
Mitophagy: Molecular Mechanisms, New Concepts on Parkin Activation and the Emerging Role of AMPK/ULK1 Axis


Abstract
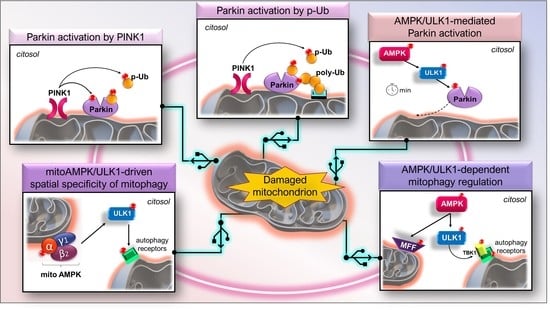
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Regulation of Mitophagy
2.1. Chemical and Natural Mitophagy Triggers
2.2. PINK1–Parkin Axis
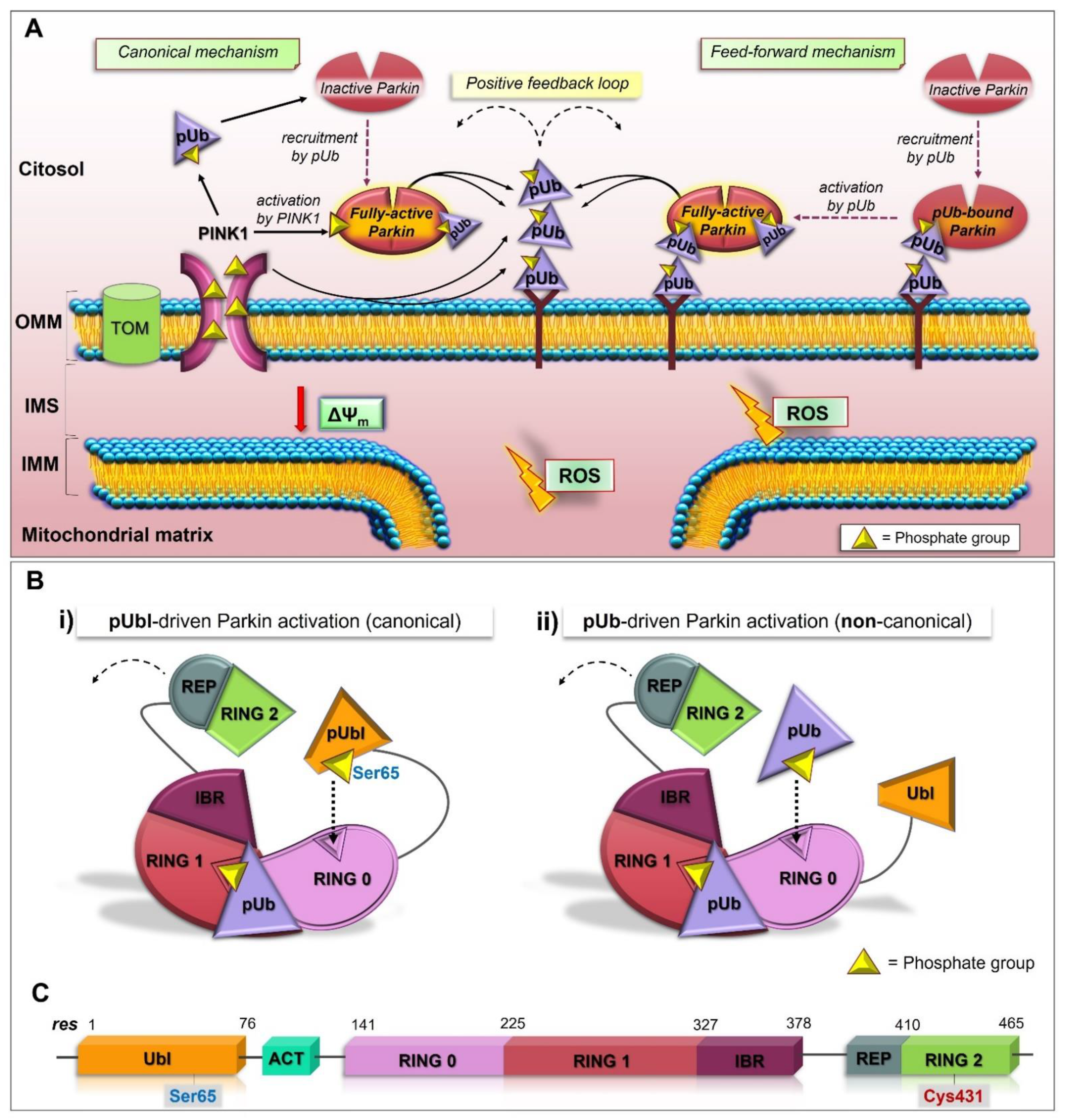
2.3. Structure and Activation of Parkin
2.4. The Feed-Forward Mechanism of Parkin Activation
2.5. Molecular Links between PINK1/Parkin-Mediated Mitophagy and Mitochondrial Dynamics
2.6. Deubiquitinating Enzymes and PTEN-L as Regulators of Mitophagy
2.7. pUbl-Independent Mechanism of Parkin Activation, the Unexpected Plasticity of RING0 Binding Site
2.8. PINK1 Processing and Stabilization
3. Functions of Mitophagy Receptors
- (a)
- BNIP3 and NIX
- (b)
- FUNDC1
- (c)
- BCL2L13, FKBP8, AMBRA-1
3.1. Other Promoters of Mitophagy: Cardiolipin and Novel E3 Ligases
3.2. Autophagy Adaptors Linking to PINK1–Parkin-Mediated Mitophagy
3.3. The Linear Sequence of Mitophagosome Synthesis
3.4. Mitophagosome Synthesis: Redundancy and Positive Feedback Signals, and the Role of mTORC1
3.5. Autophagosome Elongation and Closure
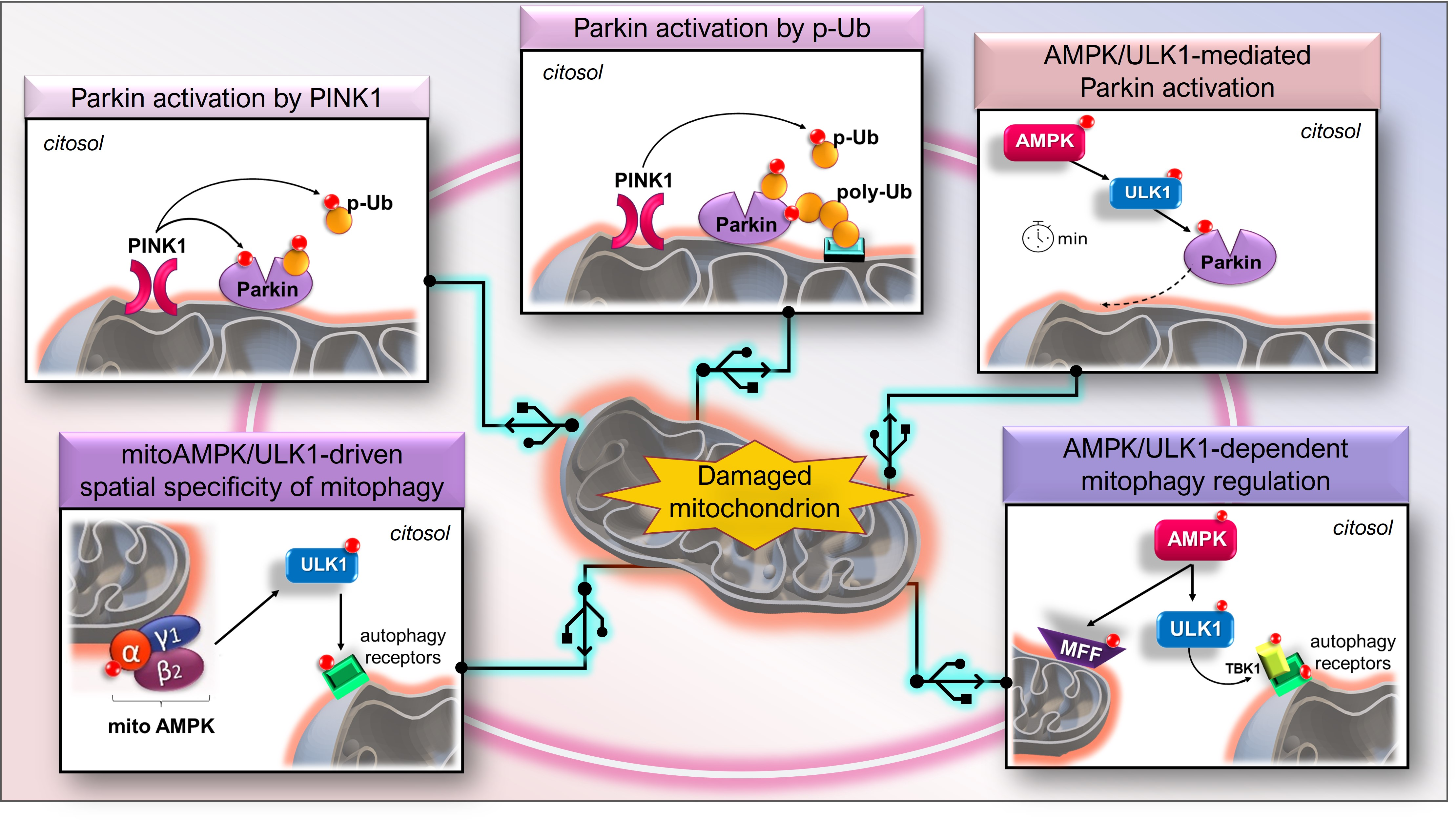
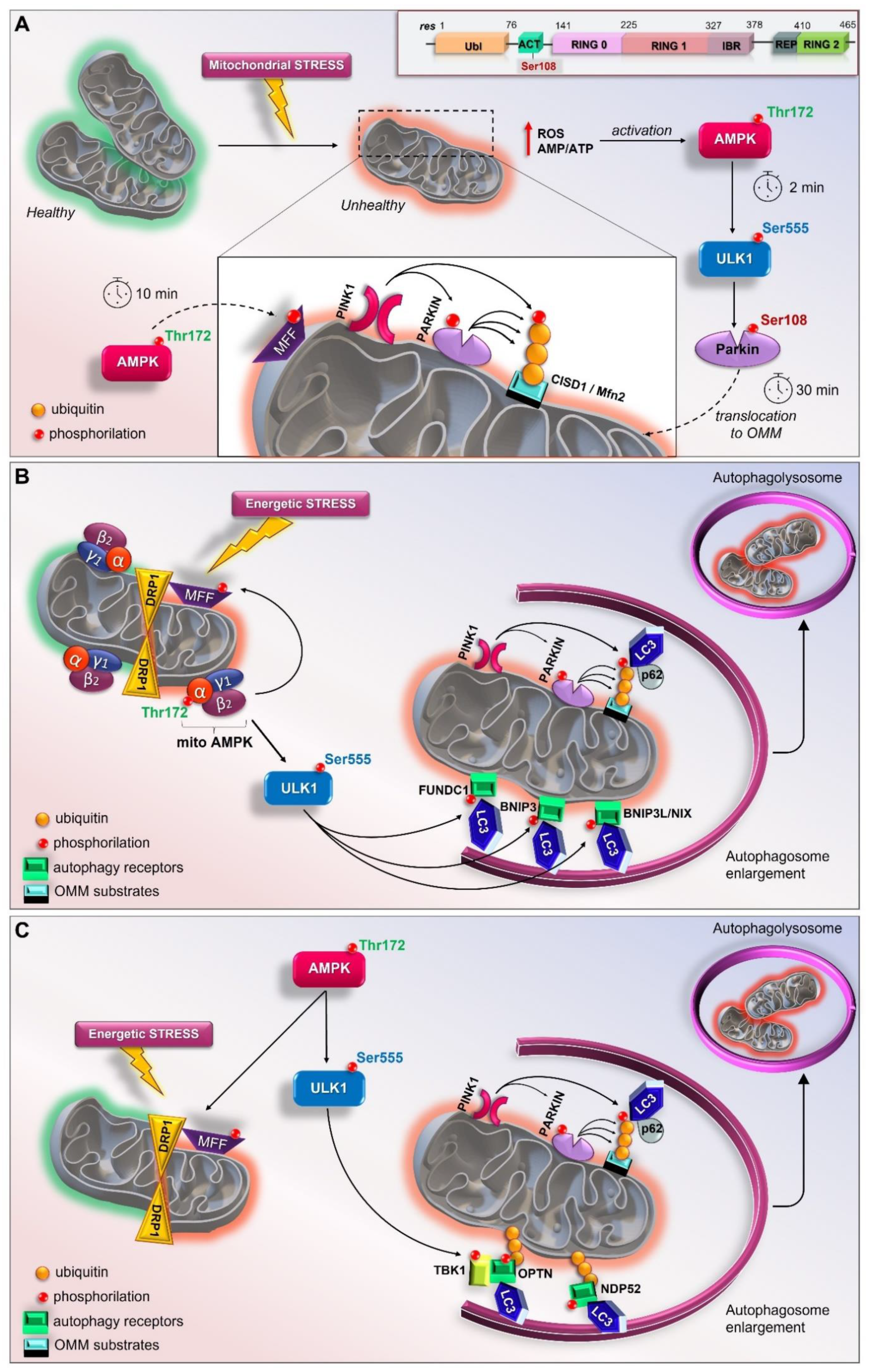
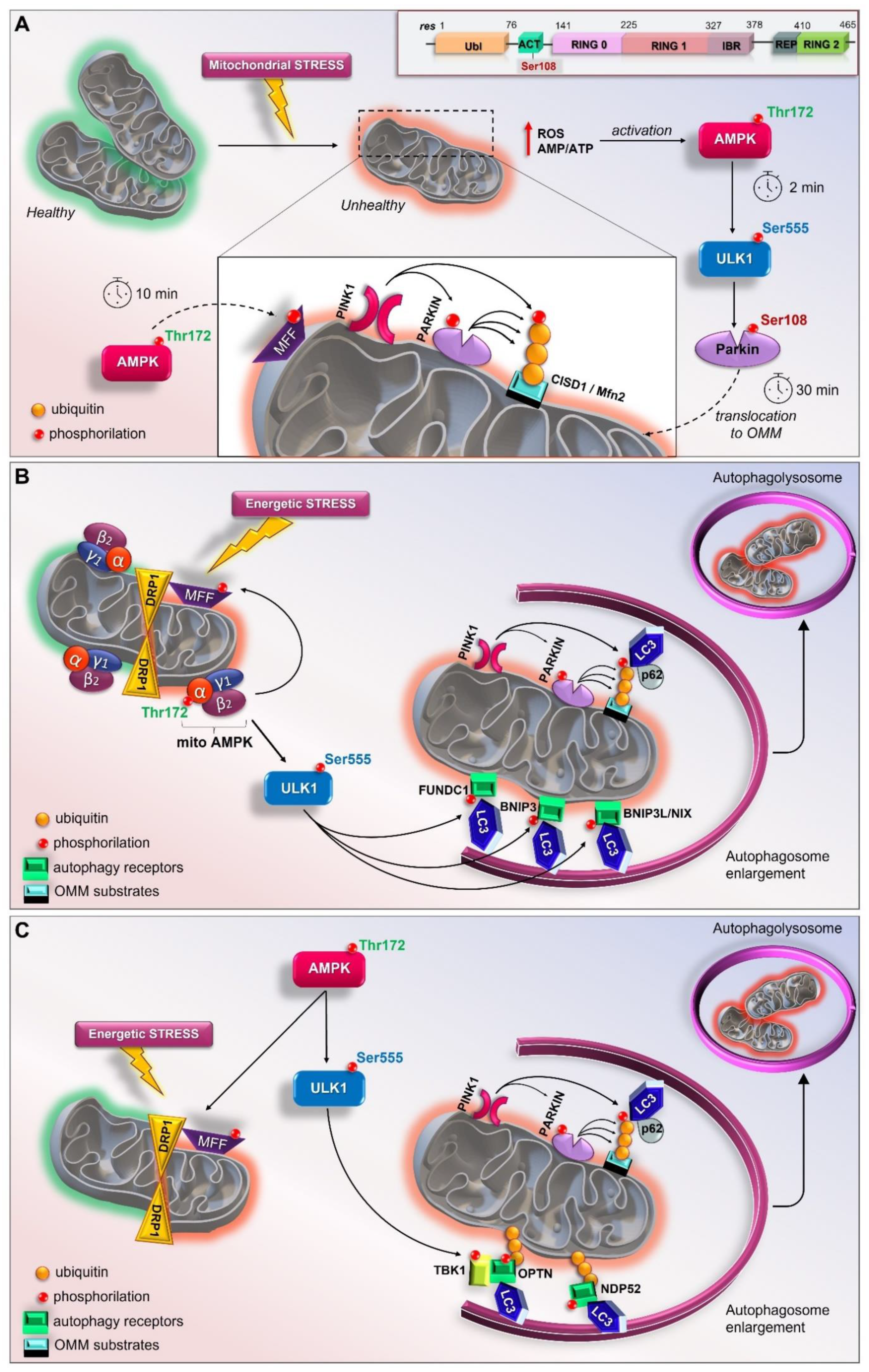
3.6. AMPK/ULK1 Axis in Mitophagy Cascade
3.7. The Early Role of the AMPK/ULK1 Axis in Triggering the Rapid Activation of Parkin
3.8. Other Scenarios of AMPK- and ULK1-Mediated Mitophagy
3.9. The Mitochondrial Pool of AMPK (mitoAMPK) Governs the Spatial Specificity of Energetic Stress-Induced Mitophagy
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Glancy, B.; Hartnell, L.M.; Combs, C.A.; Femnou, A.; Sun, J.; Murphy, E.; Subramaniam, S.; Balaban, R.S. Power Grid Protection of the Muscle Mitochondrial Reticulum. Cell Rep. 2017, 19, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum–Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Lackner, L.L. The Expanding and Unexpected Functions of Mitochondria Contact Sites. Trends Cell Biol. 2019, 29, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.-W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic Mapping of Mitochondria in Living Cells via Spatially Restricted Enzymatic Tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, H.-S.; Dighe, P.A.; Mezera, V.; Monternier, P.-A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Lu, Y.; Saredy, J.; Wang, X.; Drummer IV, C.; Shao, Y.; Saaoud, F.; Xu, K.; Liu, M.; Yang, W.Y.; et al. ROS systems are a new integrated network for sensing homeostasis and alarming stresses in organelle metabolic processes. Redox Biol. 2020, 37, 101696. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M.; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2015, 25, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Bohovych, I.; Chan, S.S.L.; Khalimonchuk, O. Mitochondrial Protein Quality Control: The Mechanisms Guarding Mitochondrial Health. Antioxid. Redox Signal. 2015, 22, 977–994. [Google Scholar] [CrossRef] [Green Version]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisner, V.; Picard, M.; Hajnóczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef]
- Petricca, S.; Flati, V.; Celenza, G.; Di Gregorio, J.; Lizzi, A.R.; Luzi, C.; Cristiano, L.; Cinque, B.; Rossi, G.; Festuccia, C.; et al. Tebuconazole and Econazole Act Synergistically in Mediating Mitochondrial Stress, Energy Imbalance, and Sequential Activation of Autophagy and Apoptosis in Mouse Sertoli TM4 Cells: Possible Role of AMPK/ULK1 Axis. Toxicol. Sci. 2019, 169, 209–223. [Google Scholar] [CrossRef]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.-F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [Green Version]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; López, B.; González, A.; Ravassa, S.; Díez, J.; et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef]
- Quiles, J.M.; Gustafsson, Å.B. Mitochondrial Quality Control and Cellular Proteostasis: Two Sides of the Same Coin. Front. Physiol. 2020, 11, 515. [Google Scholar] [CrossRef] [PubMed]
- Münch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Carvajal, F.; Sanhueza, M. The Mitochondrial Unfolded Protein Response: A Hinge Between Healthy and Pathological Aging. Front. Aging Neurosci. 2020, 12, 300. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.D.; Harley, M.E.; Kemp, A.J.; Wills, J.; Lee, M.; Arends, M.; von Kriegsheim, A.; Behrends, C.; Wilkinson, S. CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev. Cell 2018, 44, 217–232.e11. [Google Scholar] [CrossRef] [PubMed]
- Ravenhill, B.J.; Boyle, K.B.; von Muhlinen, N.; Ellison, C.J.; Masson, G.R.; Otten, E.G.; Foeglein, A.; Williams, R.; Randow, F. The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol. Cell 2019, 74, 320–329.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turco, E.; Witt, M.; Abert, C.; Bock-Bierbaum, T.; Su, M.-Y.; Trapannone, R.; Sztacho, M.; Danieli, A.; Shi, X.; Zaffagnini, G.; et al. FIP200 Claw Domain Binding to p62 Promotes Autophagosome Formation at Ubiquitin Condensates. Mol. Cell 2019, 74, 330–346.e11. [Google Scholar] [CrossRef] [Green Version]
- Le Guerroué, F.; Eck, F.; Jung, J.; Starzetz, T.; Mittelbronn, M.; Kaulich, M.; Behrends, C. Autophagosomal Content Profiling Reveals an LC3C-Dependent Piecemeal Mitophagy Pathway. Mol. Cell 2017, 68, 786–796.e6. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.G.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.K.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef]
- Sun, N.; Malide, D.; Liu, J.; Rovira, I.I.; Combs, C.A.; Finkel, T. A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nat. Protoc. 2017, 12, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Sanchez-Martinez, A.; Zarate, A.M.; Benincá, C.; Mayor, U.; Clague, M.J.; Whitworth, A.J. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J. Cell Biol. 2018, 217, 1613–1622. [Google Scholar] [CrossRef] [Green Version]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.K.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449.e5. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban-Martínez, L.; Sierra-Filardi, E.; McGreal, R.S.; Salazar-Roa, M.; Mariño, G.; Seco, E.; Durand, S.; Enot, D.; Graña, O.; Malumbres, M.; et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 2017, 36, 1688–1706. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Martínez, L.; Boya, P. BNIP3L/NIX-dependent mitophagy regulates cell differentiation via metabolic reprogramming. Autophagy 2018, 14, 915–917. [Google Scholar] [CrossRef] [PubMed]
- Ney, P.A. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta—Mol. Cell Res. 2015, 1853, 2775–2783. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Ma, Q.; Siraj, S.; Ney, P.A.; Liu, J.; Liao, X.; Yuan, Y.; Li, W.; Liu, L.; Chen, Q. Nix-mediated mitophagy regulates platelet activation and life span. Blood Adv. 2019, 3, 2342–2354. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Sato, M. Multiple ways to prevent transmission of paternal mitochondrial DNA for maternal inheritance in animals. J. Biochem. 2017, 162, 247–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, J.C.; Laker, R.C.; Wilson, R.J.; Zhang, M.; Yan, Z. Exercise-induced mitophagy in skeletal muscle occurs in the absence of stabilization of Pink1 on mitochondria. Cell Cycle 2019, 18, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Nah, J.; Oka, S.; Mukai, R.; Monden, Y.; Maejima, Y.; Ikeda, Y.; Sciarretta, S.; Liu, T.; Li, H.; et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J. Clin. Investig. 2019, 129, 802–819. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef]
- Yamaguchi, O.; Murakawa, T.; Nishida, K.; Otsu, K. Receptor-mediated mitophagy. J. Mol. Cell. Cardiol. 2016, 95, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Chen, C.; Wang, J.; Liu, L.; He, Y.; Chen, Q. Mitophagy in Cardiomyocytes and in Platelets: A Major Mechanism of Cardioprotection Against Ischemia/Reperfusion Injury. Physiology 2018, 33, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, Q. Hypoxia Activation of Mitophagy and Its Role in Disease Pathogenesis. Antioxid. Redox Signal. 2015, 22, 1032–1046. [Google Scholar] [CrossRef]
- Davis, C.O.; Marsh-Armstrong, N. Discovery and implications of transcellular mitophagy. Autophagy 2014, 10, 2383–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, J.G.; Robinson, M.B. Regulation of mitochondrial dynamics in astrocytes: Mechanisms, consequences, and unknowns. Glia 2018, 66, 1213–1234. [Google Scholar] [CrossRef]
- Morales, I.; Sanchez, A.; Puertas-Avendaño, R.; Rodriguez-Sabate, C.; Perez-Barreto, A.; Rodriguez, M. Neuroglial transmitophagy and Parkinson’s disease. Glia 2020, 68, 2277–2299. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, F.A.; Palmiter, R.D. Parkin-deficient mice are not a robust model of parkinsonism. Proc. Natl. Acad. Sci. USA 2005, 102, 2174–2179. [Google Scholar] [CrossRef] [Green Version]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.-H.; Becker, D.; Voos, W.; Leuner, K.; Müller, W.E.; Kudin, A.P.; et al. Parkinson Phenotype in Aged PINK1-Deficient Mice Is Accompanied by Progressive Mitochondrial Dysfunction in Absence of Neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef]
- Villa, E.; Marchetti, S.; Ricci, J.-E. No Parkin Zone: Mitophagy without Parkin. Trends Cell Biol. 2018, 28, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doblado, L.; Lueck, C.; Rey, C.; Samhan-Arias, A.K.; Prieto, I.; Stacchiotti, A.; Monsalve, M. Mitophagy in Human Diseases. Int. J. Mol. Sci. 2021, 22, 3903. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, P.-Y. Mitophagy in the Pathogenesis of Liver Diseases. Cells 2020, 9, 831. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Liao, X.; Wu, H.; Li, Y.; Zhu, Y.; Chen, Q. Mitophagy and Its Contribution to Metabolic and Aging-Associated Disorders. Antioxid. Redox Signal. 2020, 32, 906–927. [Google Scholar] [CrossRef]
- Macleod, K.F. Mitophagy and Mitochondrial Dysfunction in Cancer. Annu. Rev. Cancer Biol. 2020, 4, 41–60. [Google Scholar] [CrossRef] [Green Version]
- Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R.; et al. Mitophagy in Cardiovascular Diseases. J. Clin. Med. 2020, 9, 892. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Tang, Y.; Lu, J.; Zhang, W.; Zhu, Y.; Zhang, S.; Ma, G.; Jiang, P.; Zhang, W. PINK1-mediated mitophagy protects against hepatic ischemia/reperfusion injury by restraining NLRP3 inflammasome activation. Free Radic. Biol. Med. 2020, 160, 871–886. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, N.D.; Wells, G.; Campanella, M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 2017, 13, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Princz, A.; Tavernarakis, N. Mitophagy Modulators. In Encyclopedia of Biomedical Gerontology; Reference Module in Biomedical Sciences; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Killackey, S.A.; Philpott, D.J.; Girardin, S.E. Mitophagy pathways in health and disease. J. Cell Biol. 2020, 219, e202004029. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Shimura, H.; Hattori, N.; Kubo, S.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.A.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L.F. Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy 2015, 11, 422–424. [Google Scholar] [CrossRef] [Green Version]
- Vargas, J.N.S.; Wang, C.; Bunker, E.; Hao, L.; Maric, D.; Schiavo, G.; Randow, F.; Youle, R.J. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol. Cell 2019, 74, 347–362.e6. [Google Scholar] [CrossRef] [Green Version]
- Yamano, K.; Kikuchi, R.; Kojima, W.; Hayashida, R.; Koyano, F.; Kawawaki, J.; Shoda, T.; Demizu, Y.; Naito, M.; Tanaka, K.; et al. Critical role of mitochondrial ubiquitination and the OPTN–ATG9A axis in mitophagy. J. Cell Biol. 2020, 219, e201912144. [Google Scholar] [CrossRef]
- Riley, B.E.; Lougheed, J.C.; Callaway, K.; Velasquez, M.; Brecht, E.; Nguyen, L.; Shaler, T.; Walker, D.; Yang, Y.; Regnstrom, K.; et al. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat. Commun. 2013, 4, 1982. [Google Scholar] [CrossRef] [Green Version]
- Trempe, J.-F.; Sauvé, V.; Grenier, K.; Seirafi, M.; Tang, M.Y.; Ménade, M.; Al-Abdul-Wahid, S.; Krett, J.; Wong, K.; Kozlov, G.; et al. Structure of Parkin Reveals Mechanisms for Ubiquitin Ligase Activation. Science 2013, 340, 1451–1455. [Google Scholar] [CrossRef]
- Wauer, T.; Komander, D. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 2013, 32, 2099–2112. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Aguirre, J.D.; Condos, T.E.; Martinez-Torres, R.J.; Chaugule, V.K.; Toth, R.; Sundaramoorthy, R.; Mercier, P.; Knebel, A.; Spratt, D.E.; et al. Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J. 2015, 34, 2506–2521. [Google Scholar] [CrossRef]
- Wenzel, D.M.; Klevit, R.E. Following Ariadne’s thread: A new perspective on RBR ubiquitin ligases. BMC Biol. 2012, 10, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndsen, C.E.; Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Gladkova, C.; Maslen, S.L.; Skehel, J.M.; Komander, D. Mechanism of parkin activation by PINK1. Nature 2018, 559, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Sauvé, V.; Sung, G.; Soya, N.; Kozlov, G.; Blaimschein, N.; Miotto, L.S.; Trempe, J.-F.; Lukacs, G.L.; Gehring, K. Mechanism of parkin activation by phosphorylation. Nat. Struct. Mol. Biol. 2018, 25, 623–630. [Google Scholar] [CrossRef]
- Gundogdu, M.; Tadayon, R.; Salzano, G.; Shaw, G.S.; Walden, H. A mechanistic review of Parkin activation. Biochim. Biophys. Acta—Gen. Subj. 2021, 1865, 129894. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M.K. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014, 460, 127–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.-M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative Proteomics Reveal a Feedforward Mechanism for Mitochondrial PARKIN Translocation and Ubiquitin Chain Synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [Green Version]
- Kazlauskaite, A.; Martínez-Torres, R.J.; Wilkie, S.; Kumar, A.; Peltier, J.; Gonzalez, A.; Johnson, C.; Zhang, J.; Hope, A.G.; Peggie, M.; et al. Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK 1-dependent phosphorylation and activation. EMBO Rep. 2015, 16, 939–954. [Google Scholar] [CrossRef] [PubMed]
- Sauvé, V.; Lilov, A.; Seirafi, M.; Vranas, M.; Rasool, S.; Kozlov, G.; Sprules, T.; Wang, J.; Trempe, J.; Gehring, K. A Ubl/ubiquitin switch in the activation of Parkin. EMBO J. 2015, 34, 2492–2505. [Google Scholar] [CrossRef]
- Wauer, T.; Simicek, M.; Schubert, A.; Komander, D. Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 2015, 524, 370–374. [Google Scholar] [CrossRef] [Green Version]
- Yamano, K.; Queliconi, B.B.; Koyano, F.; Saeki, Y.; Hirokawa, T.; Tanaka, K.; Matsuda, N. Site-specific Interaction Mapping of Phosphorylated Ubiquitin to Uncover Parkin Activation. J. Biol. Chem. 2015, 290, 25199–25211. [Google Scholar] [CrossRef] [Green Version]
- Okatsu, K.; Koyano, F.; Kimura, M.; Kosako, H.; Saeki, Y.; Tanaka, K.; Matsuda, N. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol. 2015, 209, 111–128. [Google Scholar] [CrossRef]
- Ordureau, A.; Heo, J.-M.; Duda, D.M.; Paulo, J.A.; Olszewski, J.L.; Yanishevski, D.; Rinehart, J.; Schulman, B.A.; Harper, J.W. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. USA 2015, 112, 6637–6642. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, C.N.; Baughman, J.M.; Phu, L.; Tea, J.S.; Yu, C.; Coons, M.; Kirkpatrick, D.S.; Bingol, B.; Corn, J.E. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol. 2015, 17, 160–169. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.M.; Isasa, M.; Ordureau, A.; Prado, M.A.; Beausoleil, S.A.; Jedrychowski, M.P.; Finley, D.J.; Harper, J.W.; Gygi, S.P. Highly Multiplexed Quantitative Mass Spectrometry Analysis of Ubiquitylomes. Cell Syst. 2016, 3, 395–403.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordureau, A.; Paulo, J.A.; Zhang, W.; Ahfeldt, T.; Zhang, J.; Cohn, E.F.; Hou, Z.; Heo, J.-M.; Rubin, L.L.; Sidhu, S.S.; et al. Dynamics of PARKIN-Dependent Mitochondrial Ubiquitylation in Induced Neurons and Model Systems Revealed by Digital Snapshot Proteomics. Mol. Cell 2018, 70, 211–227.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayne, A.N.; Trempe, J.-F. Mechanisms of PINK1, ubiquitin and Parkin interactions in mitochondrial quality control and beyond. Cell. Mol. Life Sci. 2019, 76, 4589–4611. [Google Scholar] [CrossRef] [PubMed]
- Shiba-Fukushima, K.; Arano, T.; Matsumoto, G.; Inoshita, T.; Yoshida, S.; Ishihama, Y.; Ryu, K.-Y.; Nukina, N.; Hattori, N.; Imai, Y. Phosphorylation of Mitochondrial Polyubiquitin by PINK1 Promotes Parkin Mitochondrial Tethering. PLoS Genet. 2014, 10, e1004861. [Google Scholar] [CrossRef]
- Okatsu, K.; Kimura, M.; Oka, T.; Tanaka, K.; Matsuda, N. Unconventional PINK1 localization mechanism to the outer membrane of depolarized mitochondria drives Parkin recruitment. J. Cell Sci. 2015, 128, 964–978. [Google Scholar] [CrossRef] [Green Version]
- Yamano, K.; Matsuda, N.; Tanaka, K. The ubiquitin signal and autophagy: An orchestrated dance leading to mitochondrial degradation. EMBO Rep. 2016, 17, 300–316. [Google Scholar] [CrossRef]
- Matsuda, N.; Yamano, K. Two sides of a coin: Physiological significance and molecular mechanisms for damage-induced mitochondrial localization of PINK1 and Parkin. Neurosci. Res. 2020, 159, 16–24. [Google Scholar] [CrossRef]
- Koyano, F.; Yamano, K.; Kosako, H.; Tanaka, K.; Matsuda, N. Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by MITOL. J. Biol. Chem. 2019, 294, 10300–10314. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin Target Miro for Phosphorylation and Degradation to Arrest Mitochondrial Motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Dorn, G.W. PINK1-Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Gong, G.; Song, M.; Csordas, G.; Kelly, D.P.; Matkovich, S.J.; Dorn, G.W. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 2015, 350, aad2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, A.C.; Thomas, R.E.; Yu, S.; Vincow, E.S.; Pallanck, L. The Mitochondrial Fusion-Promoting Factor Mitofusin Is a Substrate of the PINK1/Parkin Pathway. PLoS ONE 2010, 5, e10054. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.E.; Cooper, J.M.; Chau, K.-Y.; Rojo, M.; Schapira, A.H.V.; Taanman, J.-W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- McLelland, G.-L.; Goiran, T.; Yi, W.; Dorval, G.; Chen, C.X.; Lauinger, N.D.; Krahn, A.I.; Valimehr, S.; Rakovic, A.; Rouiller, I.; et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 2018, 7, e32866. [Google Scholar] [CrossRef]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, T.L. Mitochondrial Trafficking in Neurons. Cold Spring Harb. Perspect. Biol. 2013, 5, a011304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Merwe, C.; Jalali Sefid Dashti, Z.; Christoffels, A.; Loos, B.; Bardien, S. Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: Parkin, PINK1 and DJ-1. Eur. J. Neurosci. 2015, 41, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Shlevkov, E.; Kramer, T.; Schapansky, J.; LaVoie, M.J.; Schwarz, T.L. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc. Natl. Acad. Sci. USA 2016, 113, E6097–E6106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Cornelissen, T.; Haddad, D.; Wauters, F.; Van Humbeeck, C.; Mandemakers, W.; Koentjoro, B.; Sue, C.; Gevaert, K.; De Strooper, B.; Verstreken, P.; et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 2014, 23, 5227–5242. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Martinez, A.; Lane, J.D.; Mayor, U.; Clague, M.J.; Urbé, S. USP 30 deubiquitylates mitochondrial Parkin substrates and restricts apoptotic cell death. EMBO Rep. 2015, 16, 618–627. [Google Scholar] [CrossRef] [Green Version]
- Niu, K.; Fang, H.; Chen, Z.; Zhu, Y.; Tan, Q.; Wei, D.; Li, Y.; Balajee, A.S.; Zhao, Y. USP33 deubiquitinates PRKN/parkin and antagonizes its role in mitophagy. Autophagy 2020, 16, 724–734. [Google Scholar] [CrossRef]
- Gersch, M.; Gladkova, C.; Schubert, A.F.; Michel, M.A.; Maslen, S.; Komander, D. Mechanism and regulation of the Lys6-selective deubiquitinase USP30. Nat. Struct. Mol. Biol. 2017, 24, 920–930. [Google Scholar] [CrossRef]
- Sato, Y.; Okatsu, K.; Saeki, Y.; Yamano, K.; Matsuda, N.; Kaiho, A.; Yamagata, A.; Goto-Ito, S.; Ishikawa, M.; Hashimoto, Y.; et al. Structural basis for specific cleavage of Lys6-linked polyubiquitin chains by USP30. Nat. Struct. Mol. Biol. 2017, 24, 911–919. [Google Scholar] [CrossRef]
- Phu, L.; Rose, C.M.; Tea, J.S.; Wall, C.E.; Verschueren, E.; Cheung, T.K.; Kirkpatrick, D.S.; Bingol, B. Dynamic Regulation of Mitochondrial Import by the Ubiquitin System. Mol. Cell 2020, 77, 1107–1123.e10. [Google Scholar] [CrossRef]
- Marcassa, E.; Kallinos, A.; Jardine, J.; Rusilowicz-Jones, E.V.; Martinez, A.; Kuehl, S.; Islinger, M.; Clague, M.J.; Urbé, S. Dual role of USP 30 in controlling basal pexophagy and mitophagy. EMBO Rep. 2018, 19, e45595. [Google Scholar] [CrossRef] [PubMed]
- Rusilowicz-Jones, E.V.; Jardine, J.; Kallinos, A.; Pinto-Fernandez, A.; Guenther, F.; Giurrandino, M.; Barone, F.G.; McCarron, K.; Burke, C.J.; Murad, A.; et al. USP30 sets a trigger threshold for PINK1–PARKIN amplification of mitochondrial ubiquitylation. Life Sci. Alliance 2020, 3, e202000768. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Tang, M.Y.; Pérusse, J.R.; Dashti, E.A.; Aguileta, M.A.; McLelland, G.; Gros, P.; Shaler, T.A.; Faubert, D.; Coulombe, B.; et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 2014, 33, 2473–2491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordureau, A.; Paulo, J.A.; Zhang, J.; An, H.; Swatek, K.N.; Cannon, J.R.; Wan, Q.; Komander, D.; Harper, J.W. Global Landscape and Dynamics of Parkin and USP30-Dependent Ubiquitylomes in iNeurons during Mitophagic Signaling. Mol. Cell 2020, 77, 1124–1142.e10. [Google Scholar] [CrossRef] [PubMed]
- Wauer, T.; Swatek, K.N.; Wagstaff, J.L.; Gladkova, C.; Pruneda, J.N.; Michel, M.A.; Gersch, M.; Johnson, C.M.; Freund, S.M.; Komander, D. Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 2015, 34, 307–325. [Google Scholar] [CrossRef]
- Wang, Y.; Serricchio, M.; Jauregui, M.; Shanbhag, R.; Stoltz, T.; Di Paolo, C.T.; Kim, P.K.; McQuibban, G.A. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 2015, 11, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, J.; Tang, Y.; Shen, H.-M. PTEN-L puts a brake on mitophagy. Autophagy 2018, 14, 2023–2025. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Cho, Y.-L.; Tang, Y.; Wang, J.; Park, J.-E.; Wu, Y.; Wang, C.; Tong, Y.; Chawla, R.; Zhang, J.; et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1–Parkin-mediated mitophagy. Cell Res. 2018, 28, 787–802. [Google Scholar] [CrossRef]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef]
- Zhuang, N.; Li, L.; Chen, S.; Wang, T. PINK1-dependent phosphorylation of PINK1 and Parkin is essential for mitochondrial quality control. Cell Death Dis. 2016, 7, e2501. [Google Scholar] [CrossRef]
- Tang, M.Y.; Vranas, M.; Krahn, A.I.; Pundlik, S.; Trempe, J.-F.; Fon, E.A. Structure-guided mutagenesis reveals a hierarchical mechanism of Parkin activation. Nat. Commun. 2017, 8, 14697. [Google Scholar] [CrossRef] [Green Version]
- Sauvé, V.; Sung, G.; MacDougall, E.; Kozlov, G.; Saran, A.; Fakih, R.; Fon, E.; Gehring, K. Structural basis for feedforward control in the PINK1/Parkin pathway. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, J.J.; Nam, H.-J.; Gao, B.; Yin, P.; Qin, B.; Yi, S.-Y.; Ham, H.; Evans, D.; Kim, S.-H.; et al. Parkin Regulates Mitosis and Genomic Stability through Cdc20/Cdh1. Mol. Cell 2015, 60, 21–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avraham, E.; Rott, R.; Liani, E.; Szargel, R.; Engelender, S. Phosphorylation of Parkin by the Cyclin-dependent Kinase 5 at the Linker Region Modulates Its Ubiquitin-Ligase Activity and Aggregation. J. Biol. Chem. 2007, 282, 12842–12850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, H.S.; Lee, Y.; Shin, J.-H.; Karuppagounder, S.S.; Gadad, B.S.; Koleske, A.J.; Pletnikova, O.; Troncoso, J.C.; Dawson, V.L.; Dawson, T.M. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin’s ubiquitination and protective function. Proc. Natl. Acad. Sci. USA 2010, 107, 16691–16696. [Google Scholar] [CrossRef] [Green Version]
- Imam, S.Z.; Zhou, Q.; Yamamoto, A.; Valente, A.J.; Ali, S.F.; Bains, M.; Roberts, J.L.; Kahle, P.J.; Clark, R.A.; Li, S. Novel Regulation of Parkin Function through c-Abl-Mediated Tyrosine Phosphorylation: Implications for Parkinson’s Disease. J. Neurosci. 2011, 31, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Lu, Q.; Rapaport, D.; Kozjak-Pavlovic, V. Tom70 Is Essential for PINK1 Import into Mitochondria. PLoS ONE 2013, 8, e58435. [Google Scholar] [CrossRef] [Green Version]
- Sekine, S.; Wang, C.; Sideris, D.P.; Bunker, E.; Zhang, Z.; Youle, R.J. Reciprocal Roles of Tom7 and OMA1 during Mitochondrial Import and Activation of PINK1. Mol. Cell 2019, 73, 1028–1043.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.Y.; Renton, A.E.M.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Youle, R.J. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Guardia-Laguarta, C.; Yin, J.; Erdjument-Bromage, H.; Martin, B.; James, M.; Jiang, X.; Przedborski, S. The Ubiquitination of PINK1 Is Restricted to Its Mature 52-kDa Form. Cell Rep. 2017, 20, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3, 1016. [Google Scholar] [CrossRef] [Green Version]
- Okatsu, K.; Uno, M.; Koyano, F.; Go, E.; Kimura, M.; Oka, T.; Tanaka, K.; Matsuda, N. A Dimeric PINK1-containing Complex on Depolarized Mitochondria Stimulates Parkin Recruitment. J. Biol. Chem. 2013, 288, 36372–36384. [Google Scholar] [CrossRef] [Green Version]
- Rasool, S.; Soya, N.; Truong, L.; Croteau, N.; Lukacs, G.L.; Trempe, J. PINK 1 autophosphorylation is required for ubiquitin recognition. EMBO Rep. 2018, 19, e44981. [Google Scholar] [CrossRef]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; De Bock, P.-J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grünewald, A.; Seibler, P.; et al. PINK1 Loss-of-Function Mutations Affect Mitochondrial Complex I Activity via NdufA10 Ubiquinone Uncoupling. Science 2014, 344, 203–207. [Google Scholar] [CrossRef]
- Tsai, P.-I.; Lin, C.-H.; Hsieh, C.-H.; Papakyrikos, A.M.; Kim, M.J.; Napolioni, V.; Schoor, C.; Couthouis, J.; Wu, R.-M.; Wszolek, Z.K.; et al. PINK1 Phosphorylates MIC60/Mitofilin to Control Structural Plasticity of Mitochondrial Crista Junctions. Mol. Cell 2018, 69, 744–756.e6. [Google Scholar] [CrossRef] [Green Version]
- Regula, K.M.; Ens, K.; Kirshenbaum, L.A. Inducible Expression of BNIP3 Provokes Mitochondrial Defects and Hypoxia-Mediated Cell Death of Ventricular Myocytes. Circ. Res. 2002, 91, 226–231. [Google Scholar] [CrossRef] [Green Version]
- Diwan, A.; Krenz, M.; Syed, F.M.; Wansapura, J.; Ren, X.; Koesters, A.G.; Li, H.; Kirshenbaum, L.A.; Hahn, H.S.; Robbins, J.; et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J. Clin. Investig. 2007, 117, 2825–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diwan, A.; Koesters, A.G.; Odley, A.M.; Pushkaran, S.; Baines, C.P.; Spike, B.T.; Daria, D.; Jegga, A.G.; Geiger, H.; Aronow, B.J.; et al. Unrestrained erythroblast development in Nix-/- mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc. Natl. Acad. Sci. USA 2007, 104, 6794–6799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diwan, A.; Matkovich, S.J.; Yuan, Q.; Zhao, W.; Yatani, A.; Brown, J.H.; Molkentin, J.D.; Kranias, E.G.; Dorn, G.W. Endoplasmic reticulum–mitochondria crosstalk in NIX-mediated murine cell death. J. Clin. Investig. 2008, 119, 203–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy Is an HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, Å.B. Microtubule-associated Protein 1 Light Chain 3 (LC3) Interacts with Bnip3 Protein to Selectively Remove Endoplasmic Reticulum and Mitochondria via Autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Zheng, Y.; Zhang, X.; Chen, Y.; Wu, X.; Wu, J.; Shen, Z.; Jiang, L.; Wang, L.; Yang, W.; et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 2017, 13, 1754–1766. [Google Scholar] [CrossRef]
- Zhu, Y.; Massen, S.; Terenzio, M.; Lang, V.; Chen-Lindner, S.; Eils, R.; Novak, I.; Dikic, I.; Hamacher-Brady, A.; Brady, N.R. Modulation of Serines 17 and 24 in the LC3-interacting Region of Bnip3 Determines Pro-survival Mitophagy versus Apoptosis. J. Biol. Chem. 2013, 288, 1099–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva Rosa, S.C.; Martens, M.D.; Field, J.T.; Nguyen, L.; Kereliuk, S.M.; Hai, Y.; Chapman, D.; Diehl-Jones, W.; Aliani, M.; West, A.R.; et al. BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy 2021, 17, 2257–2272. [Google Scholar] [CrossRef]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [Green Version]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Rogov, V.V.; Suzuki, H.; Marinković, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubašić, M.; Šprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1131. [Google Scholar] [CrossRef] [PubMed]
- Marinković, M.; Šprung, M.; Novak, I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 2021, 17, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.P.; Bock-Hughes, A.; Berardi, D.E.; Macleod, K.F. ULK1 promotes mitophagy via phosphorylation and stabilization of BNIP3. Sci. Rep. 2021, 11, 20526. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, H.-Y.; Hanna, R.A.; Gustafsson, Å.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xue, L.; Li, L.; Tang, C.; Wan, Z.; Wang, R.; Tan, J.; Tan, Y.; Han, H.; Tian, R.; et al. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J. Biol. Chem. 2016, 291, 21616–21629. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Chen, D.; Si, J.; Hu, Q.; Qin, Z.; Fang, M.; Wang, G. The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum. Mol. Genet. 2015, 24, 2528–2538. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.-X.; Ni, H.-M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W.; Yin, X.-M. Nix Is Critical to Two Distinct Phases of Mitophagy, Reactive Oxygen Species-mediated Autophagy Induction and Parkin-Ubiquitin-p62-mediated Mitochondrial Priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, L.; Cheng, Q.; Li, Y.; Wu, H.; Zhang, W.; Wang, Y.; Sehgal, S.A.; Siraj, S.; Wang, X.; et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC 1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017, 18, 495–509. [Google Scholar] [CrossRef]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A Regulatory Signaling Loop Comprising the PGAM5 Phosphatase and CK2 Controls Receptor-Mediated Mitophagy. Mol. Cell 2014, 54, 362–377. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Xue, D.; Chen, G.; Han, Z.; Huang, L.; Zhu, C.; Wang, X.; Jin, H.; Wang, J.; Zhu, Y.; et al. The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy 2014, 10, 1712–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampert, M.A.; Orogo, A.M.; Najor, R.H.; Hammerling, B.C.; Leon, L.J.; Wang, B.J.; Kim, T.; Sussman, M.A.; Gustafsson, Å.B. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 2019, 15, 1182–1198. [Google Scholar] [CrossRef] [PubMed]
- Murakawa, T.; Yamaguchi, O.; Hashimoto, A.; Hikoso, S.; Takeda, T.; Oka, T.; Yasui, H.; Ueda, H.; Akazawa, Y.; Nakayama, H.; et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 2015, 6, 7527. [Google Scholar] [CrossRef] [Green Version]
- Murakawa, T.; Okamoto, K.; Omiya, S.; Taneike, M.; Yamaguchi, O.; Otsu, K. A Mammalian Mitophagy Receptor, Bcl2-L-13, Recruits the ULK1 Complex to Induce Mitophagy. Cell Rep. 2019, 26, 338–345.e6. [Google Scholar] [CrossRef]
- Bhujabal, Z.; Birgisdottir, Å.B.; Sjøttem, E.; Brenne, H.B.; Øvervatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; De Strooper, B.; Vandenberghe, W. Parkin Interacts with Ambra1 to Induce Mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef] [Green Version]
- Di Rita, A.; D’Acunzo, P.; Simula, L.; Campello, S.; Strappazzon, F.; Cecconi, F. AMBRA1-Mediated Mitophagy Counteracts Oxidative Stress and Apoptosis Induced by Neurotoxicity in Human Neuroblastoma SH-SY5Y Cells. Front. Cell. Neurosci. 2018, 12, 92. [Google Scholar] [CrossRef]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-X.; Tsoi, B.; Li, Y.-F.; Kurihara, H.; He, R.-R. Cardiolipin and Its Different Properties in Mitophagy and Apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; St-Pierre, P.; Shankar, J.; Wang, P.T.C.; Joshi, B.; Nabi, I.R. Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol. Biol. Cell 2013, 24, 1153–1162. [Google Scholar] [CrossRef]
- Ambivero, C.T.; Cilenti, L.; Main, S.; Zervos, A.S. Mulan E3 ubiquitin ligase interacts with multiple E2 conjugating enzymes and participates in mitophagy by recruiting GABARAP. Cell. Signal. 2014, 26, 2921–2929. [Google Scholar] [CrossRef]
- Yun, J.; Puri, R.; Yang, H.; Lizzio, M.A.; Wu, C.; Sheng, Z.-H.; Guo, M. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. eLife 2014, 3, e01958. [Google Scholar] [CrossRef] [PubMed]
- Szargel, R.; Shani, V.; Abd Elghani, F.; Mekies, L.N.; Liani, E.; Rott, R.; Engelender, S. The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum. Mol. Genet. 2016, 25, 3476–3490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, E.; Proïcs, E.; Rubio-Patiño, C.; Obba, S.; Zunino, B.; Bossowski, J.P.; Rozier, R.M.; Chiche, J.; Mondragón, L.; Riley, J.S.; et al. Parkin-Independent Mitophagy Controls Chemotherapeutic Response in Cancer Cells. Cell Rep. 2017, 20, 2846–2859. [Google Scholar] [CrossRef] [Green Version]
- Zachari, M.; Gudmundsson, S.R.; Li, Z.; Manifava, M.; Cugliandolo, F.; Shah, R.; Smith, M.; Stronge, J.; Karanasios, E.; Piunti, C.; et al. Selective Autophagy of Mitochondria on a Ubiquitin-Endoplasmic-Reticulum Platform. Dev. Cell 2019, 50, 627–643.e5. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.-M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [Green Version]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the Autophagy Receptor Optineurin Restricts Salmonella Growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itakura, E.; Kishi-Itakura, C.; Koyama-Honda, I.; Mizushima, N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J. Cell Sci. 2012, 125, 1488–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karanasios, E.; Walker, S.A.; Okkenhaug, H.; Manifava, M.; Hummel, E.; Zimmermann, H.; Ahmed, Q.; Domart, M.-C.; Collinson, L.; Ktistakis, N.T. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat. Commun. 2016, 7, 12420. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burman, C.; Ktistakis, N.T. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010, 584, 1302–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanao, T.; Koike, M.; Yamaguchi, J.; Sasaki, M.; Uchiyama, Y. Cellular localization and tissue distribution of endogenous DFCP1 protein. Biomed. Res. 2015, 36, 121–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polson, H.E.J.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbé, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [Green Version]
- Dooley, H.C.; Razi, M.; Polson, H.E.J.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 Links LC3 Conjugation with PI3P, Autophagosome Formation, and Pathogen Clearance by Recruiting Atg12–5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Zhen, Y.; Spangenberg, H.; Munson, M.J.; Brech, A.; Schink, K.O.; Tan, K.-W.; Sørensen, V.; Wenzel, E.M.; Radulovic, M.; Engedal, N.; et al. ESCRT-mediated phagophore sealing during mitophagy. Autophagy 2020, 16, 826–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente, C.; Hendrickson, R.C.; Jiang, X. Nutrient-regulated Phosphorylation of ATG13 Inhibits Starvation-induced Autophagy. J. Biol. Chem. 2016, 291, 6026–6035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.G.; Hoffman, G.R.; Poulogiannis, G.; Buel, G.R.; Jang, Y.J.; Lee, K.W.; Kim, B.-Y.; Erikson, R.L.; Cantley, L.C.; Choo, A.Y.; et al. Metabolic Stress Controls mTORC1 Lysosomal Localization and Dimerization by Regulating the TTT-RUVBL1/2 Complex. Mol. Cell 2013, 49, 172–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolomé, A.; García-Aguilar, A.; Asahara, S.-I.; Kido, Y.; Guillén, C.; Pajvani, U.B.; Benito, M. MTORC1 Regulates both General Autophagy and Mitophagy Induction after Oxidative Phosphorylation Uncoupling. Mol. Cell Biol. 2017, 37, e00441-17. [Google Scholar] [CrossRef] [Green Version]
- Bartolome, A.; Kimura-Koyanagi, M.; Asahara, S.-I.; Guillen, C.; Inoue, H.; Teruyama, K.; Shimizu, S.; Kanno, A.; Garcia-Aguilar, A.; Koike, M.; et al. Pancreatic -Cell Failure Mediated by mTORC1 Hyperactivity and Autophagic Impairment. Diabetes 2014, 63, 2996–3008. [Google Scholar] [CrossRef] [Green Version]
- Bordi, M.; Darji, S.; Sato, Y.; Mellén, M.; Berg, M.J.; Kumar, A.; Jiang, Y.; Nixon, R.A. mTOR hyperactivation in Down Syndrome underlies deficits in autophagy induction, autophagosome formation, and mitophagy. Cell Death Dis. 2019, 10, 563. [Google Scholar] [CrossRef] [Green Version]
- Vadysirisack, D.D.; Ellisen, L.W. MTOR Activity Under Hypoxia. In mTOR; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2012; pp. 45–58. [Google Scholar]
- Dou, Q.; Chen, H.-N.; Wang, K.; Yuan, K.; Lei, Y.; Li, K.; Lan, J.; Chen, Y.; Huang, Z.; Xie, N.; et al. Ivermectin Induces Cytostatic Autophagy by Blocking the PAK1/Akt Axis in Breast Cancer. Cancer Res. 2016, 76, 4457–4469. [Google Scholar] [CrossRef] [Green Version]
- Hirota, Y.; Yamashita, S.; Kurihara, Y.; Jin, X.; Aihara, M.; Saigusa, T.; Kang, D.; Kanki, T. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 2015, 11, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Padman, B.S.; Nguyen, T.N.; Uoselis, L.; Skulsuppaisarn, M.; Nguyen, L.K.; Lazarou, M. LC3/GABARAPs drive ubiquitin-independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat. Commun. 2019, 10, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamano, K.; Wang, C.; Sarraf, S.A.; Münch, C.; Kikuchi, R.; Noda, N.N.; Hizukuri, Y.; Kanemaki, M.T.; Harper, W.; Tanaka, K.; et al. Endosomal Rab cycles regulate Parkin-mediated mitophagy. eLife 2018, 7, e31326. [Google Scholar] [CrossRef] [PubMed]
- Yamano, K.; Fogel, A.I.; Wang, C.; van der Bliek, A.M.; Youle, R.J. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 2014, 3, e01612. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Orgaz, A.; Kvainickas, A.; Nägele, H.; Denner, J.; Eimer, S.; Dengjel, J.; Steinberg, F. Control of RAB7 activity and localization through the retromer-TBC1D5 complex enables RAB7-dependent mitophagy. EMBO J. 2018, 37, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.-M.; Ordureau, A.; Swarup, S.; Paulo, J.A.; Shen, K.; Sabatini, D.M.; Harper, J.W. RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci. Adv. 2018, 4, eaav0443. [Google Scholar] [CrossRef] [Green Version]
- Tsuboyama, K.; Koyama-Honda, I.; Sakamaki, Y.; Koike, M.; Morishita, H.; Mizushima, N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 2016, 354, 1036–1041. [Google Scholar] [CrossRef]
- Lee, J.W.; Park, S.; Takahashi, Y.; Wang, H.-G. The Association of AMPK with ULK1 Regulates Autophagy. PLoS ONE 2010, 5, e15394. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-Activated Protein Kinase Connects Energy Sensing to Mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. USA 2011, 108, 4788–4793. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J.; et al. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab. 2015, 22, 922–935. [Google Scholar] [CrossRef] [Green Version]
- Gan, W.; Zhang, C.; Siu, K.Y.; Satoh, A.; Tanner, J.A.; Yu, S. ULK1 phosphorylates Sec23A and mediates autophagy-induced inhibition of ER-to-Golgi traffic. BMC Cell Biol. 2017, 18, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsaadi, R.M.; Losier, T.T.; Tian, W.; Jackson, A.; Guo, Z.; Rubinsztein, D.C.; Russell, R.C. ULK1-mediated phosphorylation of ATG16L1 promotes xenophagy, but destabilizes the ATG16L1 Crohn’s mutant. EMBO Rep. 2019, 20, e46885. [Google Scholar] [CrossRef]
- Rajesh, S.; Bago, R.; Odintsova, E.; Muratov, G.; Baldwin, G.; Sridhar, P.; Rajesh, S.; Overduin, M.; Berditchevski, F. Binding to Syntenin-1 Protein Defines a New Mode of Ubiquitin-based Interactions Regulated by Phosphorylation. J. Biol. Chem. 2011, 286, 39606–39614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaesu, G.; Kobayashi, T.; Yoshimura, A. TGF -activated kinase 1 (TAK1)-binding proteins (TAB) 2 and 3 negatively regulate autophagy. J. Biochem. 2012, 151, 157–166. [Google Scholar] [CrossRef]
- Dunlop, E.A.; Hunt, D.K.; Acosta-Jaquez, H.A.; Fingar, D.C.; Tee, A.R. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy 2011, 7, 737–747. [Google Scholar] [CrossRef]
- Löffler, A.S.; Alers, S.; Dieterle, A.M.; Keppeler, H.; Franz-Wachtel, M.; Kundu, M.; Campbell, D.G.; Wesselborg, S.; Alessi, D.R.; Stork, B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011, 7, 696–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.-Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.-L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Chun, M.G.H.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.-C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-M.; Jung, C.H.; Seo, M.; Otto, N.M.; Grunwald, D.; Kim, K.H.; Moriarity, B.; Kim, Y.-M.; Starker, C.; Nho, R.S.; et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 2016, 12, 547–564. [Google Scholar] [CrossRef]
- Joo, J.H.; Wang, B.; Frankel, E.; Ge, L.; Xu, L.; Iyengar, R.; Li-Harms, X.; Wright, C.; Shaw, T.I.; Lindsten, T.; et al. The Noncanonical Role of ULK/ATG1 in ER-to-Golgi Trafficking Is Essential for Cellular Homeostasis. Mol. Cell 2016, 62, 491–506. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Xu, Z.-X.; Ding, Z.; Lu, Y.; Yu, Q.; Werle, K.D.; Zhou, G.; Park, Y.-Y.; Peng, G.; Gambello, M.J.; et al. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat. Commun. 2015, 6, 7926. [Google Scholar] [CrossRef]
- Tian, W.; Li, W.; Chen, Y.; Yan, Z.; Huang, X.; Zhuang, H.; Zhong, W.; Chen, Y.; Wu, W.; Lin, C.; et al. Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett. 2015, 589, 1847–1854. [Google Scholar] [CrossRef] [Green Version]
- Kundu, M.; Lindsten, T.; Yang, C.-Y.; Wu, J.; Zhao, F.; Zhang, J.; Selak, M.A.; Ney, P.A.; Thompson, C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008, 112, 1493–1502. [Google Scholar] [CrossRef] [Green Version]
- Honda, S.; Arakawa, S.; Nishida, Y.; Yamaguchi, H.; Ishii, E.; Shimizu, S. Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat. Commun. 2014, 5, 4004. [Google Scholar] [CrossRef] [PubMed]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef]
- Hung, C.-M.; Lombardo, P.S.; Malik, N.; Brun, S.N.; Hellberg, K.; Van Nostrand, J.L.; Garcia, D.; Baumgart, J.; Diffenderfer, K.; Asara, J.M.; et al. AMPK/ULK1-mediated phosphorylation of Parkin ACT domain mediates an early step in mitophagy. Sci. Adv. 2021, 7, eabg4544. [Google Scholar] [CrossRef] [PubMed]
- Seabright, A.P.; Fine, N.H.F.; Barlow, J.P.; Lord, S.O.; Musa, I.; Gray, A.; Bryant, J.A.; Banzhaf, M.; Lavery, G.G.; Hardie, D.G.; et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J. 2020, 34, 6284–6301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seabright, A.P.; Lai, Y.-C. Regulatory Roles of PINK1-Parkin and AMPK in Ubiquitin-Dependent Skeletal Muscle Mitophagy. Front. Physiol. 2020, 11, 608474. [Google Scholar] [CrossRef]
- Drake, J.C.; Wilson, R.J.; Laker, R.C.; Guan, Y.; Spaulding, H.R.; Nichenko, A.S.; Shen, W.; Shang, H.; Dorn, M.V.; Huang, K.; et al. Mitochondria-localized AMPK responds to local energetics and contributes to exercise and energetic stress-induced mitophagy. Proc. Natl. Acad. Sci. USA 2021, 118, e2025932118. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villén, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A Tissue-Specific Atlas of Mouse Protein Phosphorylation and Expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.-H.; Chen, C.; Wang, D.W. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Kim, J.J.; Han, S.-A.; Fan, Y.; Guo, L.-S.; Aziz, K.; Nowsheen, S.; Kim, S.S.; Park, S.-Y.; Luo, Q.; et al. The AMPK–Parkin axis negatively regulates necroptosis and tumorigenesis by inhibiting the necrosome. Nat. Cell Biol. 2019, 21, 940–951. [Google Scholar] [CrossRef]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 2018, 23, 86–100.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Xuan, W.; Li, J.; Yao, H.; Huang, C.; Li, J. AMPK protects against alcohol-induced liver injury through UQCRC2 to up-regulate mitophagy. Autophagy 2021, 17, 3622–3643. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.E.; Eom, J.-I.; Jeung, H.-K.; Cheong, J.-W.; Lee, J.Y.; Kim, J.S.; Min, Y.H. Targeting AMPK-ULK1-mediated autophagy for combating BET inhibitor resistance in acute myeloid leukemia stem cells. Autophagy 2017, 13, 761–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Zong, Y.; Zhang, C.-S.; Li, M.; Wang, W.; Wang, Z.; Hawley, S.A.; Ma, T.; Feng, J.-W.; Tian, X.; Qi, Q.; et al. Hierarchical activation of compartmentalized pools of AMPK depends on severity of nutrient or energy stress. Cell Res. 2019, 29, 460–473. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iorio, R.; Celenza, G.; Petricca, S. Mitophagy: Molecular Mechanisms, New Concepts on Parkin Activation and the Emerging Role of AMPK/ULK1 Axis. Cells 2022, 11, 30. https://doi.org/10.3390/cells11010030
Iorio R, Celenza G, Petricca S. Mitophagy: Molecular Mechanisms, New Concepts on Parkin Activation and the Emerging Role of AMPK/ULK1 Axis. Cells. 2022; 11(1):30. https://doi.org/10.3390/cells11010030
Chicago/Turabian StyleIorio, Roberto, Giuseppe Celenza, and Sabrina Petricca. 2022. "Mitophagy: Molecular Mechanisms, New Concepts on Parkin Activation and the Emerging Role of AMPK/ULK1 Axis" Cells 11, no. 1: 30. https://doi.org/10.3390/cells11010030
APA StyleIorio, R., Celenza, G., & Petricca, S. (2022). Mitophagy: Molecular Mechanisms, New Concepts on Parkin Activation and the Emerging Role of AMPK/ULK1 Axis. Cells, 11(1), 30. https://doi.org/10.3390/cells11010030