
Isolation and Propagation of Human Corneal Stromal Keratocytes for Tissue Engineering and Cell Therapy

 ,
,
Abstract
:1. Introduction
2. Materials and Equipment
2.1. Equipment Required
- (1)
- Tissue culture hood;
- (2)
- Germinator (Innotech Bioscience IS-350 Sterilizer; Inotech Bioscience, Rockville, MD, USA);
- (3)
- Rotator (BOECO Rotator Multi Bio RS-24; Boeckel + Co, Hamburg, Germany);
- (4)
- Mortar and pestle;
- (5)
- Mayo scissors (Electron Microscopy Sciences, Hatfield, PA, USA, cat. no. 72996-11) *;
- (6)
- Forceps (Electron Microscopy Sciences, cat. no. 78266-04) *;
- (7)
- Spatula (VWR Scientific, Radnor, PA, USA, cat. no. 82027-528) *;(* Note: Autoclave mortar and pestle, scissors, forceps, and spatula before the experiment.);
- (8)
- Surgical blade no. 10 (FEATHER, Osaka, Japan, cat. no. 504169);
- (9)
- Dewar flask;
- (10)
- Weighing balance;
- (11)
- Centrifuge;
- (12)
- Scalpel handle #3 (Electron Microscopy Sciences, cat. no. 72040-03);
- (13)
- Dissecting microscope (Carl Zeiss Stemi 500; Carl Zeiss, Oberkochen, Germany);
- (14)
- Humidified CO2 incubator;
- (15)
- Liquid nitrogen tank;
- (16)
- 40 μm cell strainer (Corning, Corning, NY, USA, cat. no. 352340);
- (17)
- 70 μm cell strainer (Corning, cat. no. 352350);
- (18)
- 15 mL centrifuge tube (Greiner, Kremsmünster, Austria, cat. no. 188271);
- (19)
- 50 mL centrifuge tube (Greiner, cat. no. 210261);
- (20)
- 0.6 mL microcentrifuge tube (Corning, cat. no. MCT-060-C);
- (21)
- 1.5 mL microcentrifuge tube (Corning, cat. no. MCT-150-C);
- (22)
- 60 mm cell culture dish (Corning, cat. no. 353002);
- (23)
- Styrofoam box;
- (24)
- 10 mL syringe (Becton Dickinson, Franklin Lakes, NJ, USA, cat. no. 302149);
- (25)
- Minisart® RC syringe filters (Sartorius, Goettingen, Germany, cat. no. 17764-ACK);
- (26)
- BioCoat™ Collagen I 24-well Clear Flat Bottom TC-treated (Corning, cat. no. 354408);Optional: BioCoat™ Collagen I 6-well Clear Flat Bottom TC-treated (Corning, cat. no. 354400);
- (27)
- Mr. FrostyTM freezing container (contains isopropanol) (Thermo Fisher, Waltham, MA, USA, cat. no. 5100-0001);
- (28)
- 2 mL cryovial (Simport, Quebec, Canada, cat. no. T301-2).
2.2. Reagents Required
- (1)
- Human amniotic membrane;
- (2)
- Liquid nitrogen;
- (3)
- Ice;
- (4)
- Cadaveric human donor tissue;
- (5)
- Autoclaved 1× phosphate-buffered saline (PBS);
- (6)
- Bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO, USA, cat. no. A9418);
- (7)
- Collagenase I (Worthington Biochemical Corporation, Lakewood, NJ, USA, cat. no. CLS-1);
- (8)
- DMEM/F-12 (Invitrogen, Waltham, MA, USA, cat. no. 11330-032);
- (9)
- Deionized water;
- (10)
- MEM insulin-transferrin-selenium (Gibco, Waltham, MA, USA, cat. no. 41400045);
- (11)
- MEM vitamin solution (Gibco, cat. no. 11120052);
- (12)
- MEM amino acids solution (Gibco, cat. no. 11130051);
- (13)
- MEM non-essential amino acids solution (Gibco, cat. no. 11140050);
- (14)
- Antibiotic-antimycotic (Gibco, cat. no. 15240112);
- (15)
- Sodium hydroxide (NaOH; Sigma-Aldrich, cat. no. S2770);
- (16)
- StemMACSTM Y-27632 (Miltenyi Biotec, Bergisch Gladbach, Germany, cat. no. 130-104-169);
- (17)
- Recombinant human insulin-like growth factor 1 (IGF-1; Gibco, cat. no. PHG0078);
- (18)
- L-Ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Sigma-Aldrich, cat. no. A8960);
- (19)
- Fetal bovine serum (FBS; Gibco, cat. no. 10082-147);
- (20)
- StemPro™ Accutase™ cell dissociation reagent (Gibco, cat. no. A1110501); Optional: TripLE Express (Gibco, cat. no. 12604013);
- (21)
- Dimethyl sulfoxide (DMSO; Sigma-Aldrich, cat. no. D2650);
- (22)
- Protein DC Assay (Bio-Rad, Hercules, CA, USA, cat. no. 23235).
3. Preparation of Stock and Working Solutions before Procedures
3.1. Digestion Buffer Stock and Working Solutions
- (1)
- Weigh 100 mg of collagenase I powder in a 15 mL centrifuge tube.
- (2)
- Add 8 mL of 1× PBS and agitate the tube to dissolve the powder.
- (3)
- Fill up the remaining 2 mL to make a final volume of 10 mL of 10 mg/mL collagenase type I.
- (4)
- Filter sterilize the collagenase I solution using Minisart® RC syringe filter that is attached to a 10 mL syringe. Aliquot sterile collagenase I at 1 mL each into 1.5 mL microcentrifuge tubes and store the tubes of stock solutions at −20 °C * (* Note: Once thawed, collagenase should be stored at 4 °C. Repeat freeze–thawing should be avoided.)
- (5)
- Prepare 10 mL of digestion buffer working solution, containing 1 mg/mL collagenase I and 0.1% BSA by adding 1 mL of 10 mg/mL collagenase (prepared in Steps 1–5 above) and 10 mg of BSA to 9 mL of CSK basal medium which preparation protocol will be described in the following section *, **.(* Note: The digestion buffer working solution should be prepared fresh before cadaveric donor corneal tissue dissection.)(** Note: Adjust the volume of the digestion buffer accordingly based on the amount required for the corneal dissection procedure on the day. A central stromal tissue from one human cornea can typically be digested in 1 mL of digestion buffer working solution.)
3.2. CSK Basal Medium
- (1)
- Add 47.45 mL of DMEM/F-12 to a 50 mL centrifuge tube.
- (2)
- Add 50 μL of 100× MEM insulin-transferrin-selenium to the tube *.
- (3)
- Add 500 μL of 100× MEM non-essential amino acids solution to the mixture *.
- (4)
- Add 500 μL of 100× MEM vitamin solution to the mixture *.
- (5)
- Add 500 of 100× antibiotic-antimycotic to the mixture *.
- (6)
- Add 1 mL of 50× MEM amino acids solution to the tube and gently mix it *.(* Note: Step 2 dilutes the reagent to 0.1x concentration. Steps 3–6 dilute the reagents to 1x concentration.)
- (7)
- Adjust pH with 1N NaOH dropwise to ~pH7 (the media will change from yellow to orange-red, similar to the stock DMEM/F-12).
3.3. 100 μg/mL IGF-1
- (1)
- Add 200 μL of 1× PBS to 20 μg of IGF-1 in a 1.5 mL microcentrifuge tube.
- (2)
- Resuspend by pipetting the solution up and down until the powder is dissolved.
- (3)
- Store at 4 °C until use.
3.4. 10 mM ROCK Inhibitor (Y-27632)
- (1)
- Dissolve 2 mg of StemMACSTM Y-27632 in 624.4 μL of DMSO in a 1.5 mL microcentrifuge tube.
- (2)
- Resuspend by pipetting the solution up and down until the powder is dissolved.
- (3)
- Aliquot 100 μL of 10 mM StemMACSTM Y-27632 into 0.6 mL microcentrifuge tubes.
- (4)
- Store at −20 °C until use *.(* Note: Once thawed, store the solution at 4 °C and avoid repeat freeze–thawing.)
3.5. 50 mM L-ascorbic 2-phosphate
- (1)
- Weigh 145 mg of L-ascorbic-2-phosphate sesquimagnesium salt hydrate powder into a 15 mL centrifuge tube.
- (2)
- Add 8 mL of deionized water and mix until the powder is dissolved.
- (3)
- Fill the tube to the 10 mL mark with deionized water.
- (4)
- Filter the L-ascorbic 2-phosphate solution using a Minisart® RC syringe filter attached to a 10 mL syringe.
- (5)
- Aliquot 1 mL of the filtered solution to 1.5 mL microcentrifuge tubes.
- (6)
- Store at −20 °C until use *.(*Note: Once thawed, store the solution at 4 °C and avoid repeat freeze–thawing.)
3.6. CSK Complete Medium
- (1)
- Add 9.6 mL of CSK basal medium in a 15 mL centrifuge tube.
- (2)
- Add 1 μL of 100 μg/mL IGF-1 to the tube to obtain a final concentration of 10 ng/mL *.
- (3)
- Add 10 μL of 10 mM Y-27632 to obtain a final concentration of 10 μM *.
- (4)
- Add 50 of heat-inactivated FBS to obtain a final concentration of 0.5% *.
- (5)
- Add 100 μL of 50 mM L-ascorbic 2-phosphate to obtain a final concentration of 0.5 mM *.
- (6)
- Add 5 μg/mL of AME and mix well (the AME preparation protocol will be elaborated in the Protocol section) *.(* Note: All supplements/growth factors should be added immediately before culture).
4. Detailed Procedure
4.1. Overview of CSK Culture Procedure
4.2. Extraction of Proteins from Amniotic Membrane
- (1)
- For fresh amniotic membrane, proceed to step 5.
- (2)
- For cryopreserved amniotic membrane, thaw the vial containing the amniotic membrane at 4 °C overnight.
- (3)
- Prepare the tissue culture hood by UV sterilization for 15 min, followed by wiping off the work surface with 70% ethanol.
- (4)
- Turn on the germinator and heat-sterilize the forceps, scissors, and spatula. Ensure that forceps, scissors, and spatula have cooled down before handling them.
- (5)
- Wash the amnion 3–5 times in 1× PBS to remove traces of blood (for fresh amnion) or glycerol (for cryopreserved amnion). More washes have to be carried out if the amnion still has traces of blood.
- (6)
- Drain away PBS by squeezing the amnion with the forceps in a downward motion. Repeat this a few times to remove as much PBS as possible for an easier grinding process later (PBS retention can cause ice formation in the amnion tissue when cooled with liquid nitrogen, leading to difficulty in grinding).
- (7)
- Place the amnion in a 60 mm cell culture dish and using mayo scissors, cut it into ~1 cm2 pieces.
- (8)
- Add liquid nitrogen to the mortar and pestle to cool it down.
- (9)
- Transfer 5–8 amnion pieces to the cooled mortar and add liquid nitrogen.
- (10)
- Grind the cooled amnion pieces using pestle into “powder” *, **, ***.(* Note: “Powder” is used as a term. The amnion will not be fully powderized. The grinding process only shears it into smaller fragments).(** Note: Due to the water retained by the membrane, the tissue will be hardened upon the addition of liquid nitrogen and challenging to grind. Allow the liquid nitrogen to evaporate before grinding to soften the amnion somewhat.)(*** Note: Before the membrane starts to become too soft, refill the mortar with liquid nitrogen. Do not let the membrane thaw fully.)
- (11)
- Transfer the amnion “powder” to a pre-weighed 50 mL centrifuge tube. Repeat Steps 8–11 until all amnion pieces have been processed.
- (12)
- Weigh the 50 mL centrifuge tube containing the amnion “powder” and mark down the weight of the “powder”.
- (13)
- Add 3 mL of 1× PBS into the tube per gram of amnion “powder”.
- (14)
- Place the tube on a rotator at a speed of 100 rpm at 4 °C for 48 h.
- (15)
- After 48 h, filter the suspension through a 70 μm cell strainer.
- (16)
- Collect the flow-through filtrate and centrifuge at 3000 rpm for 15 min at 4 °C.
- (17)
- Aliquot the supernatant into 1.5 mL microcentrifuge tubes and centrifuge the tubes at 12,000× g for 15 min at 4 °C.
- (18)
- Collect the clear supernatant (containing AME) and aliquot the supernatant 1 mL each into new 1.5 mL microcentrifuge tubes *.(* Note: Remove 200 μL of the AME filtrate from one of the microcentrifuge tubes for protein assay. Protein concentration can be measured with a Bio-Rad Protein DC Assay according to the manufacturer’s protocol).
- (19)
- Store the AME aliquots at −80 °C until use.
4.3. Isolation of CSKs from Cadaveric Donor Cornea
- (1)
- Place the forceps and surgical blade no. 10 with its holder in the tissue culture hood.
- (2)
- UV-sterilize the hood for 15 min and clean the work surface with 70% ethanol.
- (3)
- Heat sterilize the surgical instruments (forceps and blade holder using germinator and cool them down before dissecting the corneal tissues).
- (4)
- Fill 3 mL of 1× PBS each into four 60 mm tissue culture dishes.
- (5)
- Place the cadaveric donor corneal tissue in one of the 60mm culture dishes.
- (6)
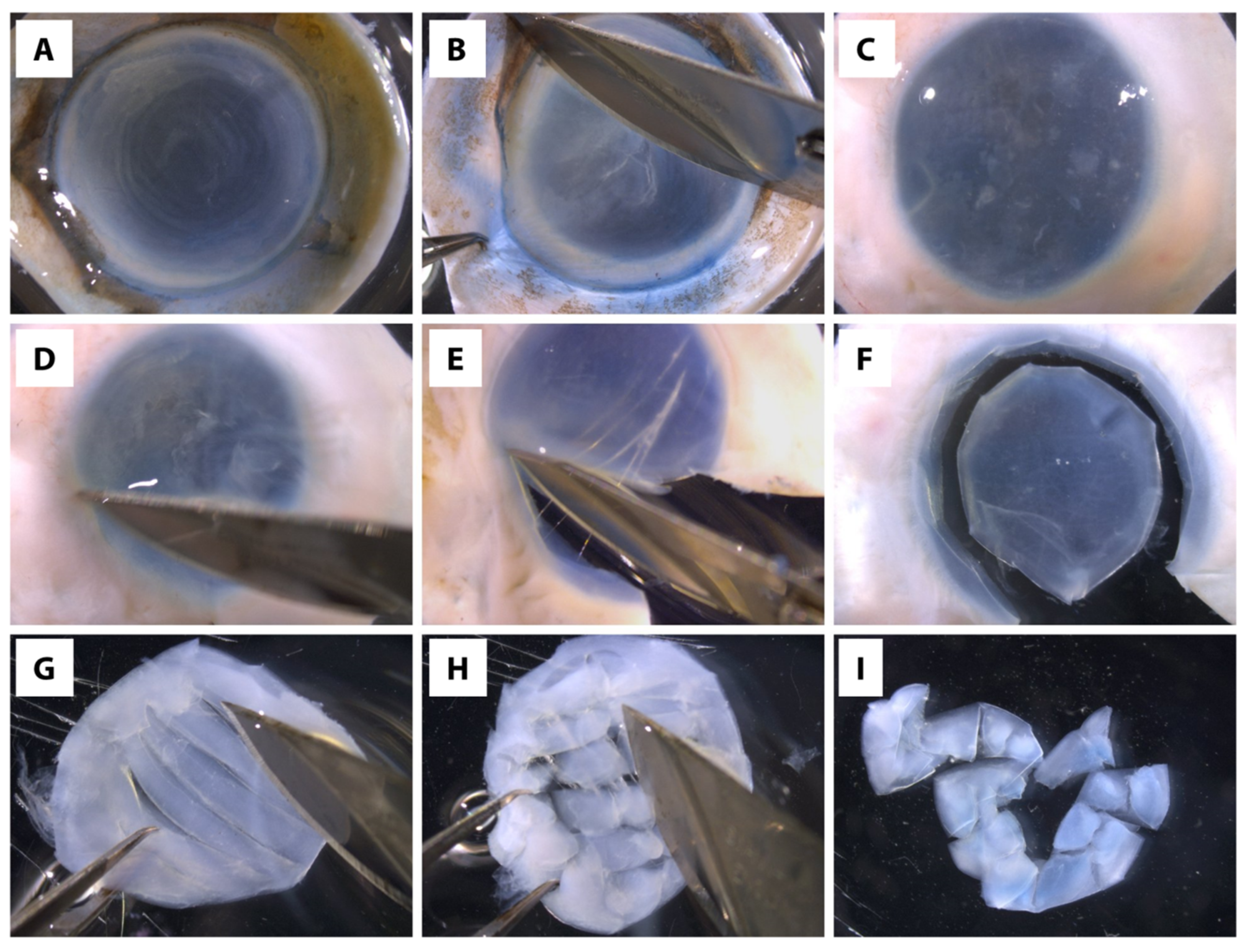
- Scrape off the corneal epithelial and endothelial cells, as well as trabecular meshwork (if any) from the donor tissue using the surgical blade (Figure 2A–D) *, **, ***.(* Note: Important to ascertain the complete removal of corneal epithelial and endothelial cells because these cells grow faster than CSK in culture, and consequently, generating culture with mixed cell types.)(** Note: If difficulties are encountered in removing the corneal epithelial cells, the epithelial scraping can be aided by dipping the cornea in 20 mg/mL dispase solution in DMEM/F-12 for 30 min at 37 °C(*** Note: If isolating CSKs from an eye globe, use mayo scissors to cut around the globe to remove the majority of the sclera, leaving ~3 mm of sclera around the cornea. Scrape top and bottom of the tissue to remove unwanted tissue/cells, such as the epithelial cells, endothelial cells, trabecular meshwork, and iris/ciliary tissues before proceeding to Step 7.)
- (7)
- Place the tissue in a new 60 mm culture dish containing 1× PBS.
- (8)
- Heat sterilize the surgical instruments and cool them down.
- (9)
- Isolate the central clear stroma (7–8 mm diameter) without including any limbal or scleral tissue (Figure 2E,F).*(* Note: Important to ascertain the complete removal of scleral tissue because the scleral fibroblasts can contaminate the CSK culture.)
- (10)
- Place the isolated stroma in a new 60 mm culture dish containing 1× PBS and continue to scrap both surfaces to ensure complete removal of epithelium and endothelium, and no tissues other than the stroma.
- (11)
- Heat sterilize the surgical instruments and cool them down.
- (12)
- Place the stromal tissue in a new 60 mm culture dish containing 1× PBS and cut the tissue into ~1 mm 2 pieces with a surgical blade no. 10 (Figure 2G–I) *.(* Note: It is recommended to leave the edges intact as this allows easier transfer of tissue to the digestion buffer and also to prevent excessive tissue tearing to preserve the CSK viability in the tissue.)
- (13)
- Transfer the cornea to a 15 mL centrifuge tube containing 1 mL of digestion buffer working solution.
- (14)
- Incubate the tube at 37 °C in a 5% CO2 humidified incubator for 10–12 h or until ~90% of tissue has been digested. Do not exceed 16 h because it can be detrimental to the cell viability.
4.4. Culture of Human CSKs
- (1)
- Filter the tissue digest from the preceding section (Isolation of CSKs from cadaveric donor cornea) using a 40 μm cell strainer into a 50 mL centrifuge tube to remove undissociated materials. Following that, wash the tube with 10 mL of 1× PBS once and pass it through the filter.
- (2)
- Add 1× PBS to at least 3 times the volume of the filtrate to dilute the collagenase I.
- (3)
- Centrifuge the solution at 350× g for 7 min at room temperature.
- (4)
- Remove the supernatant *.(* Note: Do not disturb the cell pellet. Because the number of CSKs is typically low, the cell pellet may not be visible.)
- (5)
- Resuspend the cell pellet and add 3 mL of 1× PBS.
- (6)
- Centrifuge the solution at 350× g for 7 min at room temperature.
- (7)
- Remove the supernatant *.(* Note: It is important to ascertain that the collagenase I solution is completely removed before culture. If necessary, repeat Steps 5–7 for complete removal of collagenase I from the system.)
- (8)
- Add CSK complete medium to the tube and disperse the cell pellet by gentle tapping of the tube.*(* Note: The volume of medium to be added depends on the number of corneas that are processed. Cells isolated from one cornea are typically cultured in one well of a BioCoat™ Collagen I 24-well plate. As a guide, 1 mL of medium is usually sufficient for a well of the culture plate.)
- (9)
- Seed the cells in the BioCoat™ Collagen I 24-well plate.
- (10)
- Incubate the cells at 37 °C in a 5% CO2 humidified incubator.
- (11)
- Check the activated CSKs under the microscope and change the CSK complete medium every 3–4 days *, **, ***, ****.(* Note: Cells may take up to 1 week to adhere and start extending their cellular processes. If cells do not grow, continue to observe for up to 3 weeks.)(** Note: For P0 cells, it may take between 2 weeks and 1 month to reach ~70% confluency.)(*** Note: During the first medium change, aspirate the medium with floating cells and transfer it into a new well. These floating cells may still be viable and would attach to the surface of the new well. Add CSK complete medium into the well and observe the cells every 3–4 days.)(**** Note: If the cell number is low, especially during the first week of culture, perform half medium change, i.e., remove 50% volume of the medium from the well and replenish with fresh CSK complete medium of the same volume.)
4.5. Passaging of Human CSKs and Preparation for Cell Therapy
- (1)
- Aspirate the medium from the culture well.
- (2)
- Wash the well with 1× PBS twice.
- (3)
- Add StemPro™ Accutase™ cell dissociation reagent to the well *.(* Note: Ensure that cell dissociation reagent fully cover the well.)
- (4)
- Incubate for 3 min at room temperature *, **.(* Note: Do not leave the cells in the StemPro™ Accutase™ cell dissociation reagent for a prolonged period. If longer incubation is needed, check under microscope every 1 min and stop the reaction once ~90% of cells are detached.)(** Note: If using TrypLE Express, incubate the cells at 37 °C in a 5% CO2 humidified incubator for 3 min.)
- (5)
- Add 1× PBS to at least 3 times the volume of the StemPro™ Accutase™ cell dissociation reagent into the well and flush the well by pipetting up and down to ensure most cells are detached and collected.
- (6)
- Pipette the solution containing activated CSKs or CSKs into a 15 mL centrifuge tube.
- (7)
- Centrifuge the tube at 350× g for 7 min at room temperature.
- (8)
- Remove the supernatant.
- (9)
- Resuspend the cell pellet in 3 mL of 1 × PBS.
- (10)
- Centrifuge the tube at 350× g for 7 min at room temperature.
- (11)
- Remove the supernatant.
- (12)
- For passaging, resuspend the activated CSK pellet in CSK complete medium. Tap the tube lightly to disperse the pellet into a single-cell suspension *.(* Note: For sub-culturing in BioCoat™ Collagen I 24-well plate, add 1 mL of medium per well. For a 6-well plate, add 1.5ml–2ml of medium per well. Proceed to Step 13 to continue the passaging procedure. Proceed to Step 16 to convert activated CSKs into CSKs for tissue engineering or cell therapy applications.)
- (13)
- Pipette the solution containing cells into the well.
- (14)
- Incubate the plate at 37 °C in a 5% CO2 humidified incubator.
- (15)
- Change the media every 2–3 days.
- (16)
- To convert into CSKs, activated CSKs at ~70% confluency at P3–P6 are maintained in the CSK complete medium (without 0.5% FBS) for 7–21 days. Observe the morphology of the cells every 3–4 days. Cells that do not assume long cellular processes and distinct cell bodies are disqualified from further applications. When the required cell number and desired cell morphology are met, single-cell suspension of CSKs can be prepared by repeating Step 1–11 above.
- (17)
- Resuspend the CSK in 1x PBS. Tap the tube lightly to disperse the pellet into a single-cell suspension *.(* Note: To prepare CSKs for cell therapy in vivo, determine the cell number with a hematocytometer or an automated cell counter and calculate the required therapeutic density of the cells before adding the 1× PBS in the tube. Apply the cells in vivo within 30 min of preparation.)
4.6. Cryopreservation of Human CSKs
- (1)
- Aspirate the culture medium from the well.
- (2)
- Wash the well with 1× PBS twice.
- (3)
- Add 200 μL (for 24-well plate) or 400 μL of StemPro™ Accutase™ cell dissociation reagent (for 6-well plate) into each well.
- (4)
- Incubate for 3 min at room temperature *, **.(* Note: Do not leave the cells in the StemPro™ Accutase™ cell dissociation reagent for a prolonged period. If longer incubation is needed, check under microscope every 1 min and stop the dissociation reaction once ~90% of cells have been detached.)(** Note: If using TrypLE Express, incubate the cells at 37 °C in a 5% CO2 humidified incubator for 3 min.)
- (5)
- Add 1× PBS at least 3 times the volume of StemPro™ Accutase™ cell dissociation reagent into the well and flush the well by pipetting up and down to detach the cells.
- (6)
- Collect the cell suspension into a 15 mL centrifuge tube.
- (7)
- Centrifuge the tube at 350× g for 7 min at room temperature.
- (8)
- Discard the supernatant.
- (9)
- Resuspend the cell pellet in 3 mL of 1× PBS.
- (10)
- Centrifuge the tube at 350× g for 7 min at room temperature.
- (11)
- Discard the supernatant.
- (12)
- Resuspend the cell pellet with 450 μL of CSK complete medium.
- (13)
- Add 50 μL of DMSO to obtain a final 10% DMSO concentration in the cell suspension.
- (14)
- Transfer the cell suspension into a 2 mL cryovial.
- (15)
- Place the cryovial in a Mr. FrostyTM freezing container and freeze it at −80 °C for at least 24 h.
- (16)
- Transfer the cryovial under liquid nitrogen for long-term storage.
5. Expected Results
5.1. Cell Morphology
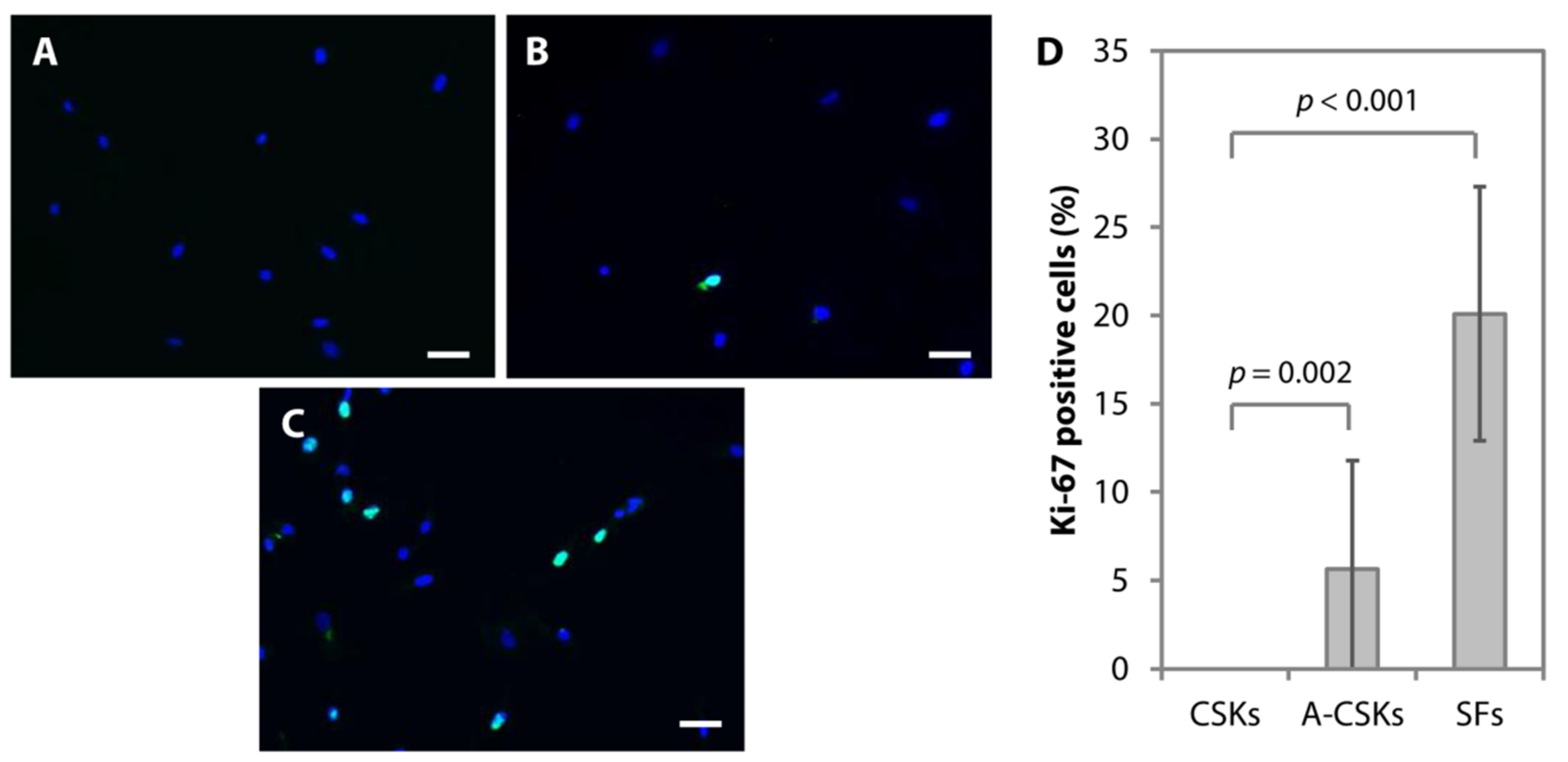
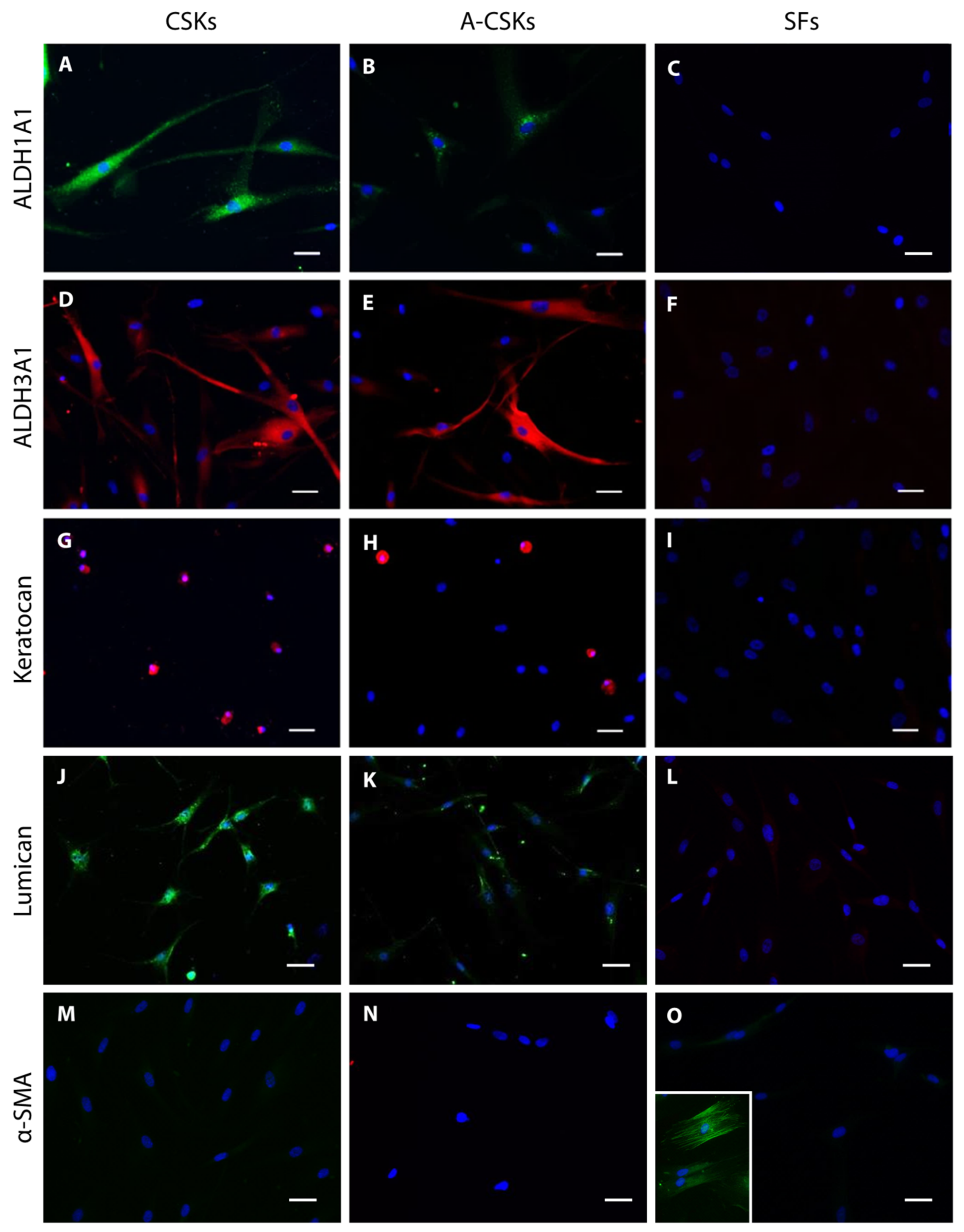
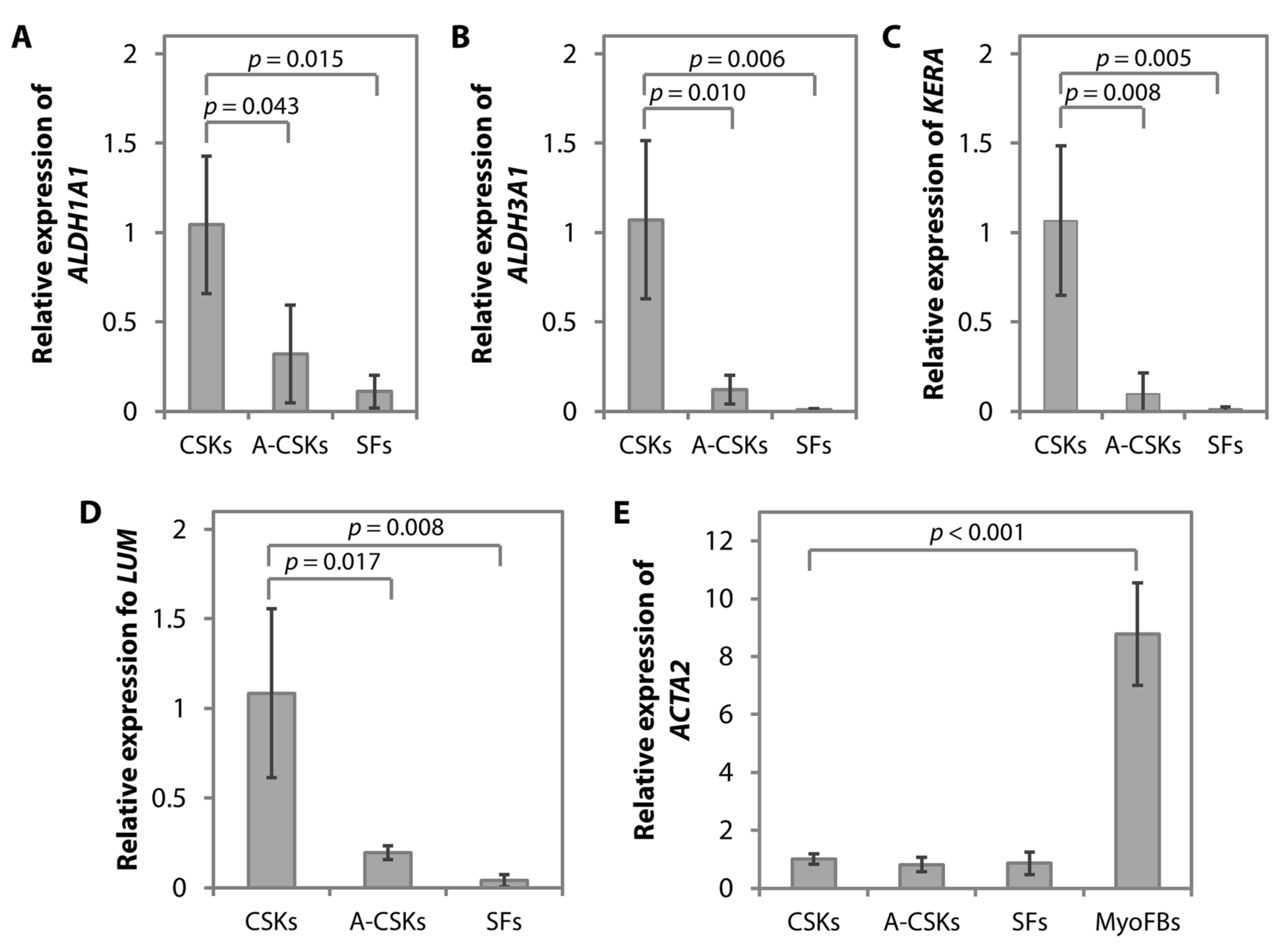
5.2. Protein and Gene Expression
5.3. Troubleshooting Guide
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McLaren, J.W.; Wacker, K.; Kane, K.M.; Patel, S.V. Measuring corneal haze by using Scheimpflug photography and confocal microscopy. Invest. Ophthalmol. Vis. Sci. 2016, 57, 227–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funderburgh, J.L.; Mann, M.M.; Funderburgh, M.L. Keratocyte phenotype mediates proteoglycan structure: A role for fibroblasts in corneal fibrosis. J. Biol. Chem. 2003, 278, 45629–45637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitcher, J.P.; Srinivasan, M.; Upadhyay, M.P. Corneal blindness: A global perspective. Bull. World Health Organ. 2001, 79, 214–221. [Google Scholar]
- Mathews, P.M.; Lindsley, K.; Aldave, A.J.; Akpek, E.K. Etiology of global corneal blindness and current practices of corneal transplantation: A focused review. Cornea 2018, 37, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.T.H.; Dart, J.K.G.; Holland, E.J.; Kinoshita, S. Corneal transplantation. Lancet 2012, 379, 1749–1761. [Google Scholar] [CrossRef]
- Griffith, M.; Polisetti, N.; Kuffova, L.; Gallar, J.; Forrester, J.; Vemuganti, G.K.; Fuchsluger, T.A. Regenerative approaches as alternatives to donor allografting for restoration of corneal function. Ocul. Surf. 2012, 10, 170–183. [Google Scholar] [CrossRef] [Green Version]
- Yam, G.H.-F.; Fuest, M.; Yusoff, N.Z.B.M.; Goh, T.-W.; Bandeira, F.; Setiawan, M.; Seah, X.-Y.; Lwin, N.-C.; Stanzel, T.P.; Ong, H.-S.; et al. Safety and feasibility of intrastromal injection of cultivated human corneal stromal keratocytes as cell-based therapy for corneal opacities. Invest. Ophthalmol. Vis. Sci. 2018, 59, 3340–3354. [Google Scholar] [CrossRef] [Green Version]
- West-Mays, J.A.; Dwivedi, D.J. The keratocyte: Corneal stromal cell with variable repair phenotypes. Int. J. Biochem. Cell Biol. 2006, 38, 1625–1631. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Du, Y.; Mann, M.M.; Funderburgh, J.L.; Wagner, W.R. Corneal stromal stem cells versus corneal fibroblasts in generating structurally appropriate corneal stromal tissue. Exp. Eye Res. 2014, 120, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Yam, G.H.F.; Riau, A.K.; Funderburgh, M.L.; Mehta, J.S.; Jhanji, V. Keratocyte biology. Exp. Eye Res. 2020, 196, 108062. [Google Scholar] [CrossRef]
- Yam, G.H.-F.; Yusoff, N.Z.B.M.; Kadaba, A.; Tian, D.; Myint, H.H.; Beuerman, R.W.; Zhou, L.; Mehta, J.S. Ex vivo propagation of human corneal stromal “activated keratocytes” for tissue engineering. Cell Transplant. 2015, 24, 1845–1861. [Google Scholar] [CrossRef]
- Riau, A.K.; Mondal, D.; Setiawan, M.; Palaniappan, A.; Yam, G.H.F.; Liedberg, B.; Venkatraman, S.S.; Mehta, J.S. Functionalization of the polymeric surface with bioceramic nanoparticles via a novel, nonthermal dip coating method. ACS Appl. Mater. Interfaces 2016, 8, 35565–35577. [Google Scholar] [CrossRef]
- Hasenzahl, M.; Müsken, M.; Mertsch, S.; Schrader, S.; Reichl, S. Cell sheet technology: Influence of culture conditions on in vitro-cultivated corneal stromal tissue for regenerative therapies of the ocular surface. J. Biomed. Mater. Res. Part. B Appl. Biomater. 2021, 109, 1488–1504. [Google Scholar] [CrossRef]
- Sidney, L.E.; Hopkinson, A. Corneal keratocyte transition to mesenchymal stem cell phenotype and reversal using serum-free medium supplemented with fibroblast growth factor-2, transforming growth factor-β3 and retinoic acid. J. Tissue Eng. Regen. Med. 2018, 12, e203–e215. [Google Scholar] [CrossRef]
- Berryhill, B.L.; Kader, R.; Kane, B.; Birk, D.E.; Feng, J.; Hassell, J.R. Partial restoration of the keratocyte phenotype to bovine keratocytes made fibroblastic by serum. Invest. Ophthalmol. Vis. Sci. 2002, 43, 3416–3421. [Google Scholar] [PubMed]
- Jester, J.V.; Budge, A.; Fisher, S.; Huang, J. Corneal keratocytes: Phenotypic and species differences in abundant protein expression and in vitro light-scattering. Invest. Ophthalmol. Vis. Sci. 2005, 46, 2369–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Bazan, H.E.P. Epidermal growth factor synergism with TGF-beta1 via PI-3 kinase activity in corneal keratocyte differentiation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2936–2945. [Google Scholar] [CrossRef] [Green Version]
- Polisetty, N.; Fatima, A.; Madhira, S.L.; Sangwan, V.S.; Vemuganti, G.K. Mesenchymal cells from limbal stroma of human eye. Mol. Vis. 2008, 14, 431–442. [Google Scholar] [PubMed]
- Branch, M.J.; Hashmani, K.; Dhillon, P.; Jones, D.R.E.; Dua, H.S.; Hopkinson, A. Mesenchymal stem cells in the human corneal limbal stroma. Invest. Ophthalmol. Vis. Sci. 2012, 53, 5109–5116. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Funderburgh, M.L.; Mann, M.M.; SundarRaj, N.; Funderburgh, J.L. Multipotent stem cells in human corneal stroma. Stem Cells 2005, 23, 1266–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funderburgh, J.L.; Funderburgh, M.L.; Du, Y. Stem cells in the limbal stroma. Ocul. Surf. 2016, 14, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Hashmani, K.; Branch, M.J.; Sidney, L.E.; Dhillon, P.S.; Verma, M.; McIntosh, O.D.; Hopkinson, A.; Dua, H.S. Characterization of corneal stromal stem cells with the potential for epithelial transdifferentiation. Stem Cell Res. Ther. 2013, 4, 75. [Google Scholar] [CrossRef] [Green Version]
- Fenner, B.J.; Yusoff, N.Z.B.M.; Fuest, M.; Zhou, L.; Bandeira, F.; Cajucom-Uy, H.Y.; Tan, H.K.; Mehta, J.S.; Yam, G.H.F. A cellular and proteomic approach to assess proteins extracted from cryopreserved human amnion in the cultivation of corneal stromal keratocytes for stromal cell therapy. Eye Vis. 2019, 6, 30. [Google Scholar] [CrossRef]
- Chen, J.; Guerriero, E.; Sado, Y.; SundarRaj, N. Rho-mediated regulation of TGF-beta1- and FGF-2-induced activation of corneal stromal keratocytes. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3662–3670. [Google Scholar] [CrossRef] [Green Version]
- Jester, J.V.; Ho-Chang, J. Modulation of cultured corneal keratocyte phenotype by growth factors/cytokines control in vitro contractility and extracellular matrix contraction. Exp. Eye Res. 2003, 77, 581–592. [Google Scholar] [CrossRef]
- Yam, G.H.-F.; Teo, E.P.-W.; Setiawan, M.; Lovatt, M.J.; Yusoff, N.Z.B.M.; Fuest, M.; Goh, B.-T.; Mehta, J.S. Postnatal periodontal ligament as a novel adult stem cell source for regenerative corneal cell therapy. J. Cell. Mol. Med. 2018, 22, 3119–3132. [Google Scholar] [CrossRef]
- Koob, T.J.; Lim, J.J.; Massee, M.; Zabek, N.; Rennert, R.; Gurtner, G.; Li, W.W. Angiogenic properties of dehydrated human amnion/chorion allografts: Therapeutic potential for soft tissue repair and regeneration. Vasc. Cell. 2014, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Dudok, D.V.; Nagdee, I.; Cheung, K.; Liu, H.; Vedovelli, L.; Ghinelli, E.; Kenyon, K.; Parapuram, S.; Hutnik, C.M. Effects of amniotic membrane extract on primary human corneal epithelial and limbal cells. Clin. Experiment. Ophthalmol. 2015, 43, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.Y.; Kim, S.E.; Cho, G.J.; Chae, S.-W.; Song, J.-J. Differential effects of amnion and chorion membrane extracts on osteoblast-like cells due to the different growth factor composition of the extracts. PLoS ONE. 2017, 12, e0182716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; He, H.; Chen, Y.-T.; Hayashida, Y.; Tseng, S.C.G. Reversal of myofibroblasts by amniotic membrane stromal extract. J. Cell. Physiol. 2008, 215, 657–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.B.; Li, D.Q.; Tan, D.T.; Meller, D.C.; Tseng, S.C. Suppression of TGF-beta signaling in both normal conjunctival fibroblasts and pterygial body fibroblasts by amniotic membrane. Curr. Eye Res. 2000, 20, 325–334. [Google Scholar] [CrossRef]
- Kawakita, T.; Espana, E.M.; He, H.; Hornia, A.; Yeh, L.-K.; Ouyang, J.; Liu, C.-Y.; Tseng, S.C.G. Keratocan expression of murine keratocytes is maintained on amniotic membrane by down-regulating transforming growth factor-beta signaling. J. Biol. Chem. 2005, 280, 27085–27092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dua, H.S.; Gomes, J.A.P.; King, A.J.; Maharajan, V.S. The amniotic membrane in ophthalmology. Surv. Ophthalmol. 2004, 49, 51–77. [Google Scholar] [CrossRef]
- Tseng, S.C.G.; Espana, E.M.; Kawakita, T.; Di Pascuale, M.A.; Li, W.; He, H.; Liu, T.-S.; Cho, T.-H.; Gao, Y.-Y.; Yeh, L.-K.; et al. How does amniotic membrane work? Ocul. Surf. 2004, 2, 177–187. [Google Scholar] [CrossRef]
- Castro-Combs, J.; Noguera, G.; Cano, M.; Yew, M.; Gehlbach, P.L.; Palmer, J.; Behrens, A. Corneal wound healing is modulated by topical application of amniotic fluid in an ex vivo organ culture model. Exp. Eye Res. 2008, 87, 56–63. [Google Scholar] [CrossRef]
- Yadav, M.K.; Go, Y.Y.; Kim, S.H.; Chae, S.-W.; Song, J.-J. Antimicrobial and antibiofilm effects of human amniotic/chorionic membrane extract on Streptococcus pneumoniae. Front. Microbiol. 2017, 8, 1948. [Google Scholar] [CrossRef] [PubMed]
- Kruse, F.E.; Joussen, A.M.; Rohrschneider, K.; You, L.; Sinn, B.; Baumann, J.; Völcker, H.E. Cryopreserved human amniotic membrane for ocular surface reconstruction. Graefes. Arch. Clin. Exp. Ophthalmol. 2000, 238, 68–75. [Google Scholar] [CrossRef]
- Lim, L.S.; Poh, R.W.Y.; Riau, A.K.; Beuerman, R.W.; Tan, D.; Mehta, J.S. Biological and ultrastructural properties of Acelagraft, a freeze-dried γ-irradiated human amniotic membrane. Arch. Ophthalmol. 2010, 128, 1303–1310. [Google Scholar] [CrossRef] [Green Version]
- Riau, A.K.; Aung, T.T.; Setiawan, M.; Yang, L.; Yam, G.H.F.; Beuerman, R.W.; Venkatraman, S.S.; Mehta, J.S. Surface immobilization of nano-silver on polymeric medical devices to prevent bacterial biofilm formation. Pathogens 2019, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Miotto, M.; Gouveia, R.; Abidin, F.Z.; Figueiredo, F.; Connon, C.J. Developing a continuous bioprocessing approach to stromal cell manufacture. ACS Appl. Mater. Interfaces 2017, 9, 41131–41142. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.N.M.; Dehsorkhi, A.; Hamley, I.W.; Connon, C.J. Supra-molecular assembly of a lumican-derived peptide amphiphile enhances its collagen-stimulating activity. Biomater. Sci. 2016, 4, 346–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Problem Encountered | Explanations | Solutions |
|---|---|---|
| Low amount of total protein in AME | Amnion proteins are degraded | • Ensure that the human amnion is placed in 4 °C or on ice before and during extraction. |
| • Store AME immediately after extraction procedure at −80 °C and thaw only when required. | ||
| Loss of proteins during processing | • Ensure that amnion is processed as soon as possible from the time of collection/harvest. | |
| The sample is diluted with a large volume of PBS after grinding | • After filtering, wash the strainer with sufficient PBS to ensure most soluble proteins are collected. | |
| • Reduce the volume of PBS added to the tube containing the AME “powder”. | ||
| The sample is obtained from the anterior part of the amnion sac where the stroma is the thinnest | • Collect protein from the more posterior part with thicker stroma; however, avoid collecting the vascularized tissue. | |
| Corneal tissue is not or only partially digested | Collagenase I might have been degraded | • Prepare collagenase I digestion fresh. |
| • Ensure proper storage of the stock solution, as well as the collagenase I powder. | ||
| Insufficient digestion | • Prolong the incubation time but do not exceed 24 h in 0.1% collagenase I. | |
| • A higher concentration of collagenase I (0.2–0.3%) can be used to digest the corneal tissue but do not subject the tissue to digestion exceeding 10 h | ||
| Epithelial cell contamination in CSK culture | Insufficient scraping of the anterior (epithelial side) of the cornea | • Check under the microscope to ensure that sufficient scraping of the corneal epithelial cells, including the limbal area, has been performed. |
| • If difficulties in removing the epithelial cells from the cornea are encountered, pre-treatment with dispase could be performed. | ||
| Bacterial contamination | • Place the surgical instruments used in the corneal dissection in the germinator for at least 30 s before each step. | |
| Epithelial cell growth in culture | • Ensure that limbal epithelial cells, if any, are completely removed from the sclera. | |
| • Cut ~2mm into the cornea, away from the sclera to ensure no contamination from the limbal epithelial cells. | ||
| • Partial trypsinization can be performed in culture. Epithelial cells generally take longer (5–7 min) to be detached from the plate in the presence of dissociation reagent, compared to the CSKs (3–5 min). Earlier termination of the dissociation reactions would allow the majority of the CSKs to be lifted, while the epithelial cells are still attached to the plate. | ||
| CSKs are not viable | Status/condition of the donor cornea is not optimal for culture (i.e., donor’s age, disease status, etc.) | • Obtain only healthy corneas from younger donors (set a cutoff age of 70 years old), if possible. The younger the donors, the higher the number of viable cells. |
| Prolonged storage of donor cornea in Optisol before CSK isolation | • Process the cornea as soon as possible upon receiving the tissue. | |
| A prolonged period of tissue digestion in collagenase I | • Do not exceed 24 h of digestion in 0.1% collagenase I. | |
| • If using higher concentrations of collagenase (0.2–0.3%), do not exceed 10 h of incubation. | ||
| Reagents used in the culture are not in optimal conditions | • Ensure that the correct concentrations of reagents are prepared. | |
| • Ensure that the reagents have not expired. | ||
| • Ensure reagents are stored correctly and according to the manufacturers’ instructions. | ||
| • Reagents may have degraded. Prepare the stock solutions fresh. | ||
| • Always prepare CSK media (with supplements) fresh. If prepared in advance, keep it at 4 °C for no more than one week. | ||
| Microbial contamination | Aseptic techniques are not observed | • Ensure that work surfaces are wiped with 70% ethanol, surgical instruments are heat-sterilized and all materials and reagents used are sterile. |
| • If contamination is minimal, remove the media and wash thrice with 1× PBS with antibiotic-antimycotic. Add media containing 2× antibiotic-antimycotic. | ||
| Donor corneal tissue is infected | • If the cornea looks cloudy/hazy, do not process. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
binte M. Yusoff, N.Z.; Riau, A.K.; Yam, G.H.F.; binte Halim, N.S.H.; Mehta, J.S. Isolation and Propagation of Human Corneal Stromal Keratocytes for Tissue Engineering and Cell Therapy. Cells 2022, 11, 178. https://doi.org/10.3390/cells11010178
binte M. Yusoff NZ, Riau AK, Yam GHF, binte Halim NSH, Mehta JS. Isolation and Propagation of Human Corneal Stromal Keratocytes for Tissue Engineering and Cell Therapy. Cells. 2022; 11(1):178. https://doi.org/10.3390/cells11010178
Chicago/Turabian Stylebinte M. Yusoff, Nur Zahirah, Andri K. Riau, Gary H. F. Yam, Nuur Shahinda Humaira binte Halim, and Jodhbir S. Mehta. 2022. "Isolation and Propagation of Human Corneal Stromal Keratocytes for Tissue Engineering and Cell Therapy" Cells 11, no. 1: 178. https://doi.org/10.3390/cells11010178
APA Stylebinte M. Yusoff, N. Z., Riau, A. K., Yam, G. H. F., binte Halim, N. S. H., & Mehta, J. S. (2022). Isolation and Propagation of Human Corneal Stromal Keratocytes for Tissue Engineering and Cell Therapy. Cells, 11(1), 178. https://doi.org/10.3390/cells11010178