
Assigning Co-Regulated Human Genes and Regulatory Gene Clusters



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genotype and Sample Quality Control
2.2. eQTL Calculation
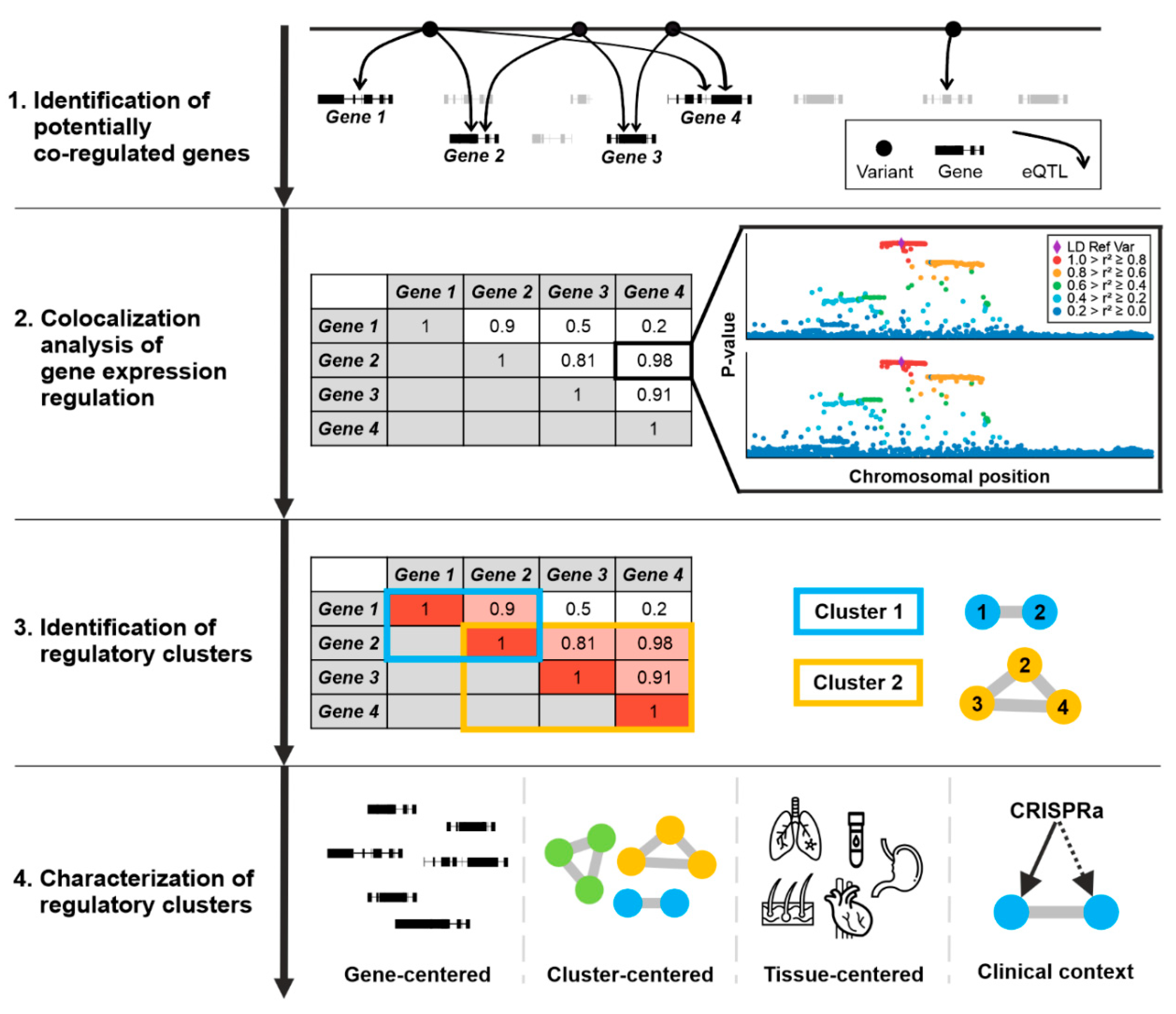
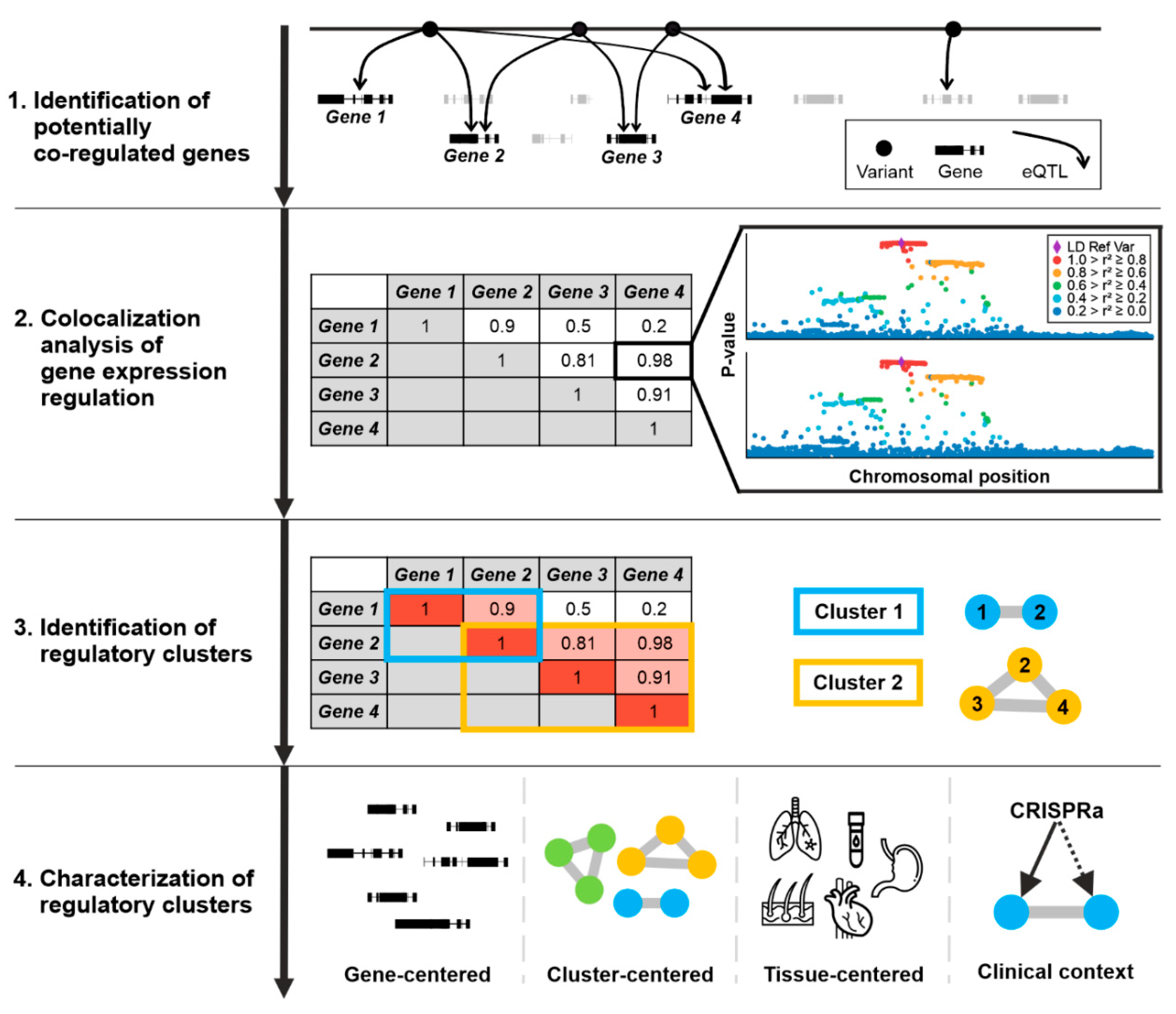
2.3. Identification of Gene Expression Regulation Cluster
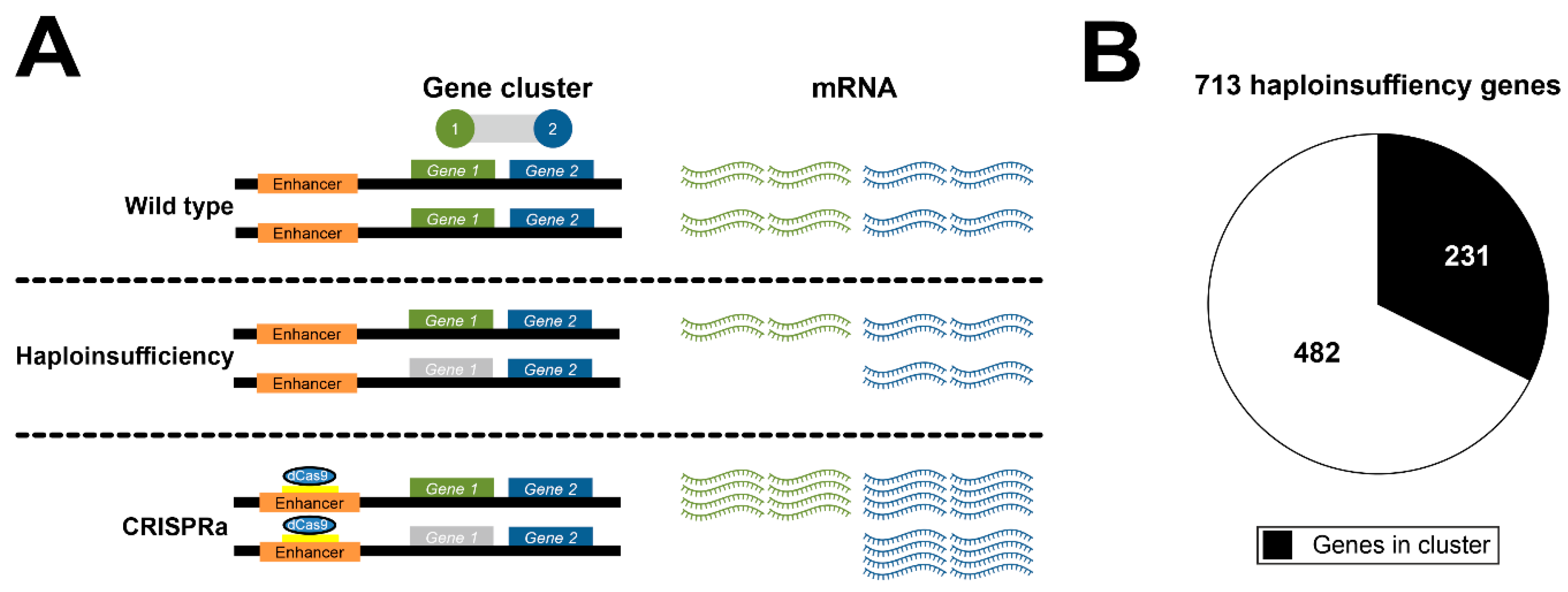
2.4. Collection of Haploinsufficiency Genes
3. Results
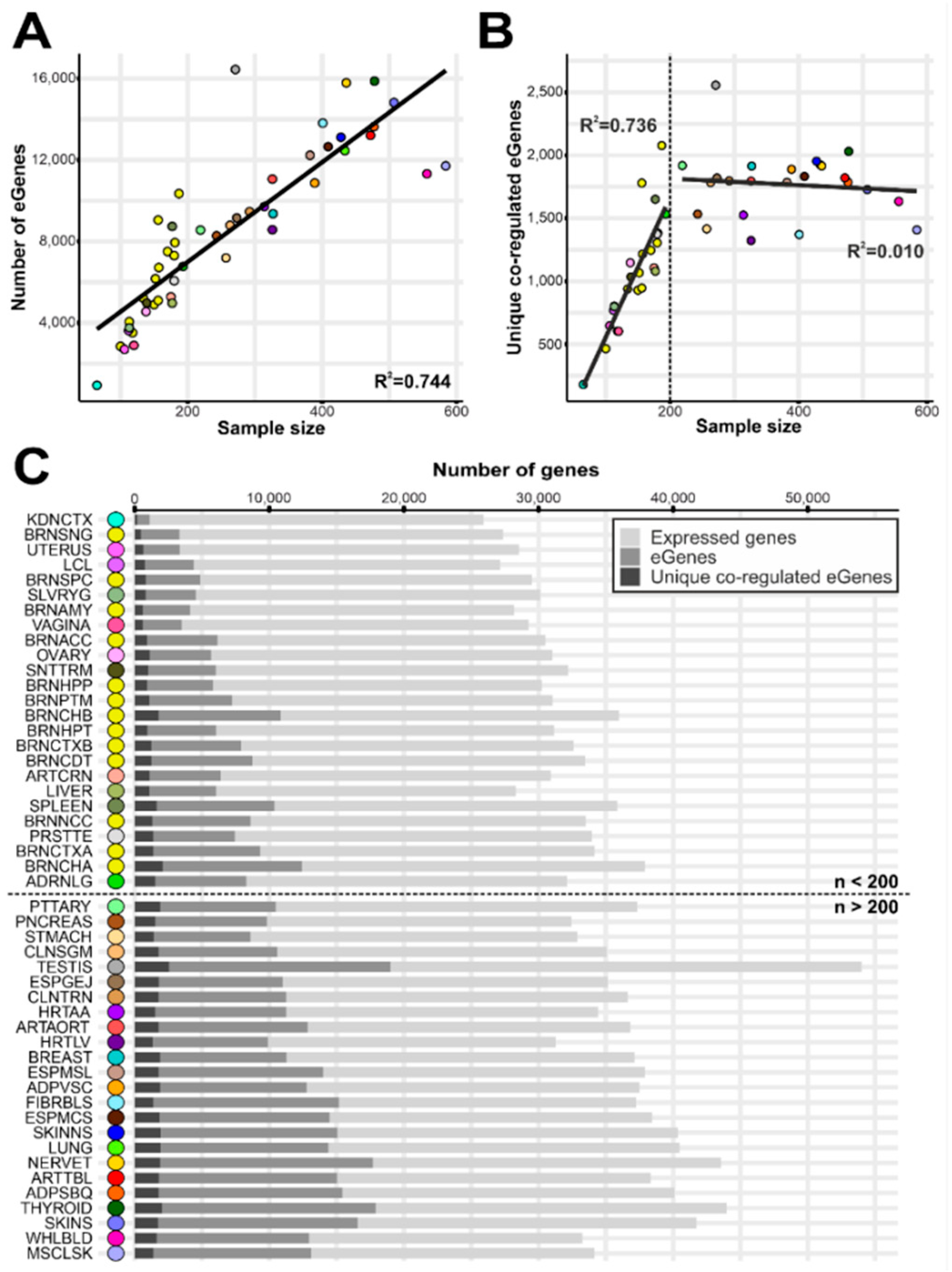
3.1. Identification of Co-Regulated Genes
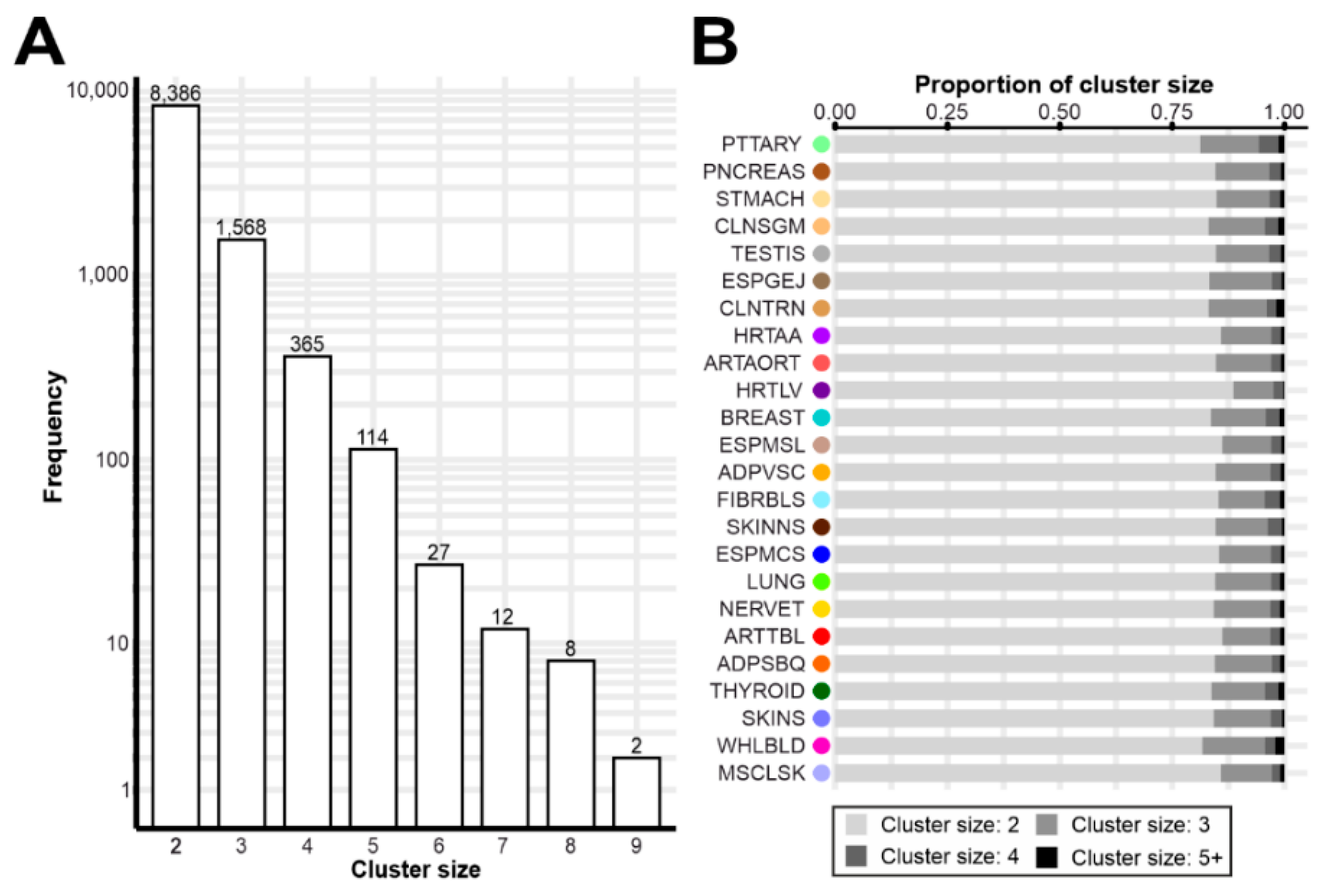
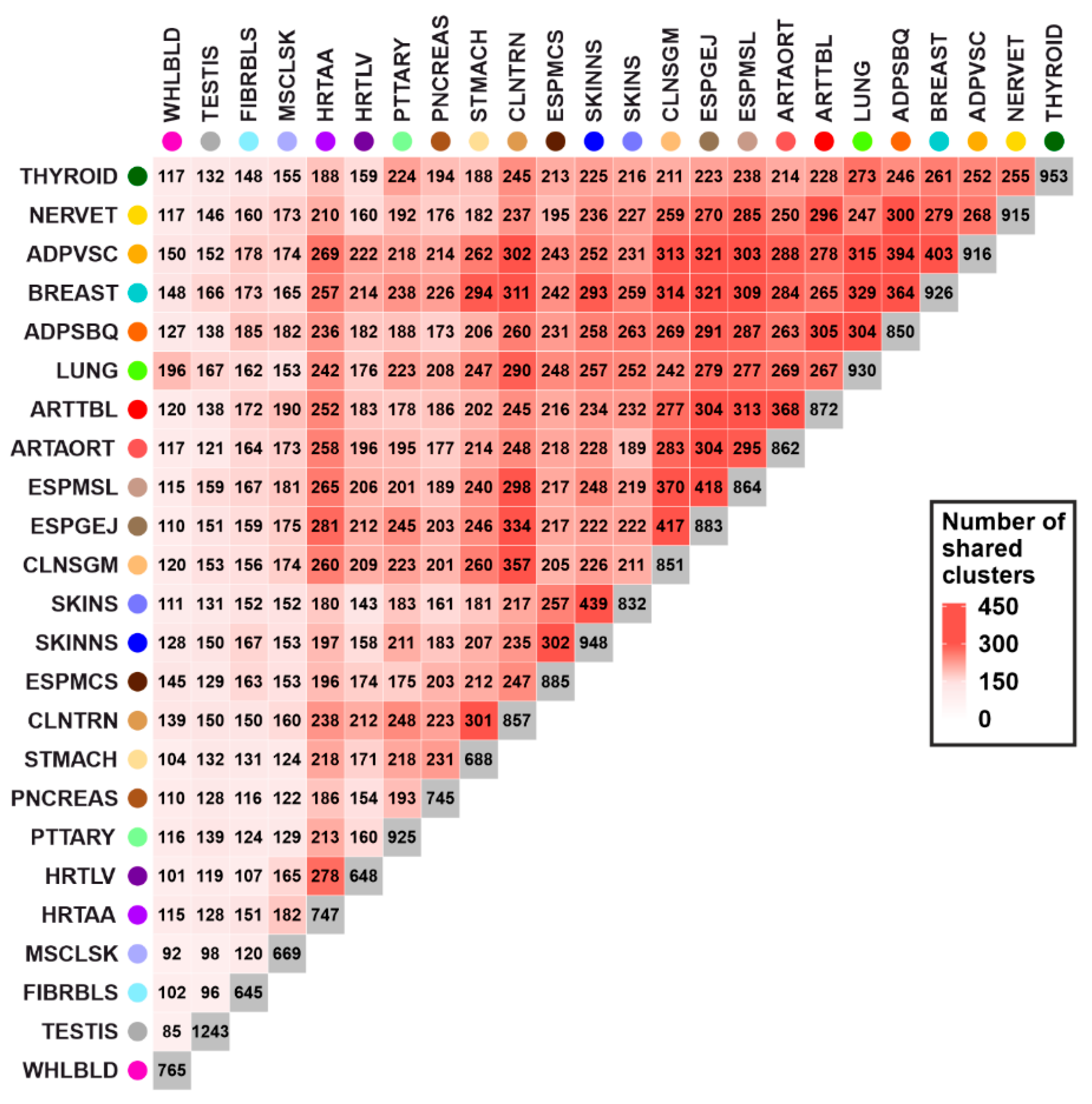
3.2. Gene Expression Regulation Cluster
3.3. Potential Clinical Consequences for Genes within Expression Regulation Clusters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- French, J.D.; Edwards, S.L. The role of noncoding variants in heritable disease. Trends Genet. 2020, 36, 880–891. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.C.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-γuided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, Z. CRISPR-Cas systems: Overview, innovations and applications in human disease research and gene therapy. Comput. Struct. Biotechnol. J. 2020, 18, 2401–2415. [Google Scholar] [CrossRef]
- Matharu, N.; Rattanasopha, S.; Tamura, S.; Maliskova, L.; Wang, Y.; Bernard, A.; Hardin, A.; Eckalbar, W.L.; Vaisse, C.; Ahituv, N. CRISPR-mediated activation of a promoter or enhancer rescues obesity caused by haploinsufficiency. Science 2019, 363, eaau0629. [Google Scholar] [CrossRef]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Denker, A.; de Laat, W. The second decade of 3C technologies: Detailed insights into nuclear organization. Genes Dev. 2016, 30, 1357–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullwood, M.J.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Mohamed, Y.B.; Orlov, Y.L.; Velkov, S.; Ho, A.; Mei, P.H.; et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, K.; Forcato, M.; Ferrari, F. Hi-C analysis: From data generation to integration. Biophys. Rev. 2019, 11, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cookson, W.; Liang, L.; Abecasis, G.; Moffatt, M.; Lathrop, M. Mapping complex disease traits with global gene expression. Nat. Rev. Genet. 2009, 10, 184–194. [Google Scholar] [CrossRef]
- Strunz, T.; Grassmann, F.; Gayán, J.; Nahkuri, S.; Souza-Costa, D.; Maugeais, C.; Fauser, S.; Nogoceke, E.; Weber, B.H.F. A mega-analysis of expression quantitative trait loci (eQTL) provides insight into the regulatory architecture of gene expression variation in liver. Sci. Rep. 2018, 8, 5865. [Google Scholar] [CrossRef] [PubMed]
- Schramm, K.; Marzi, C.; Schurmann, C.; Carstensen, M.; Reinmaa, E.; Biffar, R.; Eckstein, G.; Gieger, C.; Grabe, H.J.; Homuth, G.; et al. Mapping the genetic architecture of gene regulation in whole blood. PLoS ONE 2014, 9, e93844. [Google Scholar] [CrossRef]
- GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Ardlie, K.G.; Deluca, D.S.; Segre, A.V.; Sullivan, T.J.; Young, T.R.; Gelfand, E.T.; Trowbridge, C.A.; Maller, J.B.; Tukiainen, T.; Lek, M.; et al. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [Green Version]
- Strunz, T.; Kiel, C.; Grassmann, F.; Ratnapriya, R.; Kwicklis, M.; Karlstetter, M.; Fauser, S.; Arend, N.; Swaroop, A.; Langmann, T.; et al. A mega-analysis of expression quantitative trait loci in retinal tissue. PLoS Genet. 2020, 16, e1008934. [Google Scholar] [CrossRef]
- Giambartolomei, C.; Vukcevic, D.; Schadt, E.E.; Franke, L.; Hingorani, A.D.; Wallace, C.; Plagnol, V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014, 10, e1004383. [Google Scholar] [CrossRef] [Green Version]
- Wallace, C. Eliciting priors and relaxing the single causal variant assumption in colocalisation analyses. PLoS Genet. 2020, 16, e1008720. [Google Scholar] [CrossRef] [Green Version]
- Hormozdiari, F.; van de Bunt, M.; Segrè, A.V.; Li, X.; Joo, J.W.J.; Bilow, M.; Sul, J.H.; Sankararaman, S.; Pasaniuc, B.; Eskin, E. Colocalization of GWAS and eQTL signals detects target genes. Am. J. Hum. Genet. 2016, 99, 1245–1260. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- R Team Core. R: A language and environment for statistical computing. R Found. Stat. Comput. Vienna Austria. 2017, 7, 2017. [Google Scholar]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef] [Green Version]
- Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; Gabriel, S.B.; et al. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Gay, N.R.; Gloudemans, M.; Antonio, M.L.; Abell, N.S.; Balliu, B.; Park, Y.; Martin, A.R.; Musharoff, S.; Rao, A.S.; Aguet, F.; et al. Impact of admixture and ancestry on eQTL analysis and GWAS colocalization in GTEx. Genome Biol. 2020, 21, 233. [Google Scholar] [CrossRef] [PubMed]
- GTEx Portal datasets. Available online: https://www.gtexportal.org/home/datasets (accessed on 7 July 2020).
- Ongen, H.; Buil, A.; Brown, A.A.; Dermitzakis, E.T.; Delaneau, O. Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics 2016, 32, 1479–1485. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex. Syst. 2006, 1695, 1–9. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Use R! Springer International Publishing: Berlin/Heidelberg, Germany, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen—The clinical genome resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [Green Version]
- Riggs, E.; Church, D.; Hanson, K.; Horner, V.; Kaminsky, E.; Kuhn, R.; Wain, K.; Williams, E.; Aradhya, S.; Kearney, H.; et al. Towards an evidence-based process for the clinical interpretation of copy number variation. Clin. Genet. 2012, 81, 403–412. [Google Scholar] [CrossRef]
- ClinGen Dosage Sensitivity Map. Available online: https://dosage.clinicalgenome.org/ (accessed on 15 January 2021).
- Ward, L.D.; Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat. Biotechnol. 2012, 30, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- Knight, J.C. Regulatory polymorphisms underlying complex disease traits. J. Mol. Med. 2005, 83, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Smemo, S.; Tena, J.J.; Kim, K.-H.H.; Gamazon, E.R.; Sakabe, N.J.; Gómez-Marín, C.; Aneas, I.; Credidio, F.L.; Sobreira, D.R.; Wasserman, N.F.; et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 2014, 507, 371–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankaran, V.G.; Menne, T.F.; Xu, J.; Akie, T.E.; Lettre, G.; Van Handel, B.; Mikkola, H.K.A.; Hirschhorn, J.N.; Cantor, A.B.; Orkin, S.H. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 2008, 322, 1839–1842. [Google Scholar] [CrossRef] [Green Version]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suvà, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schöpflin, R.; Kraft, K.; Kempfer, R.; Jerković, I.; Chan, W.L.; et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef]
- Szabo, Q.; Bantignies, F.; Cavalli, G. Principles of genome folding into topologically associating domains. Sci. Adv. 2019, 5, eaaw1668. [Google Scholar] [CrossRef] [Green Version]
- Kim-Hellmuth, S.; Aguet, F.; Oliva, M.; Muñoz-Aguirre, M.; Kasela, S.; Wucher, V.; Castel, S.E.; Hamel, A.R.; Viñuela, A.; Roberts, A.L.; et al. Cell type-specific genetic regulation of gene expression across human tissues. Science 2020, 369, eaaz8528. [Google Scholar] [CrossRef] [PubMed]
- Glastonbury, C.A.; Couto Alves, A.; El-Sayed Moustafa, J.S.; Small, K.S. Cell-type heterogeneity in adipose tissue is associated with complex traits and reveals disease-relevant cell-specific eQTLs. Am. J. Hum. Genet. 2019, 104, 1013–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montavon, T.; Soshnikova, N.; Mascrez, B.; Joye, E.; Thevenet, L.; Splinter, E.; De Laat, W.; Spitz, F.; Duboule, D. A regulatory archipelago controls hox genes transcription in digits. Cell 2011, 147, 1132–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakany, J.; Duboule, D. The role of Hox genes during vertebrate limb development. Curr. Opin. Genet. Dev. 2007, 17, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.G.; Brower, C.S.; Wang, H.; Davydov, I.V.; Sheng, J.; Zhou, J.; Yong, T.K.; Varshavsky, A. Arginyltransferase, its specificity, putative substrates, bidirectional promoter, and splicing-derived isoforms. J. Biol. Chem. 2006, 281, 32559–32573. [Google Scholar] [CrossRef] [Green Version]
- Kalinina, M.; Skvortsov, D.; Kalmykova, S.; Ivanov, T.; Dontsova, O.; Pervouchine, D.D. Multiple competing RNA structures dynamically control alternative splicing in the human ATE1 gene. Nucleic Acids Res. 2021, 49, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.J.D.; Irvine, A.D.; Terron-Kwiatkowski, A.; Sandilands, A.; Campbell, L.E.; Zhao, Y.; Liao, H.; Evans, A.T.; Goudie, D.R.; Lewis-Jones, S.; et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat. Genet. 2006, 38, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Meyer-Hoffert, U.; Reithmayer, K.; Paus, R.; Hansmann, B.; He, Y.; Bartels, J.; Gläser, R.; Harder, J.; Schröder, J.M. Highly complex peptide aggregates of the S100 fused-type protein hornerin are present in human skin. J. Investig. Dermatol. 2009, 129, 1446–1458. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Park, J.; Kim, J.S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef] [Green Version]
- Fennell, T.; Isik, M.; Wang, T.; Gotta, G.; Zhang, D.; Wilson, C.J.; Marco, E. CALITAS: A CRISPR/Cas-aware ALigner for In silico off-TArget Search. Cris. J. 2021, 4, 264–274. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strunz, T.; Kellner, M.; Kiel, C.; Weber, B.H.F. Assigning Co-Regulated Human Genes and Regulatory Gene Clusters. Cells 2021, 10, 2395. https://doi.org/10.3390/cells10092395
Strunz T, Kellner M, Kiel C, Weber BHF. Assigning Co-Regulated Human Genes and Regulatory Gene Clusters. Cells. 2021; 10(9):2395. https://doi.org/10.3390/cells10092395
Chicago/Turabian StyleStrunz, Tobias, Martin Kellner, Christina Kiel, and Bernhard H. F. Weber. 2021. "Assigning Co-Regulated Human Genes and Regulatory Gene Clusters" Cells 10, no. 9: 2395. https://doi.org/10.3390/cells10092395
APA StyleStrunz, T., Kellner, M., Kiel, C., & Weber, B. H. F. (2021). Assigning Co-Regulated Human Genes and Regulatory Gene Clusters. Cells, 10(9), 2395. https://doi.org/10.3390/cells10092395