad. Sci. USA 2019, 116, 11418–11427. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Wang, C.-W.; Chen, C.-L.; Wang, C.-K.; Chang, Y.-J.; Jian, J.-Y.; Lin, C.-S.; Tai, C.-J.; Tai, C.-J. Cisplatin-, Doxorubicin-, and Docetaxel-Induced Cell Death Promoted by the Aqueous Extract of Solanum nigrum in Human Ovarian Carcinoma Cells. Integr. Cancer Ther. 2015, 14, 546–555. [Google Scholar] [CrossRef]
- Medici, D.; Razzaque, M.S.; Deluca, S.; Rector, T.L.; Hou, B.; Kang, K.; Goetz, R.; Mohammadi, M.; Kuro-o, M.; Olsen, B.R.; et al. FGF-23-Klotho signaling stimulates proliferation and prevents vitamin D-induced apoptosis. J. Cell Biol. 2008, 182, 459–465. [Google Scholar] [CrossRef]
- Andrukhova, O.; Zeitz, U.; Goetz, R.; Mohammadi, M.; Lanske, B.; Erben, R.G. FGF23 acts directly on renal proximal tubules to induce phosphaturia through activation of the ERK1/2-SGK1 signaling pathway. Bone 2012, 51, 621–628. [Google Scholar] [CrossRef]
- Bai, J.-A.; Xu, G.-F.; Yan, L.-J.; Zeng, W.-W.; Ji, Q.-Q.; Wu, J.-D.; Tang, Q.-Y. SGK1 inhibits cellular apoptosis and promotes proliferation via the MEK/ERK/p53 pathway in colitis. World J. Gastroenterol. 2015, 21, 6180–6193. [Google Scholar] [CrossRef]
- Chang, H.-M.; Peng, K.-Y.; Chan, C.-K.; Sun, C.-Y.; Chen, Y.-Y.; Chang, H.-M.; Huang, C.-L.; Liu, P.-C.; Chen, P.-Y.; Wang, K.-C.; et al. FGF23 ameliorates ischemia-reperfusion induced acute kidney injury via modulation of endothelial progenitor cells: Targeting SDF-1/CXCR4 signaling. Cell Death Dis. 2021, 12, 409. [Google Scholar] [CrossRef]
- Feng, S.; Wang, J.; Zhang, Y.; Creighton, C.J.; Ittmann, M. FGF23 promotes prostate cancer progression. Oncotarget 2015, 6, 17291–17301. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Kim, J.S.; Lee, J.H.; Yoon, W.J.; Lee, D.S.; Ko, M.S.; Kwon, B.S.; Choi, D.H.; Cho, H.R.; Lee, B.J.; et al. NF-kappaB activation is required for cisplatin-induced apoptosis in head and neck squamous carcinoma cells. FEBS Lett. 2006, 580, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Huang, L.; Su, X.-L.; Gu, Q.-H.; Hu, C.-P. Inhibition of nuclear factor-κB activity enhanced chemosensitivity to cisplatin in human lung adeno-carcinoma A549 cells under chemical hypoxia conditions. Chin. Med. J. 2013, 126, 3276–3282. [Google Scholar] [PubMed]
- Ozkok, A.; Ravichandran, K.; Wang, Q.; Ljubanovic, D.; Edelstein, C.L. NF-κB transcriptional inhibition ameliorates cisplatin-induced acute kidney injury (AKI). Toxicol. Lett. 2016, 240, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Esparza-López, J.; Medina-Franco, H.; Escobar-Arriaga, E.; León-Rodríguez, E.; Zentella-Dehesa, A.; Ibarra-Sánchez, M.J. Doxorubicin induces atypical NF-κB activation through c-Abl kinase activity in breast cancer cells. J. Cancer Res. Clin. Oncol. 2013, 139, 1625–1635. [Google Scholar] [CrossRef]
- Wang, S.; Kotamraju, S.; Konorev, E.; Kalivendi, S.; Joseph, J.; Kalyanaraman, B. Activation of nuclear factor-kappaB during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: The role of hydrogen peroxide. Biochem. J. 2002, 367, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Mason, E.F.; Rathmell, J.C. Cell metabolism: An essential link between cell growth and apoptosis. Biochim. Biophys. Acta 2011, 1813, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Wallach, D.; Kovalenko, A. Keeping inflammation at bay. eLife 2014, 3, 645. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Münz, S.; Feger, M.; Edemir, B.; Föller, M. Up-Regulation of Fibroblast Growth Factor 23 Gene Expression in UMR106 Osteoblast-like Cells with Reduced Viability. Cells 2022, 11, 40. https://doi.org/10.3390/cells11010040
Münz S, Feger M, Edemir B, Föller M. Up-Regulation of Fibroblast Growth Factor 23 Gene Expression in UMR106 Osteoblast-like Cells with Reduced Viability. Cells. 2022; 11(1):40. https://doi.org/10.3390/cells11010040
Chicago/Turabian StyleMünz, Sina, Martina Feger, Bayram Edemir, and Michael Föller. 2022. "Up-Regulation of Fibroblast Growth Factor 23 Gene Expression in UMR106 Osteoblast-like Cells with Reduced Viability" Cells 11, no. 1: 40. https://doi.org/10.3390/cells11010040
APA StyleMünz, S., Feger, M., Edemir, B., & Föller, M. (2022). Up-Regulation of Fibroblast Growth Factor 23 Gene Expression in UMR106 Osteoblast-like Cells with Reduced Viability. Cells, 11(1), 40. https://doi.org/10.3390/cells11010040