
Ang II-Induced Hypertension Exacerbates the Pathogenesis of Tuberculosis

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
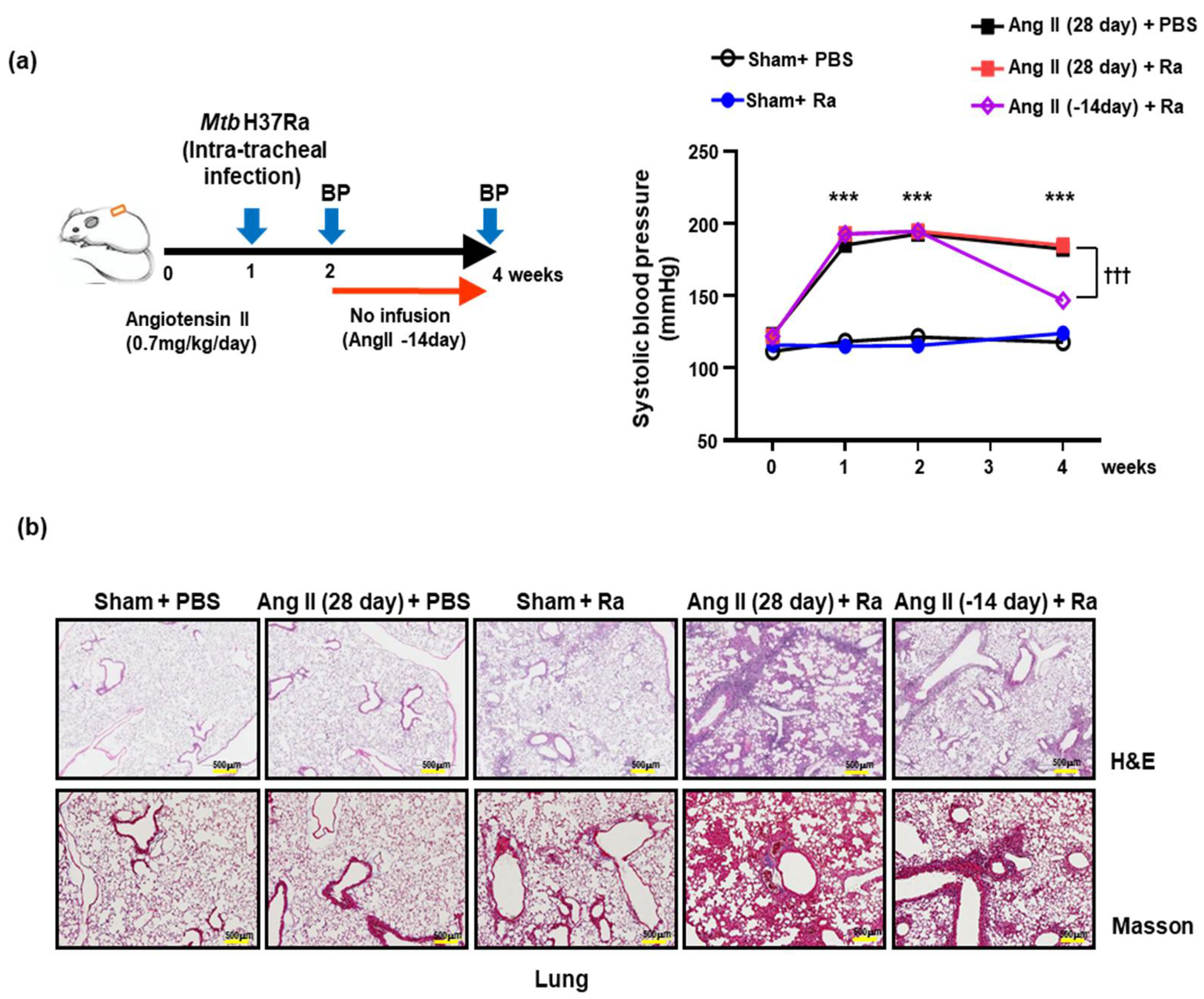
2.2. Hypertensive Animal Model and Blood Pressure Measurement
2.3. Histopathological Analysis and Scanning Microscopy
2.4. Hematoxylin and Eosin Stain
2.5. Acid-Fast Bacillus Stain
2.6. Immunohistochemistry (IHC)
2.7. Oil Red O Stain
2.8. Histological Analysis
2.9. Cell Culture and Bacterial Infection
2.10. Cytotoxicity Analysis
2.11. FACS Analysis for CD36 Surface Expression
2.12. LDH Cytotoxicity Detection
2.13. Western Blotting Analysis
2.14. Intracellular Survival Assay
2.15. Statistical Analysis
3. Results
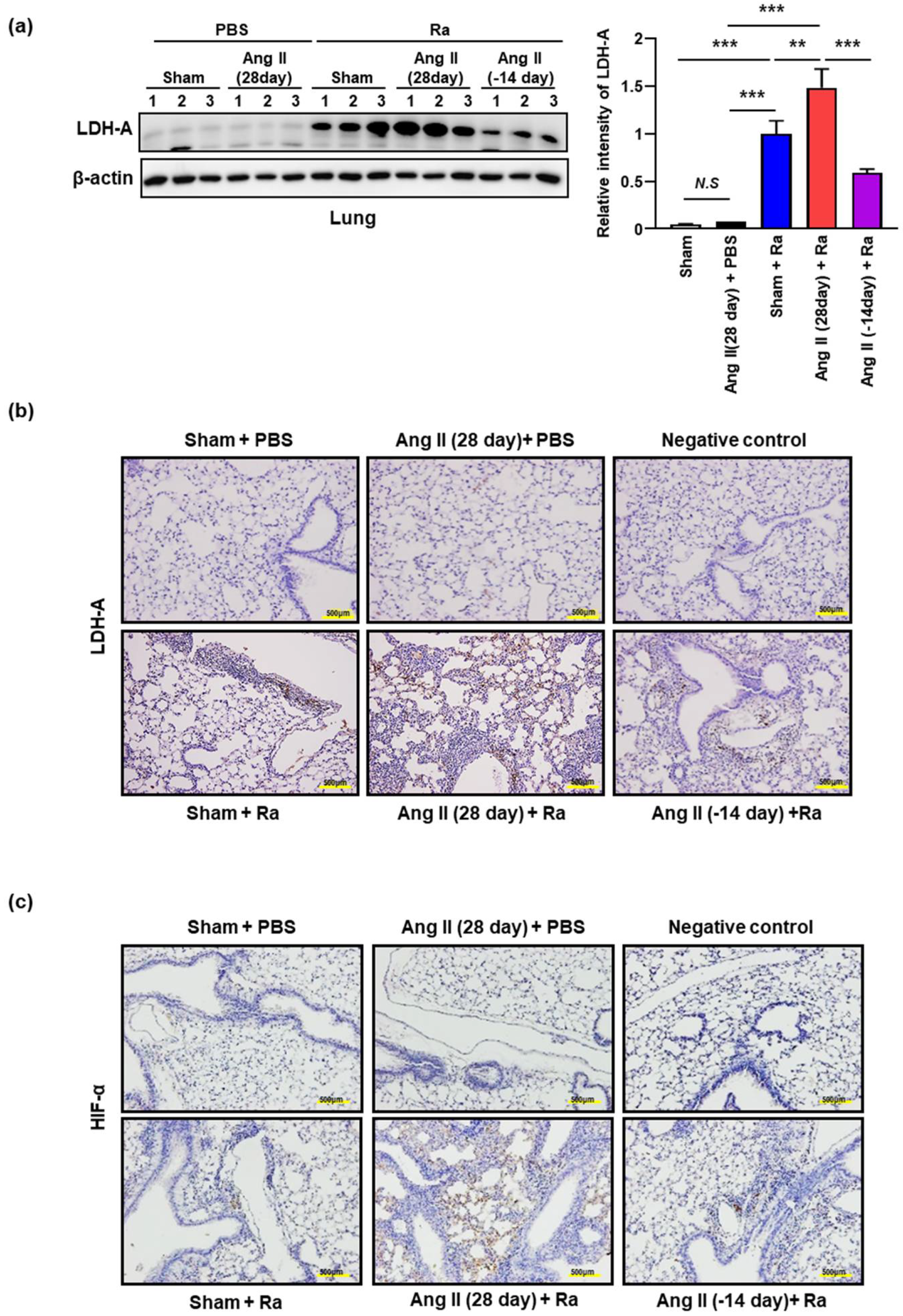
3.1. Ang II-Induced Hypertension Increases the Cell Death in Mtb Infected Mice Lungs
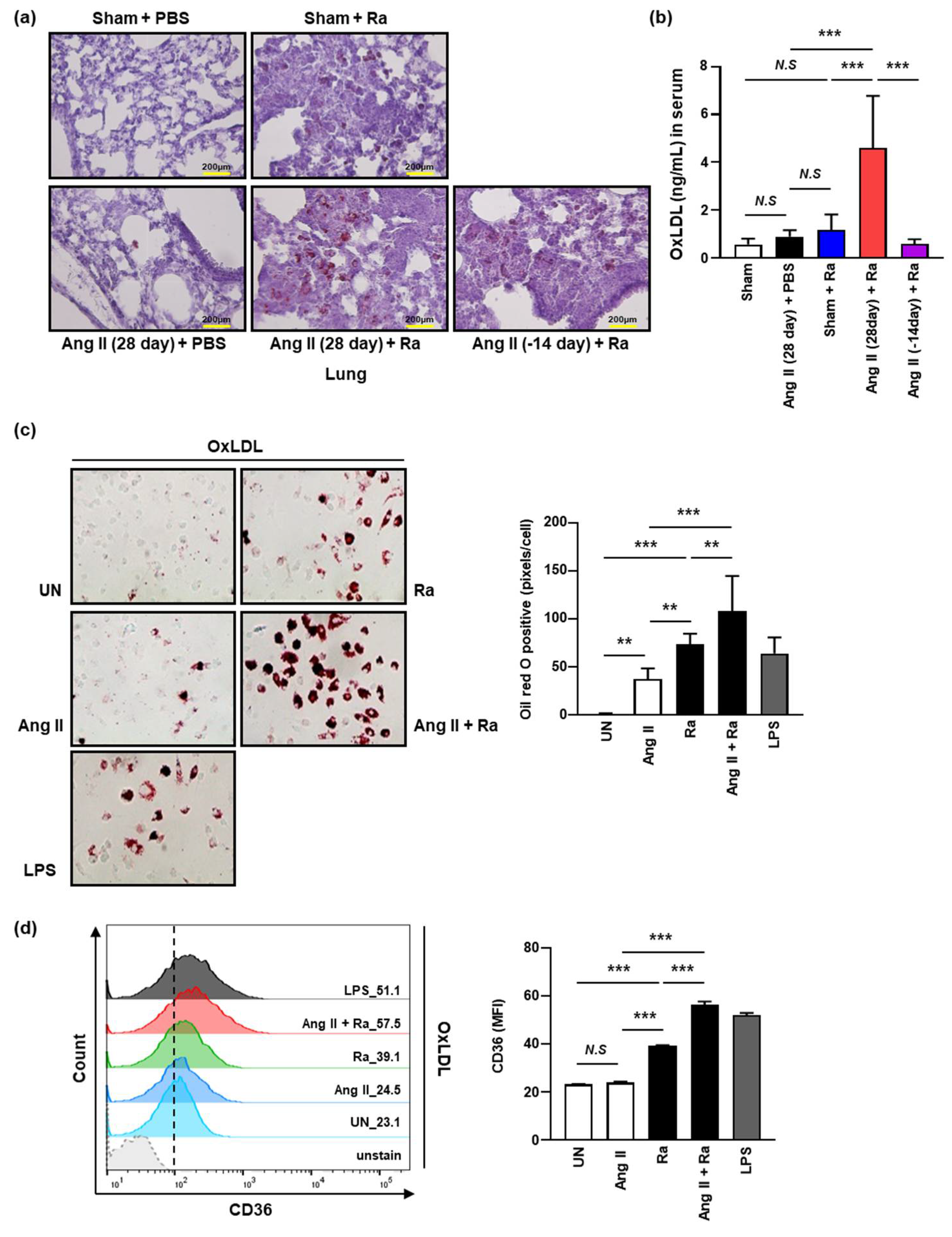
3.2. Ang II-Induced Hypertension Enhances the Accumulation of Intracellular Lipids by Increasing the Expression of CD36 on the Macrophage Surface during Mtb H37Ra Infection
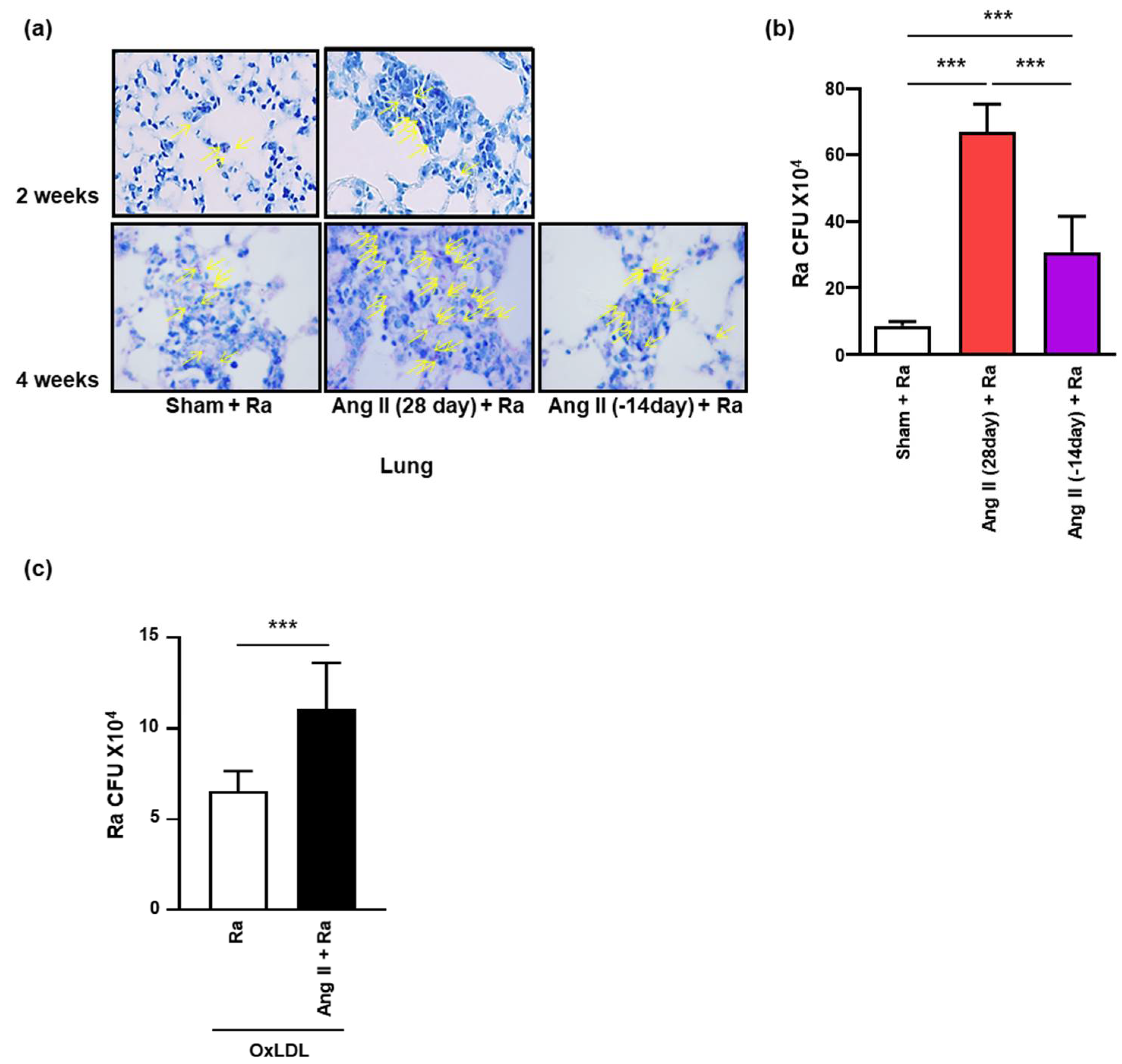
3.3. Ang II-Induced Hypertension Contributes to Intracellular Survival of Mtb in the Mice Lungs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zaman, K. Tuberculosis: A Global Health Problem. J. Heal. Popul. Nutr. 2010, 28, 111–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, D.F. Mycobacterium tuberculosis Metabolism. Cold Spring Harb. Perspect. Med. 2014, 5, a021121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seegert, A.B.; Rudolf, F.; Wejse, C.; Neupane, D. Tuberculosis and hypertension—A systematic review of the literature. Int. J. Infect. Dis. 2017, 56, 54–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, Y.S.; Park, J.H.; Lee, J.K.; Heo, E.Y.; Chung, H.S.; Kim, D.K. Risk factors for pulmonary arterial hypertension in patients with tu-berculosis-destroyed lungs and their clinical characteristics compared with patients with chronic obstructive pulmonary dis-ease. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokes, J., 3rd; Kannel, W.B.; Wolf, P.A.; D’Agostino, R.B.; Cupples, L.A. Blood pressure as a risk factor for cardiovascular disease. The Framingham Study--30 years of follow-up. Hypertension 1989, 13 (Suppl. 5), I13–I18. [Google Scholar] [CrossRef] [Green Version]
- Kreatsoulas, C.; Anand, S.S. The impact of social determinants on cardiovascular disease. Can. J. Cardiol. 2010, 26, 8C–13C. [Google Scholar] [CrossRef] [Green Version]
- Pauletto, P.; Rattazzi, M. Inflammation and hypertension: The search for a link. Nephrol. Dial. Transplant. 2006, 21, 850–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willerson, J.T.; Ridker, P.M. Inflammation as a cardiovascular risk factor. Circulation 2004, 109 (Suppl. 1), II-2–II-10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fyhrquist, F.; Metsarinne, K.; Tikkanen, I. Role of angiotensin II in blood pressure regulation and in the pathophysiology of car-diovascular disorders. J. Hum. Hypertens. 1995, 9 (Suppl. 5), S19–S24. [Google Scholar] [PubMed]
- Fliser, D.; Buchholz, K.; Haller, H. Antiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammation. Circulation 2004, 110, 1103–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieffer, B.; Bunte, C.; Witte, J.; Hoeper, K.; Boger, R.H.; Schwedhelm, E.; Drexler, H. Comparative effects of AT1-antagonism and angiotensin-converting enzyme inhibition on markers of inflammation and platelet aggregation in patients with coronary ar-tery disease. J. Am. Coll. Cardiol. 2004, 44, 362–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz-Rosa, D.; Oubina, M.P.; Cediel, E.; de Las Heras, N.; Vegazo, O.; Jimenez, J.; Lahera, V.; Cachofeiro, V. Effect of AT1 receptor antagonism on vascular and circulating inflammatory mediators in SHR: Role of NF-kappaB/IkappaB system. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H111–H115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.J.; Vapaatalo, H.; Mervaala, E. Angiotensin II and vascular inflammation. Med. Sci. Monit. 2005, 11, RA194–RA205. [Google Scholar]
- Khademi, F.; Vaez, H.; Momtazi, A.A.; Majnooni, A.; Banach, M.; Sahebkar, A. Bacterial infections are associated with cardiovascular disease in Iran: A meta-analysis. Arch. Med. Sci. 2019, 15, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.A.; Rosenfeld, M.E. Infection and Atherosclerosis Development. Arch. Med Res. 2015, 46, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Vercellotti, G.M. Overview of infections and cardiovascular diseases. J. Allergy Clin. Immunol. 2001, 108, S117–S120. [Google Scholar] [CrossRef]
- Mattila, K.J.; Valtonen, V.V.; Nieminen, M.S.; Asikainen, S. Role of infection as a risk factor for atherosclerosis, myocardial infarc-tion, and stroke. Clin. Infect. Dis. 1998, 26, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huaman, M.A.; Henson, D.; Ticona, E.; Sterling, T.R.; Garvy, B.A. Tuberculosis and cardiovascular disease: Linking the epidemics. Trop. Dis. Travel Med. Vaccines 2015, 1, 1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basham, C.; Smith, S.J.; Romanowski, K.; Johnston, J.C. Cardiovascular morbidity and mortality among persons diagnosed with tuberculosis: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0235821. [Google Scholar] [CrossRef]
- Vahdat, K.; Pourbehi, M.R.; Ostovar, A.; Hadavand, F.; Bolkheir, A.; Farrokhnia, M.; Assadi, M.; Nabipour, I. Association of Pathogen Burden and Hypertension: The Persian Gulf Healthy Heart Study. Am. J. Hypertens. 2013, 26, 1140–1147. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wu, J.; Sun, X.; Xue, H.; Shao, J.; Cai, W.; Jing, Y.; Yue, M.; Dong, C. Association of hypertension with the severity and fatal-ity of SARS-CoV-2 infection: A meta-analysis. Epidemiol. Infect. 2020, 148, e106. [Google Scholar] [CrossRef]
- Van Eeden, S.; Leipsic, J.; Paul Man, S.F.; Sin, D.D. The relationship between lung inflammation and cardiovascular disease. Am. J. Respir. Crit. Care Med. 2012, 186, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Haanen, C.; Vermes, I. Apoptosis and inflammation. Mediat. Inflamm. 1995, 4, 5–15. [Google Scholar] [CrossRef]
- Anderton, H.; Wicks, I.P.; Silke, J. Cell death in chronic inflammation: Breaking the cycle to treat rheumatic disease. Nat. Rev. Rheumatol. 2020, 16, 496–513. [Google Scholar] [CrossRef]
- Butler, R.E.; Brodin, P.; Jang, J.; Jang, M.-S.; Robertson, B.D.; Gicquel, B.; Stewart, G.R. The Balance of Apoptotic and Necrotic Cell Death in Mycobacterium tuberculosis Infected Macrophages Is Not Dependent on Bacterial Virulence. PLoS ONE 2012, 7, e47573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behar, S.M.; Martin, C.J.; Booty, M.G.; Nishimura, T.; Zhao, X.; Gan, H.X.; Divangahi, M.; Remold, H.G. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal. Immunol. 2011, 4, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Sasindran, S.J.; Torrelles, J.B. Mycobacterium Tuberculosis Infection and Inflammation: What is Beneficial for the Host and for the Bacterium? Front. Microbiol. 2011, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voskuil, M.I.; Bartek, I.L.; Evisconti, K.; Schoolnik, G.K. The Response of Mycobacterium Tuberculosis to Reactive Oxygen and Nitrogen Species. Front. Microbiol. 2011, 2, 105. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Farhana, A.; Guidry, L.; Saini, V.; Hondalus, M.; Steyn, A.J. Redox homeostasis in mycobacteria: The key to tuberculosis control? Expert Rev. Mol. Med. 2011, 13, e39. [Google Scholar] [CrossRef] [Green Version]
- Domingo-Gonzalez, R.; Prince, O.; Cooper, A.; Khader, S.A. Cytokines and Chemokines in Mycobacterium tuberculosis Infection. Microbiol. Spectr. 2016, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.M. Cell-Mediated Immune Responses in Tuberculosis. Annu. Rev. Immunol. 2009, 27, 393–422. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Hu, Y.; Coates, A. Sudden cardiac death and tuberculosis—How much do we know? Tuberculosis 2012, 92, 307–313. [Google Scholar] [CrossRef]
- Santucci, M.B.; Amicosante, M.; Cicconi, R.; Montesano, C.; Casarini, M.; Giosuè, S.; Bisetti, A.; Colizzi, V.; Fraziano, M. Mycobacterium tuberculosis–Induced Apoptosis in Monocytes/Macrophages: Early Membrane Modifications and Intracellular Mycobacterial Viability. J. Infect. Dis. 2000, 181, 1506–1509. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Song, C.-H. Effect of Reactive Oxygen Species on the Endoplasmic Reticulum and Mitochondria during Intracellular Pathogen Infection of Mammalian Cells. Antioxidants 2021, 10, 872. [Google Scholar] [CrossRef]
- Lim, Y.J.; Choi, J.A.; Choi, H.H.; Cho, S.N.; Kim, H.J.; Jo, E.K.; Park, J.K.; Song, C.H. Endoplasmic Reticulum Stress Pathway-Mediated Apoptosis in Macrophages Contributes to the Survival of Mycobacterium tuberculosis. PLoS ONE 2011, 6, e28531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, C.N. Endoplasmic reticulum stress in the pathogenesis of hypertension. Exp. Physiol. 2017, 102, 869–884. [Google Scholar] [CrossRef]
- Chan, F.K.-M.; Moriwaki, K.; De Rosa, M.J. Detection of Necrosis by Release of Lactate Dehydrogenase Activity. Stem Cells Aging 2013, 979, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Prosser, G.; Brandenburg, J.; Reiling, N.; Barry, C.E., 3rd; Wilkinson, R.J.; Wilkinson, K.A. The bacillary and macrophage response to hypoxia in tuberculosis and the consequences for T cell antigen recognition. Microbes Infect. 2017, 19, 177–192. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.G.; Han, Z.T.; He, S.H.; Wu, X.D.; Chen, T.R.; Shao, C.H.; Chen, D.L.; Su, N.; Chen, Y.M.; Wang, T.; et al. HIF1/2alpha mediates hy-poxia-induced LDHA expression in human pancreatic cancer cells. Oncotarget 2017, 8, 24840–24852. [Google Scholar] [CrossRef]
- Singh, V.; Jamwal, S.; Jain, R.; Verma, P.; Gokhale, R.; Rao, K.V. Mycobacterium tuberculosis-driven targeted recalibration of mac-rophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe 2012, 12, 669–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, D.; Kim, H.; Shin, S.J. Mycobacterium tuberculosis Infection-Driven Foamy Macrophages and Their Implications in Tu-berculosis Control as Targets for Host-Directed Therapy. Front. Immunol. 2020, 11, 910. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Harrison, D.G. Inflammation in Hypertension. Can. J. Cardiol. 2020, 36, 635–647. [Google Scholar] [CrossRef]
- Frostegard, J.; Ulfgren, A.-K.; Nyberg, P.; Hedin, U.; Swedenborg, J.; Andersson, U.; Hansson, G.K. Cytokine expression in advanced human atherosclerotic plaques: Dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis 1999, 145, 33–43. [Google Scholar] [CrossRef]
- Smith, J.D.; Trogan, E.; Ginsberg, M.; Grigaux, C.; Tian, J.; Miyata, M. Decreased atherosclerosis in mice deficient in both macro-phage colony-stimulating factor (op) and apolipoprotein E. Proc. Natl. Acad. Sci. USA 1995, 92, 8264–8268. [Google Scholar] [CrossRef] [Green Version]
- Agostini, C.; Gurrieri, C. Chemokine/cytokine cocktail in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 357–363. [Google Scholar] [CrossRef]
- Warsinske, H.C.; DiFazio, R.; Linderman, J.J.; Flynn, J.L.; Kirschner, D.E. Identifying mechanisms driving formation of granuloma-associated fibrosis during Mycobacterium tuberculosis infection. J. Theor. Biol. 2017, 429, 1–17. [Google Scholar] [CrossRef]
- Rios, F.J.; Koga, M.M.; Pecenin, M.; Ferracini, M.; Gidlund, M.; Jancar, S. Oxidized LDL Induces Alternative Macrophage Phenotype through Activation of CD36 and PAFR. Mediat. Inflamm. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Rios, F.J.O.; Ferracini, M.; Pecenin, M.; Koga, M.M.; Wang, Y.; Ketelhuth, D.F.J.; Jancar, S. Uptake of oxLDL and IL-10 Production by Macrophages Requires PAFR and CD36 Recruitment into the Same Lipid Rafts. PLoS ONE 2013, 8, e76893. [Google Scholar] [CrossRef]
- Maggi, E.; Marchesi, E.; Ravetta, V.; Falaschi, F.; Finardi, G.; Bellomo, G. Low-density lipoprotein oxidation in essential hyperten-sion. J. Hypertens. 1993, 11, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G.; Cardona, P.-J.; Kim, M.-J.; Allain, S.; Altare, F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 2009, 10, 943–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, S.; Havelka, A.M.; Ueno, T.; Shoshan, M.C. Determining tumor apoptosis and necrosis in patient serum using cytokeratin 18 as a biomarker. Cancer Lett. 2004, 214, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Quist, J.; Hill, A.R. Serum lactate dehydrogenase (LDH) in Pneumocystis carinii pneumonia, tuberculosis, and bacterial pneu-monia. Chest 1995, 108, 415–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, C.J.; Booty, M.G.; Rosebrock, T.R.; Nunes-Alves, C.; Desjardins, D.M.; Keren, I.; Fortune, S.M.; Remold, H.G.; Behar, S.M. Efferocy-tosis Is an Innate Antibacterial Mechanism. Cell Host Microbe 2012, 12, 289–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, G.; Schroeder, R.; Stahl, R.A. Angiotensin II induces hypoxia-inducible factor-1 alpha in PC 12 cells through a posttran-scriptional mechanism: Role of AT2 receptors. Am. J. Nephrol. 2004, 24, 415–421. [Google Scholar] [CrossRef]
- Divangahi, M.; Chen, M.; Gan, H.; Desjardins, D.; Hickman, T.T.; Lee, D.M.; Fortune, S.; Behar, S.M.; Remold, H.G. Mycobacterium tu-berculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 2009, 10, 899–906. [Google Scholar] [CrossRef] [Green Version]
- Pedruzzi, G.; Das, P.N.; Rao, K.V.; Chatterjee, S. Understanding PGE2, LXA4 and LTB4 balance during Mycobacterium tuberculo-sis infection through mathematical model. J. Theor. Biol. 2016, 389, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Gago, G.; Diacovich, L.; Gramajo, H. Lipid metabolism and its implication in mycobacteria–host interaction. Curr. Opin. Microbiol. 2018, 41, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Nazarova, E.V.; Russell, D.G. Mycobacterium tuberculosis: Bacterial Fitness within the Host Macrophage. Microbiol. Spectr. 2019, 7, 127–138. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, S.-N.; Choi, J.-A.; Lee, J.; Son, S.-H.; Lee, S.-A.; Nguyen, T.-D.; Choi, S.-Y.; Song, C.-H. Ang II-Induced Hypertension Exacerbates the Pathogenesis of Tuberculosis. Cells 2021, 10, 2478. https://doi.org/10.3390/cells10092478
Cho S-N, Choi J-A, Lee J, Son S-H, Lee S-A, Nguyen T-D, Choi S-Y, Song C-H. Ang II-Induced Hypertension Exacerbates the Pathogenesis of Tuberculosis. Cells. 2021; 10(9):2478. https://doi.org/10.3390/cells10092478
Chicago/Turabian StyleCho, Soo-Na, Ji-Ae Choi, Junghwan Lee, Sang-Hun Son, Seong-Ahn Lee, Tam-Doan Nguyen, Song-Yi Choi, and Chang-Hwa Song. 2021. "Ang II-Induced Hypertension Exacerbates the Pathogenesis of Tuberculosis" Cells 10, no. 9: 2478. https://doi.org/10.3390/cells10092478
APA StyleCho, S.-N., Choi, J.-A., Lee, J., Son, S.-H., Lee, S.-A., Nguyen, T.-D., Choi, S.-Y., & Song, C.-H. (2021). Ang II-Induced Hypertension Exacerbates the Pathogenesis of Tuberculosis. Cells, 10(9), 2478. https://doi.org/10.3390/cells10092478