
Stem Cell-Based Disease Models for Inborn Errors of Immunity



Abstract
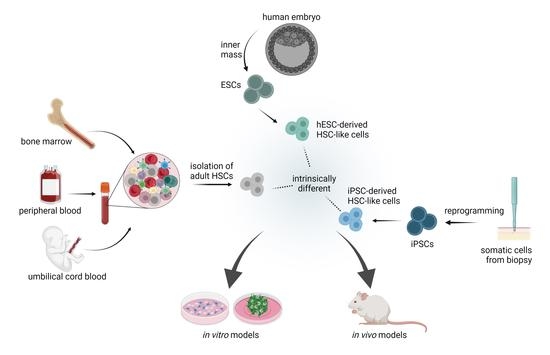
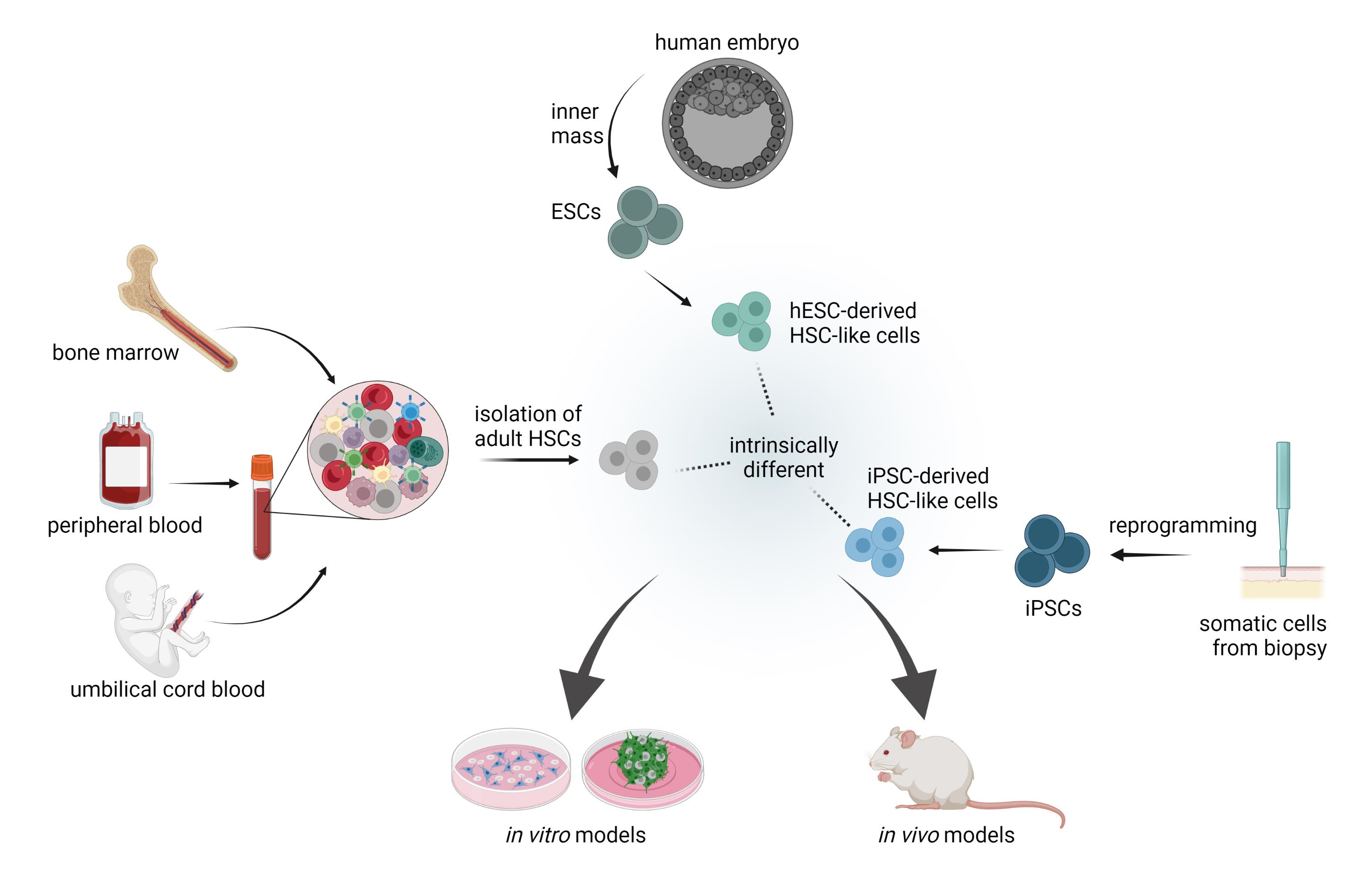
1. Introduction
2. Paradigm of HSCs Origin and Potency: Implications for Disease Modeling
3. HSC-Based Immune Disease Modelling
3.1. Source of Stem Cells: Important Considerations
3.1.1. Adult Hematopoeitic Stem Cells
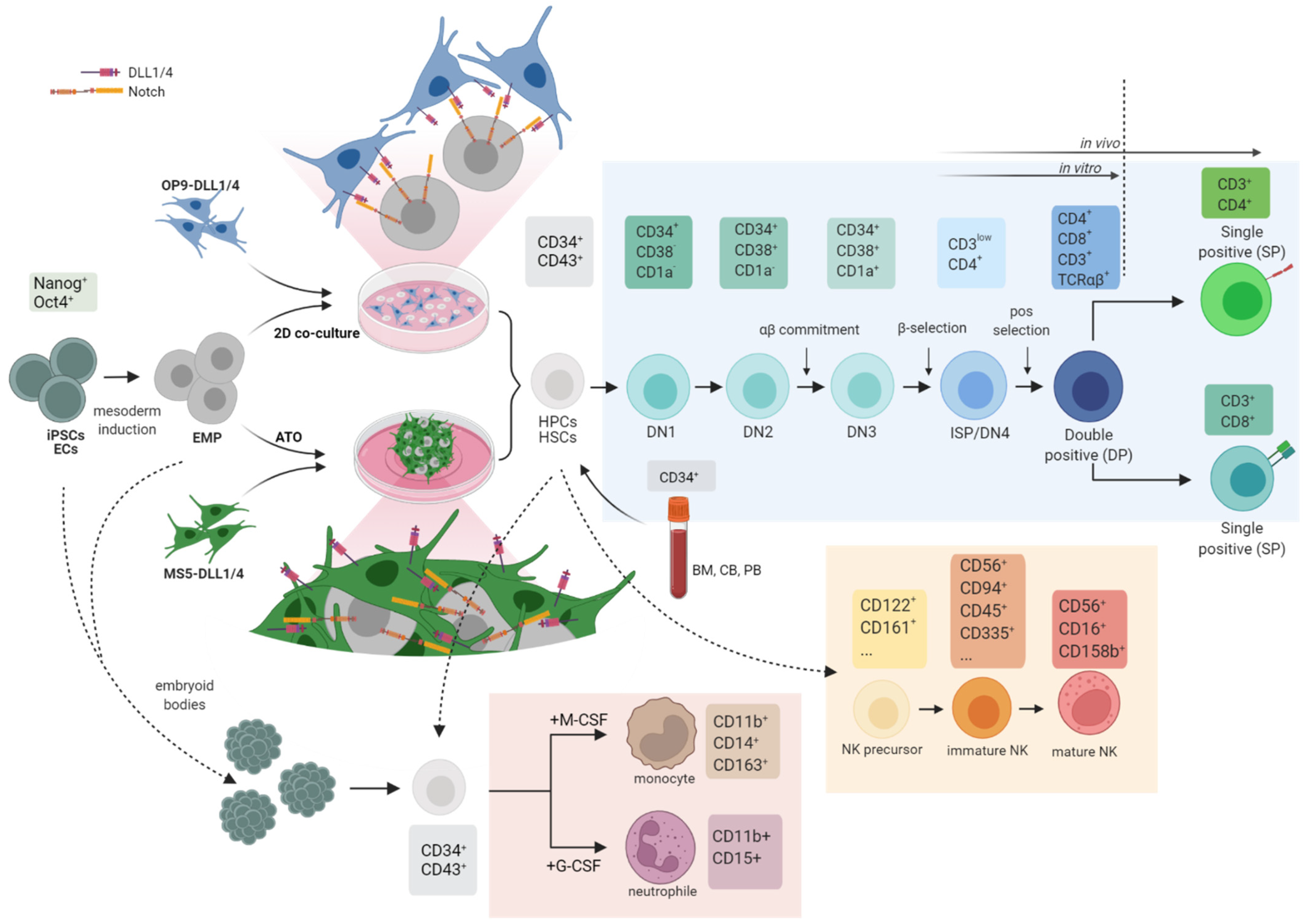
3.1.2. Differentiation Potential and De Novo Generation of HSCs from iPSCs
3.2. In Vitro Inborn Errors of Immunity (IEI) Disease Models
3.3. In Vivo Disease Models: Towards a Fully Hummanized Immune System
4. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2020, 40, 24–64. [Google Scholar] [CrossRef] [PubMed]
- Copelan, E.A. Hematopoietic Stem-Cell Transplantation. N. Engl. J. Med. 2006, 354, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Duarte, R.F.; Labopin, M.; Bader, P.; Basak, G.W.; Bonini, C.; Chabannon, C.; Corbacioglu, S.; Dreger, P.; Dufour, C.; Gennery, A.R.; et al. Indications for haematopoietic stem cell transplantation for haematological diseases, solid tumours and immune disorders: Current practice in Europe, 2019. Bone Marrow Transplant. 2019, 54, 1525–1552. [Google Scholar] [CrossRef] [PubMed]
- Jagasia, M.; Arora, M.; Flowers, M.E.D.; Chao, N.J.; McCarthy, P.L.; Cutler, C.S.; Urbano-Ispizua, A.; Pavletic, S.Z.; Haagenson, M.D.; Zhang, M.-J.; et al. Risk factors for acute GVHD and survival after hematopoietic cell transplantation. Blood 2012, 119, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Cring, M.R.; Sheffield, V.C. Gene therapy and gene correction: Targets, progress, and challenges for treating human diseases. Gene Ther. 2020, 1–10. [Google Scholar] [CrossRef]
- Eaves, C.J. Hematopoietic stem cells: Concepts, definitions, and the new reality. Blood 2015, 125, 2605–2613. [Google Scholar] [CrossRef]
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. WIREs Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef]
- Demirci, S.; Leonard, A.; Tisdale, J.F. Hematopoietic stem cells from pluripotent stem cells: Clinical potential, challenges, and future perspectives. Stem Cells Transl. Med. 2020, 9, 1549–1557. [Google Scholar] [CrossRef]
- Palis, J.; Robertson, S.; Kennedy, M.; Wall, C.; Keller, G. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development 1999, 126, 5073–5084. [Google Scholar] [CrossRef]
- Lin, Y.; Yoder, M.; Yoshimoto, M. Lymphoid Progenitor Emergence in the Murine Embryo and Yolk Sac Precedes Stem Cell Detection. Stem Cells Dev. 2014, 23, 1168–1177. [Google Scholar] [CrossRef]
- McGrath, K.E.; Frame, J.M.; Fegan, K.H.; Bowen, J.R.; Conway, S.J.; Catherman, S.C.; Kingsley, P.D.; Koniski, A.D.; Palis, J. Distinct Sources of Hematopoietic Progenitors Emerge before HSCs and Provide Functional Blood Cells in the Mammalian Embryo. Cell Rep. 2015, 11, 1892–1904. [Google Scholar] [CrossRef]
- Müller, A.M.; Medvinsky, A.; Strouboulis, J.; Grosveld, F.; Dzierzakt, E. Development of hematopoietic stem cell activity in the mouse embryo. Immunity 1994, 1, 291–301. [Google Scholar] [CrossRef]
- Medvinsky, A.; Dzierzak, E. Definitive Hematopoiesis Is Autonomously Initiated by the AGM Region. Cell 1996, 86, 897–906. [Google Scholar] [CrossRef]
- Moore, M.A.S.; Metcalf, D. Ontogeny of the Haemopoietic System: Yolk Sac Origin of In Vivo and In Vitro Colony Forming Cells in the Developing Mouse Embryo. Br. J. Haematol. 1970, 18, 279–296. [Google Scholar] [CrossRef]
- Gekas, C.; Dieterlen-Lièvre, F.; Orkin, S.H.; Mikkola, H.K. The Placenta Is a Niche for Hematopoietic Stem Cells. Dev. Cell 2005, 8, 365–375. [Google Scholar] [CrossRef]
- Cumano, A.; Dieterlen-Lievre, F.; Godin, I. Lymphoid Potential, Probed before Circulation in Mouse, Is Restricted to Caudal Intraembryonic Splanchnopleura. Cell 1996, 86, 907–916. [Google Scholar] [CrossRef]
- Cumano, A.; Ferraz, J.C.; Klaine, M.; Di Santo, J.; Godin, I. Intraembryonic, but Not Yolk Sac Hematopoietic Precursors, Isolated before Circulation, Provide Long-Term Multilineage Reconstitution. Immunity 2001, 15, 477–485. [Google Scholar] [CrossRef]
- Samokhvalov, I.M.; Samokhvalova, N.I.; Nishikawa, S.-I. Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature 2007, 446, 1056–1061. [Google Scholar] [CrossRef]
- Tanaka, Y.; Hayashi, M.; Kubota, Y.; Nagai, H.; Sheng, G.; Nishikawa, S.-I.; Samokhvalov, I.M. Early ontogenic origin of the hematopoietic stem cell lineage. Proc. Natl. Acad. Sci. USA 2012, 109, 4515–4520. [Google Scholar] [CrossRef]
- Lee, L.K.; Ghorbanian, Y.; Wang, W.; Wang, Y.; Kim, Y.J.; Weissman, I.L.; Inlay, M.A.; Mikkola, H.K. LYVE1 Marks the Divergence of Yolk Sac Definitive Hemogenic Endothelium from the Primitive Erythroid Lineage. Cell Rep. 2016, 17, 2286–2298. [Google Scholar] [CrossRef]
- Wittamer, V.; Bertrand, J.Y. Yolk sac hematopoiesis: Does it contribute to the adult hematopoietic system? Cell. Mol. Life Sci. 2020, 77, 4081–4091. [Google Scholar] [CrossRef]
- Senserrich, J.; Batsivari, A.; Rybtsov, S.; Gordon-Keylock, S.; Souilhol, C.; Buchholz, F.; Hills, D.; Zhao, S.; Medvinsky, A. Analysis of Runx1 Using Induced Gene Ablation Reveals Its Essential Role in Pre-liver HSC Development and Limitations of an In Vivo Approach. Stem Cell Rep. 2018, 11, 784–794. [Google Scholar] [CrossRef]
- Taoudi, S.; Medvinsky, A. Functional identification of the hematopoietic stem cell niche in the ventral domain of the embryonic dorsal aorta. Proc. Natl. Acad. Sci. USA 2007, 104, 9399–9403. [Google Scholar] [CrossRef]
- Kumaravelu, P.; Hook, L.; Morrison, A.M.; Ure, J.; Zhao, S.; Zuyev, S.; Ansell, J.; Medvinsky, A. Quantitative developmental anatomy of definitive haematopoietic stem cells/long-term repopulating units (HSC/RUs): Role of the aorta-gonad-mesonephros (AGM) region and the yolk sac in colonisation of the mouse embryonic liver. Development 2002, 129, 4891–4899. [Google Scholar] [CrossRef] [PubMed]
- Rybtsov, S.; Sobiesiak, M.; Taoudi, S.; Souilhol, C.; Senserrich, J.; Liakhovitskaia, A.; Ivanovs, A.; Frampton, J.; Zhao, S.; Medvinsky, A. Hierarchical organization and early hematopoietic specification of the developing HSC lineage in the AGM region. J. Exp. Med. 2011, 208, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Liu, F. Fetal liver: An ideal niche for hematopoietic stem cell expansion. Sci. China Life Sci. 2018, 61, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Ema, H.; Nakauchi, H. Expansion of hematopoietic stem cells in the developing liver of a mouse embryo. Blood 2000, 95, 2284–2288. [Google Scholar] [CrossRef] [PubMed]
- Yvernogeau, L.; Gautier, R.; Petit, L.; Khoury, H.; Relaix, F.; Ribes, V.; Sang, H.; Charbord, P.; Souyri, M.; Robin, C.; et al. In vivo generation of haematopoietic stem/progenitor cells from bone marrow-derived haemogenic endothelium. Nat. Cell Biol. 2019, 21, 1334–1345. [Google Scholar] [CrossRef] [PubMed]
- Beaudin, A.E.; Boyer, S.W.; Perez-Cunningham, J.; Hernandez, G.E.; Derderian, S.C.; Jujjavarapu, C.; Aaserude, E.; MacKenzie, T.; Forsberg, E.C. A Transient Developmental Hematopoietic Stem Cell Gives Rise to Innate-like B and T Cells. Cell Stem Cell 2016, 19, 768–783. [Google Scholar] [CrossRef]
- Fraser, S.T.; Ogawa, M.; Yu, R.T.; Nishikawa, S.; Yoder, M.C.; Nishikawa, S.-I. Definitive hematopoietic commitment within the embryonic vascular endothelial-cadherin+ population. Exp. Hematol. 2002, 30, 1070–1078. [Google Scholar] [CrossRef]
- Zovein, A.C.; Hofmann, J.J.; Lynch, M.; French, W.J.; Turlo, K.A.; Yang, Y.; Becker, M.S.; Zanetta, L.; Dejana, E.; Gasson, J.C.; et al. Fate Tracing Reveals the Endothelial Origin of Hematopoietic Stem Cells. Cell Stem Cell 2008, 3, 625–636. [Google Scholar] [CrossRef]
- Boisset, J.-C.; Van Cappellen, W.; Andrieu-Soler, C.; Galjart, N.; Dzierzak, E.; Robin, C. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature 2010, 464, 116–120. [Google Scholar] [CrossRef]
- Ivanovs, A.; Rybtsov, S.; Anderson, R.A.; Turner, M.L.; Medvinsky, A. Identification of the Niche and Phenotype of the First Human Hematopoietic Stem Cells. Stem Cell Rep. 2014, 2, 449–456. [Google Scholar] [CrossRef]
- Bertrand, J.; Giroux, S.; Golub, R.; Klaine, M.; Jalil, A.; Boucontet, L.; Godin, I.; Cumano, A. Characterization of purified intraembryonic hematopoietic stem cells as a tool to define their site of origin. Proc. Natl. Acad. Sci. USA 2004, 102, 134–139. [Google Scholar] [CrossRef]
- de Bruijn, M.F.; Ma, X.; Robin, C.; Ottersbach, K.; Sánchez, M.J.; Dzierzak, E. Hematopoietic Stem Cells Localize to the Endothelial Cell Layer in the Midgestation Mouse Aorta. Immunity 2002, 16, 673–683. [Google Scholar] [CrossRef]
- Taoudi, S.; Morrison, A.M.; Inoue, H.; Gribi, R.; Ure, J.; Medvinsky, A. Progressive divergence of definitive haematopoietic stem cells from the endothelial compartment does not depend on contact with the foetal liver. Development 2005, 132, 4179–4191. [Google Scholar] [CrossRef]
- Yoder, M.C.; Hiatt, K.; Dutt, P.; Mukherjee, P.; Bodine, D.M.; Orlic, D. Characterization of Definitive Lymphohematopoietic Stem Cells in the Day 9 Murine Yolk Sac. Immunity 1997, 7, 335–344. [Google Scholar] [CrossRef]
- Yoder, M.C.; Hiatt, K.; Mukherjee, P. In vivo repopulating hematopoietic stem cells are present in the murine yolk sac at day 9.0 postcoitus. Proc. Natl. Acad. Sci. USA 1997, 94, 6776–6780. [Google Scholar] [CrossRef]
- Taoudi, S.; Gonneau, C.; Moore, K.; Sheridan, J.M.; Blackburn, C.C.; Taylor, E.; Medvinsky, A. Extensive Hematopoietic Stem Cell Generation in the AGM Region via Maturation of VE-Cadherin+CD45+ Pre-Definitive HSCs. Cell Stem Cell 2008, 3, 99–108. [Google Scholar] [CrossRef]
- Hadland, B.K.; Varnum-Finney, B.; Poulos, M.G.; Moon, R.; Butler, J.M.; Rafii, S.; Bernstein, I.D. Endothelium and NOTCH specify and amplify aorta-gonad-mesonephros–derived hematopoietic stem cells. J. Clin. Investig. 2015, 125, 2032–2045. [Google Scholar] [CrossRef]
- Rybtsov, S.; Batsivari, A.; Bilotkach, K.; Paruzina, D.; Senserrich, J.; Nerushev, O.; Medvinsky, A. Tracing the Origin of the HSC Hierarchy Reveals an SCF-Dependent, IL-3-Independent CD43− Embryonic Precursor. Stem Cell Rep. 2014, 3, 489–501. [Google Scholar] [CrossRef]
- Lange, L.; Morgan, M.; Schambach, A. The hemogenic endothelium: A critical source for the generation of PSC-derived hematopoietic stem and progenitor cells. Cell. Mol. Life Sci. 2021, 78, 4143–4160. [Google Scholar] [CrossRef]
- Mende, N.; Bastos, H.P.; Santoro, A.; Sham, K.; Mahbubani, K.T.; Curd, A.; Takizawa, H.; Wilson, N.K.; Göttgens, B.; Saeb-Parsy, K. Quantitative and molecular differences distinguish adult human medullary and extramedullary haematopoietic stem and progenitor cell landscapes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Coppin, E.; Florentin, J.; Vasamsetti, S.B.; Arunkumar, A.; Sembrat, J.; Rojas, M.; Dutta, P. Splenic hematopoietic stem cells display a pre-activated phenotype. Immunol. Cell Biol. 2018, 96, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Dor, F.J.; Ramirez, M.L.; Parmar, K.; Altman, E.L.; Huang, C.A.; Down, J.D.; Cooper, D.K. Primitive hematopoietic cell populations reside in the spleen: Studies in the pig, baboon, and human. Exp. Hematol. 2006, 34, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Golden-Mason, L.; Curry, M.P.; Nolan, N.; Traynor, O.; McEntee, G.; Kelly, J.; Hegarty, J.E.; O’Farrelly, C. Differential expression of lymphoid and myeloid markers on differentiating hematopoietic stem cells in normal and tumor-bearing adult human liver. Hepatology 2000, 31, 1251–1256. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Lo, C.M.; Chen, L.; Cheung, C.K.; Yang, Z.F.; Chen, Y.X.; Ng, M.N.; Yu, W.C.; Ming, X.; Zhang, W.; et al. Hematopoietic chimerism in liver transplantation patients and hematopoietic stem/progenitor cells in adult human liver. Hepatology 2012, 56, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Zuber, J.; Martinez, M.; Shonts, B.; Obradovic, A.; Wang, H.; Lau, S.-P.; Xia, A.; Waffarn, E.E.; Frangaj, K.; et al. Human Intestinal Allografts Contain Functional Hematopoietic Stem and Progenitor Cells that Are Maintained by a Circulating Pool. Cell Stem Cell 2018, 24, 227–239.e8. [Google Scholar] [CrossRef]
- Lynch, L.; O’Donoghue, D.; Dean, J.; O’Sullivan, J.; O’Farrelly, C.; Golden-Mason, L. Detection and Characterization of Hemopoietic Stem Cells in the Adult Human Small Intestine. J. Immunol. 2006, 176, 5199–5204. [Google Scholar] [CrossRef]
- Wright, D.E.; Wagers, A.J.; Gulati, A.P.; Johnson, F.L.; Weissman, I.L. Physiological Migration of Hematopoietic Stem and Progenitor Cells. Science 2001, 294, 1933–1936. [Google Scholar] [CrossRef]
- Takizawa, H.; Fritsch, K.; Kovtonyuk, L.V.; Saito, Y.; Yakkala, C.; Jacobs, K.; Ahuja, A.K.; Lopes, M.; Hausmann, A.; Hardt, W.-D.; et al. Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell 2017, 21, 225–240.e5. [Google Scholar] [CrossRef]
- Massberg, S.; Schaerli, P.; Knezevic-Maramica, I.; Köllnberger, M.; Tubo, N.; Moseman, E.A.; Huff, I.V.; Junt, T.; Wagers, A.J.; Mazo, I.B.; et al. Immunosurveillance by Hematopoietic Progenitor Cells Trafficking through Blood, Lymph, and Peripheral Tissues. Cell 2007, 131, 994–1008. [Google Scholar] [CrossRef]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef]
- Asakura, A. Side population cells from diverse adult tissues are capable of in vitro hematopoietic differentiation. Exp. Hematol. 2002, 30, 1339–1345. [Google Scholar] [CrossRef]
- Taniguchi, H.; Toyoshima, T.; Fukao, K.; Nakauchi, H. Presence of hematopoietic stem cells in the adult liver. Nat. Med. 1996, 2, 198–203. [Google Scholar] [CrossRef]
- Lefrancais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.; David, T.; Coughlin, T.D.S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef]
- Krishnan, S.; Wemyss, K.; Prise, I.E.; McClure, F.A.; O’Boyle, C.; Bridgeman, H.M.; Shaw, T.N.; Grainger, J.R.; Konkel, J.E. Hematopoietic stem and progenitor cells are present in healthy gingiva tissue. J. Exp. Med. 2021, 218, e20200737. [Google Scholar] [CrossRef]
- McKinney-Freeman, S.L.; Majka, S.M.; Jackson, K.A.; Norwood, K.; Hirschi, K.K.; Goodell, M. Altered phenotype and reduced function of muscle-derived hematopoietic stem cells. Exp. Hematol. 2003, 31, 806–814. [Google Scholar] [CrossRef]
- Sun, J.; Ramos, A.; Chapman, B.; Johnnidis, J.B.; Le, L.; Ho, Y.-J.; Klein, A.M.; Hofmann, O.; Camargo, F.D. Clonal dynamics of native haematopoiesis. Nature 2014, 514, 322–327. [Google Scholar] [CrossRef]
- Brugman, M.H.; Wiekmeijer, A.-S.; van Eggermond, M.; Wolvers-Tettero, I.; Langerak, A.W.; de Haas, E.F.E.; Bystrykh, L.V.; van Rood, J.J.; de Haan, G.; Fibbe, W.E.; et al. Development of a diverse human T-cell repertoire despite stringent restriction of hematopoietic clonality in the thymus. Proc. Natl. Acad. Sci. USA 2015, 112, E6020–E6027. [Google Scholar] [CrossRef]
- Bryder, D.; Rossi, D.J.; Weissman, I.L. Hematopoietic Stem Cells: The Paradigmatic Tissue-Specific Stem Cell. Am. J. Pathol. 2006, 169, 338–346. [Google Scholar] [CrossRef]
- Carrelha, J.; Meng, Y.; Kettyle, L.M.; Luis, T.C.; Norfo, R.; Alcolea, V.; Boukarabila, H.; Grasso, F.; Gambardella, A.; Grover, A.; et al. Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature 2018, 554, 106–111. [Google Scholar] [CrossRef]
- Laurenti, E.; Frelin, C.; Xie, S.; Ferrari, R.; Dunant, C.; Zandi, S.; Neumann, A.; Plumb, I.; Doulatov, S.; Chen, J.; et al. CDK6 Levels Regulate Quiescence Exit in Human Hematopoietic Stem Cells. Cell Stem Cell 2015, 16, 302–313. [Google Scholar] [CrossRef]
- Scala, S.; Basso-Ricci, L.; Dionisio, F.; Pellin, D.; Giannelli, S.; Salerio, F.A.; Leonardelli, L.; Cicalese, M.P.; Ferrua, F.; Aiuti, A.; et al. Dynamics of genetically engineered hematopoietic stem and progenitor cells after autologous transplantation in humans. Nat. Med. 2018, 24, 1683–1690. [Google Scholar] [CrossRef]
- Civin, C.I.; Strauss, L.C.; Brovall, C.; Fackler, M.J.; Schwartz, J.F.; Shaper, J.H. Antigenic analysis of hematopoiesis. III. A hematopoietic progenitor cell surface antigen defined by a monoclonal antibody raised against KG-1a cells. J. Immunol. 1984, 133, 157–165. [Google Scholar]
- Martins, A.; Han, J.; Kim, S.O. The multifaceted effects of granulocyte colony-stimulating factor in immunomodulation and potential roles in intestinal immune homeostasis. IUBMB Life 2010, 62, 611–617. [Google Scholar] [CrossRef]
- Tajer, P.; Pike-Overzet, K.; Arias, S.; Havenga, M.; Staal, F.J. Ex Vivo Expansion of Hematopoietic Stem Cells for Therapeutic Purposes: Lessons from Development and the Niche. Cells 2019, 8, 169. [Google Scholar] [CrossRef]
- Hess, N.J.; Lindner, P.N.; Vazquez, J.; Grindel, S.; Hudson, A.W.; Stanic, A.K.; Ikeda, A.; Hematti, P.; Gumperz, J.E. Different Human Immune Lineage Compositions Are Generated in Non-Conditioned NBSGW Mice Depending on HSPC Source. Front. Immunol. 2020, 11, 573406. [Google Scholar] [CrossRef]
- Dmytrus, J.; Matthes-Martin, S.; Pichler, H.; Worel, N.; Geyeregger, R.; Frank, N.; Frech, C.; Fritsch, G. Multi-color immune-phenotyping of CD34 subsets reveals unexpected differences between various stem cell sources. Bone Marrow Transplant. 2016, 51, 1093–1100. [Google Scholar] [CrossRef][Green Version]
- Seggewiss, R.; Einsele, H. Immune reconstitution after allogeneic transplantation and expanding options for immunomodulation: An update. Blood 2010, 115, 3861–3868. [Google Scholar] [CrossRef]
- Ogonek, J.; Juric, M.K.; Ghimire, S.; Varanasi, P.; Holler, E.; Greinix, H.; Weissinger, E. Immune Reconstitution after Allogeneic Hematopoietic Stem Cell Transplantation. Front. Immunol. 2016, 7, 507. [Google Scholar] [CrossRef] [PubMed]
- Salzmann-Manrique, E.; Bremm, M.; Huenecke, S.; Stech, M.; Orth, A.; Eyrich, M.; Schulz, A.; Esser, R.; Klingebiel, T.; Bader, P.; et al. Joint Modeling of Immune Reconstitution Post Haploidentical Stem Cell Transplantation in Pediatric Patients with Acute Leukemia Comparing CD34+-Selected to CD3/CD19-Depleted Grafts in a Retrospective Multicenter Study. Front. Immunol. 2018, 9, 1841. [Google Scholar] [CrossRef] [PubMed]
- Kasow, K.A.; Sims-Poston, L.; Eldridge, P.; Hale, G.A. CD34+ Hematopoietic Progenitor Cell Selection of Bone Marrow Grafts for Autologous Transplantation in Pediatric Patients. Biol. Blood Marrow Transplant. 2007, 13, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, W.I. Allogeneic transplantation. Curr. Opin. Oncol. 2012, 24, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Panch, S.R.; Szymanski, J.; Savani, B.N.; Stroncek, D.F. Sources of Hematopoietic Stem and Progenitor Cells and Methods to Optimize Yields for Clinical Cell Therapy. Biol. Blood Marrow Transplant. 2017, 23, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Blache, C.; Chauvin, J.-M.; Marie-Cardine, A.; Contentin, N.; Pommier, P.; Dedreux, I.; François, S.; Jacquot, S.; Bastit, D.; Boyer, O. Reduced Frequency of Regulatory T Cells in Peripheral Blood Stem Cell Compared to Bone Marrow Transplantations. Biol. Blood Marrow Transplant. 2010, 16, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Holtick, U.; Albrecht, M.; Chemnitz, J.M.; Theurich, S.; Shimabukuro-Vornhagen, A.; Skoetz, N.; Scheid, C.; von Bergwelt-Baildon, M. Comparison of bone marrow versus peripheral blood allogeneic hematopoietic stem cell transplantation for hematological malignancies in adults—a systematic review and meta-analysis. Crit. Rev. Oncol. 2015, 94, 179–188. [Google Scholar] [CrossRef]
- Magenau, J.; Runaas, L.; Reddy, P. Advances in understanding the pathogenesis of graft-versus-host disease. Br. J. Haematol. 2016, 173, 190–205. [Google Scholar] [CrossRef]
- Miyara, M.; Sakaguchi, S. Human FoxP3 + CD4 + regulatory T cells: Their knowns and unknowns. Immunol. Cell Biol. 2011, 89, 346–351. [Google Scholar] [CrossRef]
- Martelli, M.F.; Di Ianni, M.; Ruggeri, L.; Falzetti, F.; Carotti, A.; Terenzi, A.; Pierini, A.; Massei, M.S.; Amico, L.; Urbani, E.; et al. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood 2014, 124, 638–644. [Google Scholar] [CrossRef]
- DiVito, S.J.; Aasebø, A.T.; Matos, T.R.; Hsieh, P.-C.; Collin, M.; Elco, C.P.; O’Malley, J.; Bækkevold, E.S.; Reims, H.M.; Gedde-Dahl, T.; et al. Peripheral host T cells survive hematopoietic stem cell transplantation and promote graft-versus-host disease. J. Clin. Investig. 2020, 130, 4624–4636. [Google Scholar] [CrossRef]
- Genovese, P.; Schiroli, G.; Escobar, G.; Di Tomaso, T.; Firrito, C.; Calabria, A.; Moi, D.; Mazzieri, R.; Bonini, M.C.; Holmes, M.C.; et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature 2014, 510, 235–240. [Google Scholar] [CrossRef]
- Radtke, S.; Pande, D.; Cui, M.; Perez, A.M.; Chan, Y.-Y.; Enstrom, M.; Schmuck, S.; Berger, A.; Eunson, T.; Adair, J.E.; et al. Purification of Human CD34+CD90+ HSCs Reduces Target Cell Population and Improves Lentiviral Transduction for Gene Therapy. Mol. Ther. Methods Clin. Dev. 2020, 18, 679–691. [Google Scholar] [CrossRef]
- Radtke, S.; Adair, J.E.; Giese, M.A.; Chan, Y.-Y.; Norgaard, Z.K.; Enstrom, M.; Haworth, K.G.; Schefter, L.E.; Kiem, H.-P. A distinct hematopoietic stem cell population for rapid multilineage engraftment in nonhuman primates. Sci. Transl. Med. 2017, 9, eaan1145. [Google Scholar] [CrossRef]
- Yamamoto, R.; Morita, Y.; Ooehara, J.; Hamanaka, S.; Onodera, M.; Rudolph, K.L.; Ema, H.; Nakauchi, H. Clonal Analysis Unveils Self-Renewing Lineage-Restricted Progenitors Generated Directly from Hematopoietic Stem Cells. Cell 2013, 154, 1112–1126. [Google Scholar] [CrossRef]
- Kim, S.; Kim, N.; Presson, A.P.; Metzger, M.E.; Bonifacino, A.C.; Sehl, M.; Chow, S.A.; Crooks, G.M.; Dunbar, C.; An, D.S.; et al. Dynamics of HSPC Repopulation in Nonhuman Primates Revealed by a Decade-Long Clonal-Tracking Study. Cell Stem Cell 2014, 14, 473–485. [Google Scholar] [CrossRef]
- Benz, C.; Copley, M.R.; Kent, D.; Wohrer, S.; Cortes, A.; Aghaeepour, N.; Ma, E.; Mader, H.; Rowe, K.; Day, C.; et al. Hematopoietic Stem Cell Subtypes Expand Differentially during Development and Display Distinct Lymphopoietic Programs. Cell Stem Cell 2012, 10, 273–283. [Google Scholar] [CrossRef]
- Naik, S.H.; Perié, L.; Swart, E.; Gerlach, C.; Van Rooij, N.; De Boer, R.; Schumacher, T. Diverse and heritable lineage imprinting of early haematopoietic progenitors. Nature 2013, 496, 229–232. [Google Scholar] [CrossRef]
- Six, E.; Guilloux, A.; Denis, A.; Lecoules, A.; Magnani, A.; Vilette, R.; Male, F.; Cagnard, N.; Delville, M.; Magrin, E.; et al. Clonal tracking in gene therapy patients reveals a diversity of human hematopoietic differentiation programs. Blood 2020, 135, 1219–1231. [Google Scholar] [CrossRef]
- Lareau, C.A.; Ludwig, L.S.; Muus, C.; Gohil, S.H.; Zhao, T.; Chiang, Z.; Pelka, K.; Verboon, J.M.; Luo, W.; Christian, E.; et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat. Biotechnol. 2020, 39, 451–461. [Google Scholar] [CrossRef]
- Pang, W.W.; Price, E.A.; Sahoo, D.; Beerman, I.; Maloney, W.J.; Rossi, D.J.; Schrier, S.L.; Weissman, I.L. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl. Acad. Sci. USA 2011, 108, 20012–20017. [Google Scholar] [CrossRef]
- Lu, R.; Czechowicz, A.; Seita, J.; Jiang, D.; Weissman, I.L. Clonal-level lineage commitment pathways of hematopoietic stem cells in vivo. Proc. Natl. Acad. Sci. USA 2019, 116, 1447–1456. [Google Scholar] [CrossRef]
- Wilkinson, A.C.; Ishida, R.; Kikuchi, M.; Sudo, K.; Morita, M.; Crisostomo, R.V.; Yamamoto, R.; Loh, K.M.; Nakamura, Y.; Watanabe, M.; et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature 2019, 571, 117–121, Erratum in Nature 2019, 571, E12. [Google Scholar] [CrossRef]
- Lacaud, G.; Kouskoff, V. Hemangioblast, hemogenic endothelium, and primitive versus definitive hematopoiesis. Exp. Hematol. 2017, 49, 19–24. [Google Scholar] [CrossRef]
- Ivanovs, A.; Rybtsov, S.; Ng, E.S.; Stanley, E.G.; Elefanty, A.G.; Medvinsky, A. Human haematopoietic stem cell development: From the embryo to the dish. Development 2017, 144, 2323–2337. [Google Scholar] [CrossRef]
- Sturgeon, C.M.; Ditadi, A.; Awong, G.; Kennedy, M.; Keller, G. Wnt signaling controls the specification of definitive and primitive hematopoiesis from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 554–561. [Google Scholar] [CrossRef]
- Ng, E.S.; Azzola, L.; Bruveris, F.F.; Calvanese, V.; Phipson, B.; Vlahos, K.; Hirst, C.; Jokubaitis, V.J.; Yu, Q.C.; Maksimovic, J.; et al. Differentiation of human embryonic stem cells to HOXA+ hemogenic vasculature that resembles the aorta-gonad-mesonephros. Nat. Biotechnol. 2016, 34, 1168–1179. [Google Scholar] [CrossRef]
- Bruveris, F.F.; Ng, E.S.; Leitoguinho, A.R.; Motazedian, A.; Vlahos, K.; Sourris, K.; Mayberry, R.; McDonald, P.; Azzola, L.; Davidson, N.M.; et al. Human yolk sac-like haematopoiesis generates RUNX1- and GFI1/1B-dependent blood and SOX17-positive endothelium. Development 2020, 147, dev193037. [Google Scholar] [CrossRef]
- Choi, K.-D.; Vodyanik, M.A.; Togarrati, P.P.; Suknuntha, K.; Kumar, A.; Samarjeet, F.; Probasco, M.D.; Tian, S.; Stewart, R.; Thomson, J.A.; et al. Identification of the Hemogenic Endothelial Progenitor and Its Direct Precursor in Human Pluripotent Stem Cell Differentiation Cultures. Cell Rep. 2012, 2, 553–567. [Google Scholar] [CrossRef]
- Vo, L.T.; Daley, G.Q. De novo generation of HSCs from somatic and pluripotent stem cell sources. Blood 2015, 125, 2641–2648. [Google Scholar] [CrossRef]
- Sandler, V.M.; Lis, R.; Liu, Y.; Kedem, A.; James, D.; Elemento, O.; Butler, J.M.; Scandura, J.; Rafii, S. Reprogramming human endothelial cells to haematopoietic cells requires vascular induction. Nat. Cell Biol. 2014, 511, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Doulatov, S.; Vo, L.T.; Chou, S.S.; Kim, P.G.; Arora, N.; Li, H.; Hadland, B.K.; Bernstein, I.D.; Collins, J.J.; Zon, L.I.; et al. Induction of Multipotential Hematopoietic Progenitors from Human Pluripotent Stem Cells via Respecification of Lineage-Restricted Precursors. Cell Stem Cell 2013, 13, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Vo, L.T.; Kinney, M.; Liu, X.; Zhang, Y.; Barragan, J.; Sousa, P.M.; Jha, D.; Han, A.; Cesana, M.; Shao, Z.; et al. Regulation of embryonic haematopoietic multipotency by EZH1. Nature 2018, 553, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, R.; Jha, D.K.; Han, A.; Soria-Valles, C.; da Rocha, E.L.; Lu, Y.-F.; Goettel, J.A.; Serrao, E.; Rowe, R.G.; Malleshaiah, M.; et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 2017, 545, 432–438. [Google Scholar] [CrossRef]
- Fidanza, A.; Forrester, L.M. Progress in the production of haematopoietic stem and progenitor cells from human pluripotent stem cells. J. Immunol. Regen. Med. 2021, 13, 100050. [Google Scholar] [CrossRef]
- Cypris, O.; Frobel, J.; Rai, S.; Franzen, J.; Sontag, S.; Goetzke, R.; De Toledo, M.A.S.; Zenke, M.; Wagner, W. Tracking of epigenetic changes during hematopoietic differentiation of induced pluripotent stem cells. Clin. Epigenetics 2019, 11, 19. [Google Scholar] [CrossRef]
- Haake, K.; Ackermann, M.; Lachmann, N. Concise Review: Towards the Clinical Translation of Induced Pluripotent Stem Cell-Derived Blood Cells— Ready for Take-Off. Stem Cells Transl. Med. 2018, 8, 332–339. [Google Scholar] [CrossRef]
- Kumrah, R.; Vignesh, P.; Patra, P.; Singh, A.; Anjani, G.; Saini, P.; Sharma, M.; Kaur, A.; Rawat, A. Genetics of severe combined immunodeficiency. Genes Dis. 2020, 7, 52–61. [Google Scholar] [CrossRef]
- Chang, C.-W.; Lai, Y.-S.; Westin, E.; Khodadadi-Jamayran, A.; Pawlik, K.M.; Lamb, L.S.; Goldman, F.D.; Townes, T.M. Modeling Human Severe Combined Immunodeficiency and Correction by CRISPR/Cas9-Enhanced Gene Targeting. Cell Rep. 2015, 12, 1668–1677. [Google Scholar] [CrossRef]
- Menon, T.; Firth, A.L.; Scripture-Adams, D.D.; Galic, Z.; Qualls, S.J.; Gilmore, W.; Ke, E.; Singer, O.; Anderson, L.; Bornzin, A.R.; et al. Lymphoid Regeneration from Gene-Corrected SCID-X1 Subject-Derived iPSCs. Cell Stem Cell 2015, 16, 367–372. [Google Scholar] [CrossRef]
- Themeli, M.; Chhatta, A.; Boersma, H.; Prins, H.J.; Cordes, M.; de Wilt, E.; Farahani, A.S.; Vandekerckhove, B.; van der Burg, M.; Hoeben, R.C.; et al. iPSC-Based Modeling of RAG2 Severe Combined Immunodeficiency Reveals Multiple T Cell Developmental Arrests. Stem Cell Rep. 2020, 14, 300–311. [Google Scholar] [CrossRef]
- Brauer, P.M.; Pessach, I.M.; Clarke, E.; Rowe, J.H.; De Bruin, L.O.; Lee, Y.N.; Dominguez-Brauer, C.; Comeau, A.M.; Awong, G.; Felgentreff, K.; et al. Modeling altered T-cell development with induced pluripotent stem cells from patients with RAG1-dependent immune deficiencies. Blood 2016, 128, 783–793. [Google Scholar] [CrossRef]
- Gardner, C.L.; Pavel-Dinu, M.; Dobbs, K.; Bosticardo, M.; Reardon, P.K.; Lack, J.; DeRavin, S.S.; Le, K.; Bello, E.; Pala, F.; et al. Gene Editing Rescues In vitro T Cell Development of RAG2-Deficient Induced Pluripotent Stem Cells in an Artificial Thymic Organoid System. J. Clin. Immunol. 2021, 41, 852–862. [Google Scholar] [CrossRef]
- Eynon, E.E.; Livak, F.; Kuida, K.; Schatz, D.G.; Flavell, R.A. Distinct effects of Jak3 signaling on alphabeta and gammadelta thymocyte development. J. Immunol. 1999, 162, 1448–1459. [Google Scholar]
- Seet, C.S.; He, C.; Bethune, M.T.; Li, S.; Chick, B.; Gschweng, E.H.; Zhu, Y.; Kim, K.; Kohn, D.B.; Baltimore, D.; et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat. Methods 2017, 14, 521–530. [Google Scholar] [CrossRef]
- Montel-Hagen, A.; Seet, C.S.; Li, S.; Chick, B.; Zhu, Y.; Chang, P.; Tsai, S.; Sun, V.; Lopez, S.; Chen, H.-C.; et al. Organoid-Induced Differentiation of Conventional T Cells from Human Pluripotent Stem Cells. Cell Stem Cell 2019, 24, 376–389.e8. [Google Scholar] [CrossRef]
- Wiekmeijer, A.-S.; Pike-Overzet, K.; Ijspeert, H.; Brugman, M.H.; Wolvers-Tettero, I.L.; Lankester, A.C.; Bredius, R.G.; Van Dongen, J.; Fibbe, W.E.; Langerak, A.W.; et al. Identification of checkpoints in human T-cell development using severe combined immunodeficiency stem cells. J. Allergy Clin. Immunol. 2015, 137, 517–526.e3. [Google Scholar] [CrossRef]
- Perez, L.G.; van Eggermond, M.; van Roon, L.; Vloemans, S.A.; Cordes, M.; Schambach, A.; Rothe, M.; Berghuis, D.; Lagresle-Peyrou, C.; Cavazzana, M.; et al. Successful Preclinical Development of Gene Therapy for Recombinase-Activating Gene-1-Deficient SCID. Mol. Ther. Methods Clin. Dev. 2020, 17, 666–682. [Google Scholar] [CrossRef]
- Laskowski, T.J.; Van Caeneghem, Y.; Pourebrahim, R.; Ma, C.; Ni, Z.; Garate, Z.; Crane, A.M.; Li, X.S.; Liao, W.; Gonzalez-Garay, M.; et al. Gene Correction of iPSCs from a Wiskott-Aldrich Syndrome Patient Normalizes the Lymphoid Developmental and Functional Defects. Stem Cell Rep. 2016, 7, 139–148. [Google Scholar] [CrossRef]
- Orange, J.S.; Ramesh, N.; Remold-O’Donnell, E.; Sasahara, Y.; Koopman, L.; Byrne, M.; Bonilla, F.A.; Rosen, F.S.; Geha, R.S.; Strominger, J.L. Wiskott-Aldrich syndrome protein is required for NK cell cytotoxicity and colocalizes with actin to NK cell-activating immunologic synapses. Proc. Natl. Acad. Sci. USA 2002, 99, 11351–11356. [Google Scholar] [CrossRef]
- Arnold, D.E.; Heimall, J.R. A Review of Chronic Granulomatous Disease. Adv. Ther. 2017, 34, 2543–2557. [Google Scholar] [CrossRef]
- Zou, J.; Sweeney, C.L.; Chou, B.-K.; Choi, U.; Pan, J.; Wang, H.; Dowey, S.N.; Cheng, L.; Malech, H. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: Functional correction by zinc finger nuclease–mediated safe harbor targeting. Blood 2011, 117, 5561–5572. [Google Scholar] [CrossRef]
- Merling, R.K.; Sweeney, C.L.; Chu, J.; Bodansky, A.; Choi, U.; Priel, D.L.; Kuhns, D.B.; Wang, H.; Vasilevsky, S.; De Ravin, S.S.; et al. An AAVS1-Targeted Minigene Platform for Correction of iPSCs From All Five Types of Chronic Granulomatous Disease. Mol. Ther. 2015, 23, 147–157. [Google Scholar] [CrossRef]
- Dreyer, A.-K.; Hoffmann, D.; Lachmann, N.; Ackermann, M.; Steinemann, D.; Timm, B.; Siler, U.; Reichenbach, J.; Grez, M.; Moritz, T.; et al. TALEN-mediated functional correction of X-linked chronic granulomatous disease in patient-derived induced pluripotent stem cells. Biomaterials 2015, 69, 191–200. [Google Scholar] [CrossRef]
- Flynn, R.; Grundmann, A.; Renz, P.; Hänseler, W.; James, W.; Cowley, S.; Moore, M.D. CRISPR-mediated genotypic and phenotypic correction of a chronic granulomatous disease mutation in human iPS cells. Exp. Hematol. 2015, 43, 838–848.e3. [Google Scholar] [CrossRef]
- Laugsch, M.; Rostovskaya, M.; Velychko, S.; Richter, C.; Zimmer, A.; Klink, B.; Schröck, E.; Haase, M.; Neumann, K.; Thieme, S.; et al. Functional Restoration of gp91phox-Oxidase Activity by BAC Transgenesis and Gene Targeting in X-linked Chronic Granulomatous Disease iPSCs. Mol. Ther. 2016, 24, 812–822. [Google Scholar] [CrossRef]
- Sweeney, C.L.; Zou, J.; Choi, U.; Merling, R.K.; Liu, A.; Bodansky, A.; Burkett, S.; Kim, J.-W.; De Ravin, S.S.; Malech, H. Targeted Repair of CYBB in X-CGD iPSCs Requires Retention of Intronic Sequences for Expression and Functional Correction. Mol. Ther. 2017, 25, 321–330. [Google Scholar] [CrossRef]
- Klatt, D.; Cheng, E.; Philipp, F.; Selich, A.; Dahlke, J.; Schmidt, R.E.; Schott, J.W.; Büning, H.; Hoffmann, D.; Thrasher, A.J.; et al. Targeted Repair of p47-CGD in iPSCs by CRISPR/Cas9: Functional Correction without Cleavage in the Highly Homologous Pseudogenes. Stem Cell Rep. 2019, 13, 590–598. [Google Scholar] [CrossRef]
- Merling, R.K.; Kuhns, U.B.; Sweeney, C.L.; Wu, X.; Burkett, S.; Chu, J.; Lee, J.; Koontz, S.; Di Pasquale, G.; Afione, S.A.; et al. Gene-edited pseudogene resurrection corrects p47phox-deficient chronic granulomatous disease. Blood Adv. 2016, 1, 270–278. [Google Scholar] [CrossRef]
- Karlsson, K.R.; Cowley, S.; Martinez, F.O.; Shaw, M.; Minger, S.L.; James, W. Homogeneous monocytes and macrophages from human embryonic stem cells following coculture-free differentiation in M-CSF and IL-3. Exp. Hematol. 2008, 36, 1167–1175. [Google Scholar] [CrossRef]
- Hänseler, W.; Sansom, S.N.; Buchrieser, J.; Newey, S.E.; Moore, C.S.; Nicholls, F.; Chintawar, S.; Schnell, C.; Antel, J.; Allen, N.; et al. A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem Cell Rep. 2017, 8, 1727–1742. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Kozaki, T.; Lee, C.; Thion, M.S.; Otsuka, M.; Lim, S.; Utami, K.H.; Fidan, K.; Park, D.S.; Malleret, B.; et al. Induced-Pluripotent-Stem-Cell-Derived Primitive Macrophages Provide a Platform for Modeling Tissue-Resident Macrophage Differentiation and Function. Immunity 2017, 47, 183–198.e6. [Google Scholar] [CrossRef] [PubMed]
- Buchrieser, J.; James, W.; Moore, M.D. Human Induced Pluripotent Stem Cell-Derived Macrophages Share Ontogeny with MYB—Independent Tissue-Resident Macrophages. Stem Cell Rep. 2017, 8, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, N.; Ackermann, M.; Frenzel, E.; Liebhaber, S.; Brennig, S.; Happle, C.; Hoffmann, D.; Klimenkova, O.; Lüttge, D.; Buchegger, T.; et al. Large-Scale Hematopoietic Differentiation of Human Induced Pluripotent Stem Cells Provides Granulocytes or Macrophages for Cell Replacement Therapies. Stem Cell Rep. 2015, 4, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Canté-Barrett, K.; Staal, F.J. An adequate human T cell repertoire from a single T cell progenitor: Lessons from an experiment of nature. EBioMedicine 2020, 60, 103015. [Google Scholar] [CrossRef]
- Kraus, H.; Kaiser, S.; Aumann, K.; Bönelt, P.; Salzer, U.; Vestweber, D.; Erlacher, M.; Kunze, M.; Burger, M.; Pieper, K.; et al. A Feeder-Free Differentiation System Identifies Autonomously Proliferating B Cell Precursors in Human Bone Marrow. J. Immunol. 2013, 192, 1044–1054. [Google Scholar] [CrossRef]
- Nehls, M.; Pfeifer, D.; Schorpp, M.; Hedrich, H.J.; Boehm, T. New member of the winged-helix protein family disrupted in mouse and rat nude mutations. Nature 1994, 372, 103–107. [Google Scholar] [CrossRef]
- Flanagan, S.P. ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet. Res. 1966, 8, 295–309. [Google Scholar] [CrossRef]
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A severe combined immunodeficiency mutation in the mouse. Nature 1983, 301, 527–530. [Google Scholar] [CrossRef]
- Bosma, M.J. B and T cell leakiness in the scid mouse mutant. Immunodefic. Rev. 1992, 3, 261–276. [Google Scholar]
- Bosma, G.C.; Fried, M.; Custer, R.P.; Carroll, A.; Gibson, D.M.; Bosma, M.J. Evidence of functional lymphocytes in some (leaky) scid mice. J. Exp. Med. 1988, 167, 1016–1033. [Google Scholar] [CrossRef]
- Mombaerts, P.; Iacomini, J.; Johnson, R.; Herrup, K.; Tonegawa, S.; Papaioannou, V. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 1992, 68, 869–877. [Google Scholar] [CrossRef]
- Shinkai, Y. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 1992, 68, 855–867. [Google Scholar] [CrossRef]
- Lagresle-Peyrou, C.; Yates, F.; Malassis-Séris, M.; Hue, C.; Morillon, E.; Garrigue, A.; Liu, A.; Hajdari, P.; Stockholm, D.; Danos, O.; et al. Long-term immune reconstitution in RAG-1-deficient mice treated by retroviral gene therapy: A balance between efficiency and toxicity. Blood 2005, 107, 63–72. [Google Scholar] [CrossRef][Green Version]
- van Til, N.P.; Sarwari, R.; Visser, T.P.; Hauer, J.; Peyrou, C.L.; van der Velden, G.; Malshetty, V.; Cortes, P.; Jollet, A.; Danos, O.; et al. Recombination-activating gene 1 (Rag1)–deficient mice with severe combined immunodeficiency treated with lentiviral gene therapy demonstrate autoimmune Omenn-like syndrome. J. Allergy Clin. Immunol. 2014, 133, 1116–1123. [Google Scholar] [CrossRef]
- Pike-Overzet, K.; Rodijk, M.; Ng, Y.-Y.; Baert, M.R.M.; Peyrou, C.L.; Schambach, A.; Zhang, F.; Hoeben, R.C.; Hacein-Bey-Abina, S.; Lankester, A.C.; et al. Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia 2011, 25, 1471–1483. [Google Scholar] [CrossRef]
- Kim, M.-S.; Lapkouski, M.; Yang, W.; Gellert, M. Crystal structure of the V(D)J recombinase RAG1–RAG2. Nature 2015, 518, 507–511. [Google Scholar] [CrossRef]
- Schwarz, K.; Gauss, G.H.; Ludwig, L.; Pannicke, U.; Li, Z.; Lindner, D.; Friedrich, W.; Seger, R.A.; Hansen-Hagge, T.E.; Desiderio, S.; et al. RAG Mutations in Human B Cell-Negative SCID. Science 1996, 274, 97–99. [Google Scholar] [CrossRef]
- De Bruin, L.O.; Yang, W.; Capuder, K.; Lee, Y.N.; Antolini, M.; Meyers, R.; Gellert, M.; Musunuru, K.; Manis, J.; Notarangelo, L. Rapid generation of novel models of RAG1 deficiency by CRISPR/Cas9-induced mutagenesis in murine zygotes. Oncotarget 2016, 7, 12962–12974. [Google Scholar] [CrossRef]
- Greiner, D.L.; Hesselton, R.A.; Shultz, L.D. SCID Mouse Models of Human Stem Cell Engraftment. Stem Cells 1998, 16, 166–177. [Google Scholar] [CrossRef]
- Di Santo, J.; Muller, W.; Guy-Grand, D.; Fischer, A.; Rajewsky, K. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc. Natl. Acad. Sci. USA 1995, 92, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Zamora, A.E.; Grossenbacher, S.K.; Aguilar, E.G.; Murphy, W.J. Models to Study NK Cell Biology and Possible Clinical Application. Curr. Protoc. Immunol. 2015, 110, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Poletti, V.; Charrier, S.; Corre, G.; Gjata, B.; Vignaud, A.; Zhang, F.; Rothe, M.; Schambach, A.; Gaspar, H.B.; Thrasher, A.J.; et al. Preclinical Development of a Lentiviral Vector for Gene Therapy of X-Linked Severe Combined Immunodeficiency. Mol. Ther. Methods Clin. Dev. 2018, 9, 257–269. [Google Scholar] [CrossRef]
- Schiroli, G.; Ferrari, S.; Conway, A.; Jacob, A.; Capo, V.; Albano, L.; Plati, T.; Castiello, M.C.; Sanvito, F.; Gennery, A.R.; et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci. Transl. Med. 2017, 9, eaan0820. [Google Scholar] [CrossRef]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K.; et al. NOD/SCID/γcnull mouse: An excellent recipient mouse model for engraftment of human cells. Blood 2002, 100, 3175–3182. [Google Scholar] [CrossRef]
- Ishikawa, F.; Yasukawa, M.; Lyons, B.; Yoshida, S.; Miyamoto, T.; Yoshimoto, G.; Watanabe, T.; Akashi, K.; Shultz, L.D.; Harada, M. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor γ chainnull mice. Blood 2005, 106, 1565–1573. [Google Scholar] [CrossRef]
- Katano, I.; Ito, R.; Eto, T.; Aiso, S.; Ito, M. Immunodeficient NOD-scid IL-2R.GAMMA.null Mice Do Not Display T and B Cell Leakiness. Exp. Anim. 2011, 60, 181–186. [Google Scholar] [CrossRef][Green Version]
- Prochazka, M.; Gaskins, H.R.; Shultz, L.D.; Leiter, E.H. The nonobese diabetic scid mouse: Model for spontaneous thymomagenesis associated with immunodeficiency. Proc. Natl. Acad. Sci. USA 1992, 89, 3290–3294. [Google Scholar] [CrossRef]
- Yahata, T.; Ando, K.; Nakamura, Y.; Ueyama, Y.; Shimamura, K.; Tamaoki, N.; Kato, S.; Hotta, T. Functional Human T Lymphocyte Development from Cord Blood CD34+Cells in Nonobese Diabetic/Shi-scid, IL-2 Receptor γ Null Mice. J. Immunol. 2002, 169, 204–209. [Google Scholar] [CrossRef]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2Rγnull mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef]
- Wiekmeijer, A.-S.; Pike-Overzet, K.; Brugman, M.; Salvatori, D.C.; Egeler, R.M.; Bredius, R.G.; Fibbe, W.E.; Staal, F.J. Sustained Engraftment of Cryopreserved Human Bone Marrow CD34+Cells in Young Adult NSG Mice. BioRes. Open Access 2014, 3, 110–116. [Google Scholar] [CrossRef]
- Peschon, J.J.; Morrissey, P.J.; Grabstein, K.H.; Ramsdell, F.J.; Maraskovsky, E.; Gliniak, B.C.; Park, L.S.; Ziegler, S.F.; Williams, D.E.; Ware, C.B.; et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J. Exp. Med. 1994, 180, 1955–1960. [Google Scholar] [CrossRef]
- Puel, A.; Leonard, W.J. Mutations in the gene for the IL-7 receptor result in T–B+NK+ severe combined immunodeficiency disease. Curr. Opin. Immunol. 2000, 12, 468–473. [Google Scholar] [CrossRef]
- Tan, Y.-T.; Ye, L.; Xie, F.; Beyer, A.I.; Muench, M.; Wang, J.; Chen, Z.; Liu, H.; Chen, S.-J.; Kan, Y.W. Respecifying human iPSC-derived blood cells into highly engraftable hematopoietic stem and progenitor cells with a single factor. Proc. Natl. Acad. Sci. USA 2018, 115, 2180–2185. [Google Scholar] [CrossRef]
- Ben-David, U.; Mayshar, Y.; Benvenisty, N. Large-Scale Analysis Reveals Acquisition of Lineage-Specific Chromosomal Aberrations in Human Adult Stem Cells. Cell Stem Cell 2011, 9, 97–102. [Google Scholar] [CrossRef]
- Nori, S.; Okada, Y.; Nishimura, S.; Sasaki, T.; Itakura, G.; Kobayashi, Y.; Renault-Mihara, F.; Shimizu, A.; Koya, I.; Yoshida, R.; et al. Long-Term Safety Issues of iPSC-Based Cell Therapy in a Spinal Cord Injury Model: Oncogenic Transformation with Epithelial-Mesenchymal Transition. Stem Cell Rep. 2015, 4, 360–373. [Google Scholar] [CrossRef]
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014, 32, 364–372. [Google Scholar] [CrossRef]
- Jangalwe, S.; Shultz, L.D.; Mathew, A.; Brehm, M.A. Improved B cell development in humanized NOD-scid IL2Rγnullmice transgenically expressing human stem cell factor, granulocyte-macrophage colony-stimulating factor and interleukin-3. Immun. Inflamm. Dis. 2016, 4, 427–440. [Google Scholar] [CrossRef]
- Sippel, T.R.; Radtke, S.; Olsen, T.M.; Kiem, H.-P.; Rongvaux, A. Human hematopoietic stem cell maintenance and myeloid cell development in next-generation humanized mouse models. Blood Adv. 2019, 3, 268–274. [Google Scholar] [CrossRef]
- Tamplin, O.J.; Durand, E.M.; Carr, L.A.; Childs, S.; Hagedorn, E.J.; Li, P.; Yzaguirre, A.D.; Speck, N.A.; Zon, L.I. Hematopoietic Stem Cell Arrival Triggers Dynamic Remodeling of the Perivascular Niche. Cell 2015, 160, 241–252. [Google Scholar] [CrossRef]
- Cosgun, K.; Rahmig, S.; Mende, N.; Reinke, S.; Hauber, I.; Schäfer, C.; Petzold, A.; Weisbach, H.; Heidkamp, G.; Purbojo, A.; et al. Kit Regulates HSC Engraftment across the Human-Mouse Species Barrier. Cell Stem Cell 2014, 15, 227–238. [Google Scholar] [CrossRef]
- Adigbli, G.; Hua, P.; Uchiyama, M.; Roberts, I.; Hester, J.; Watt, S.M.; Issa, F. Development of LT-HSC-Reconstituted Non-Irradiated NBSGW Mice for the Study of Human Hematopoiesis In Vivo. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Coppin, E.; Sundarasetty, B.S.; Rahmig, S.; Blume, J.; Verheyden, N.A.; Bahlmann, F.; Ravens, S.; Schubert, U.; Schmid, J.; Ludwig, S.; et al. Enhanced differentiation of functional human T cells in NSGW41 mice with tissue-specific expression of human interleukin-7. Leukemia 2021, 1–7. [Google Scholar] [CrossRef]
- Blümich, S.; Zdimerova, H.; Münz, C.; Kipar, A.; Pellegrini, G. Human CD34+ Hematopoietic Stem Cell–Engrafted NSG Mice: Morphological and Immunophenotypic Features. Veter Pathol. 2020, 58, 161–180. [Google Scholar] [CrossRef]
- Janke, L.J.; Imai, D.M.; Tillman, H.; Doty, R.; Hoenerhoff, M.J.; Xu, J.J.; Freeman, Z.T.; Allen, P.; Fowlkes, N.W.; Iacobucci, I.; et al. Development of Mast Cell and Eosinophil Hyperplasia and HLH/MAS-Like Disease in NSG-SGM3 Mice Receiving Human CD34+ Hematopoietic Stem Cells or Patient-Derived Leukemia Xenografts. Veter Pathol. 2020, 58, 181–204. [Google Scholar] [CrossRef]
- Chandra, S.; Cristofori, P.; Fonck, C.; O’Neill, C.A. Ex Vivo Gene Therapy: Graft-versus-host Disease (GVHD) in NSG™ (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) Mice Transplanted with CD34+ Human Hematopoietic Stem Cells. Toxicol. Pathol. 2019, 47, 656–660. [Google Scholar] [CrossRef]
- Adigbli, G.; Ménoret, S.; Cross, A.R.; Hester, J.; Issa, F.; Anegon, I. Humanization of Immunodeficient Animals for the Modeling of Transplantation, Graft Versus Host Disease, and Regenerative Medicine. Transplantation 2020, 104, 2290–2306. [Google Scholar] [CrossRef]
- Boettcher, A.N.; Loving, C.L.; Cunnick, J.E.; Tuggle, C.K. Development of Severe Combined Immunodeficient (SCID) Pig Models for Translational Cancer Modeling: Future Insights on How Humanized SCID Pigs Can Improve Preclinical Cancer Research. Front. Oncol. 2018, 8, 559. [Google Scholar] [CrossRef]
- Graves, S.S.; Storb, R. Evolution of haematopoietic cell transplantation for canine blood disorders and a platform for solid organ transplantation. Veter Med. Sci. 2021, 7, 2156–2171. [Google Scholar] [CrossRef]
- Shultz, L.D.; Schweitzer, P.A.; Christianson, S.W.; Gott, B.; Schweitzer, I.B.; Tennent, B.; McKenna, S.; Mobraaten, L.; Rajan, T.V.; Greiner, D.L.; et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 1995, 154, 180–191. [Google Scholar] [PubMed]
- Ménoret, S.; Ouisse, L.-H.; Tesson, L.; Delbos, F.; Garnier, D.; Remy, S.; Usal, C.; Concordet, J.-P.; Giovannangeli, C.; Chenouard, V.; et al. Generation of Immunodeficient Rats with Rag1 and Il2rg Gene Deletions and Human Tissue Grafting Models. Transplantation 2018, 102, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Ménoret, S.; Ouisse, L.-H.; Tesson, L.; Remy, S.; Usal, C.; Guiffes, A.; Chenouard, V.; Royer, P.-J.; Evanno, G.; Vanhove, B.; et al. In Vivo Analysis of Human Immune Responses in Immunodeficient Rats. Transplantation 2020, 104, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhou, J.; He, J.; Liu, J.; Wang, H.; Liu, Y.; Jiang, T.; Zhang, Q.; Fu, X.; Xu, Y. An Immune System-Modified Rat Model for Human Stem Cell Transplantation Research. Stem Cell Rep. 2018, 11, 514–521. [Google Scholar] [CrossRef]
- Dawson, H.D.; Loveland, J.E.; Pascal, G.; Gilbert, J.G.; Uenishi, H.; Mann, K.M.; Sang, Y.; Zhang, J.; Carvalho-Silva, D.; Hunt, T.; et al. Structural and functional annotation of the porcine immunome. BMC Genom. 2013, 14, 332. [Google Scholar] [CrossRef]
- Roth, J.A.; Tuggle, C.K. Livestock Models in Translational Medicine. ILAR J. 2015, 56, 1–6. [Google Scholar] [CrossRef]
- Boettcher, A.N.; Li, Y.; Ahrens, A.P.; Kiupel, M.; Byrne, K.A.; Loving, C.L.; Cino-Ozuna, A.G.; Wiarda, J.; Adur, M.; Schultz, B.; et al. Novel Engraftment and T Cell Differentiation of Human Hematopoietic Cells in ART−/−IL2RG−/Y SCID Pigs. Front. Immunol. 2020, 11, 100. [Google Scholar] [CrossRef]
- Justice, M.J.; Dhillon, P. Using the mouse to model human disease: Increasing validity and reproducibility. Dis. Model. Mech. 2016, 9, 101–103. [Google Scholar] [CrossRef]
- Bosticardo, M.; Pala, F.; Calzoni, E.; Delmonte, O.M.; Dobbs, K.; Gardner, C.L.; Sacchetti, N.; Kawai, T.; Garabedian, E.K.; Draper, D.; et al. Artificial thymic organoids represent a reliable tool to study T-cell differentiation in patients with severe T-cell lymphopenia. Blood Adv. 2020, 4, 2611–2616. [Google Scholar] [CrossRef]
- Parent, A.V.; Russ, H.A.; Khan, I.; LaFlam, T.N.; Metzger, T.C.; Anderson, M.S.; Hebrok, M. Generation of Functional Thymic Epithelium from Human Embryonic Stem Cells that Supports Host T Cell Development. Cell Stem Cell 2013, 13, 219–229. [Google Scholar] [CrossRef]
- Chhatta, A.R.; Cordes, M.; Hanegraaf, M.A.; Vloemans, S.; Cupedo, T.; Cornelissen, J.J.; Carlotti, F.; Salvatori, D.; Pike-Overzet, K.; Fibbe, W.E.; et al. De novo generation of a functional human thymus from induced pluripotent stem cells. J. Allergy Clin. Immunol. 2019, 144, 1416–1419.e7. [Google Scholar] [CrossRef]
- Otsuka, R.; Wada, H.; Tsuji, H.; Sasaki, A.; Murata, T.; Itoh, M.; Baghdadi, M.; Seino, K.-I. Efficient generation of thymic epithelium from induced pluripotent stem cells that prolongs allograft survival. Sci. Rep. 2020, 10, 224. [Google Scholar] [CrossRef]
- Ramos, S.A.; Morton, J.J.; Yadav, P.; Reed, B.; Alizadeh, S.I.; Shilleh, A.H.; Perrenoud, L.; Jaggers, J.; Kappler, J.; Jimeno, A.; et al. Generation of functional human thymic cells from induced pluripotent stem cells. J. Allergy Clin. Immunol. 2021, in press. [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Adult Stem Cells | hESCs | hiPSCs | |
|---|---|---|---|
| Stem cell type |
PBSC
BM
UCB
|
|
|
| |||
| In vitro IEI models | In vivo IEI models | ||
| Model Architecture |
|
| |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zbinden, A.; Canté-Barrett, K.; Pike-Overzet, K.; Staal, F.J.T. Stem Cell-Based Disease Models for Inborn Errors of Immunity. Cells 2022, 11, 108. https://doi.org/10.3390/cells11010108
Zbinden A, Canté-Barrett K, Pike-Overzet K, Staal FJT. Stem Cell-Based Disease Models for Inborn Errors of Immunity. Cells. 2022; 11(1):108. https://doi.org/10.3390/cells11010108
Chicago/Turabian StyleZbinden, Aline, Kirsten Canté-Barrett, Karin Pike-Overzet, and Frank J. T. Staal. 2022. "Stem Cell-Based Disease Models for Inborn Errors of Immunity" Cells 11, no. 1: 108. https://doi.org/10.3390/cells11010108
APA StyleZbinden, A., Canté-Barrett, K., Pike-Overzet, K., & Staal, F. J. T. (2022). Stem Cell-Based Disease Models for Inborn Errors of Immunity. Cells, 11(1), 108. https://doi.org/10.3390/cells11010108