1. Introduction
Profilin is a monomeric (G-)actin-binding protein needed in all non-muscle cells to maintain a G-actin pool necessary for the fast actin dynamics of these cells. Compared to other G-actin-binding proteins, profilin has certain unique functions that have raised interest since its discovery more than 40 years ago [
1]. Profilin has a catalytic activity that increases the exchange rate of ADP with ATP on G-actin by 1000 times [
2], a function that is fundamental to generate ATP-loaded G-actin subunits that facilitate actin filament growth (F-actin) through efficient addition of the actin monomer to the filaments’ barbed end [
3,
4]. In addition, profilin interacts with phosphatidylinositol 4,5-bisphosphate at the plasma membrane [
5], possibly to transport polymerization-competent actin monomers to the site of filament growth. Profilin’s amino-acid sequence is not particularly well conserved during evolution, but its structure and actin-binding properties, as well as the poly-
l-proline (PLP)-interacting domain, are preserved across the animal kingdom and down to unicellular organisms. Already in
D. discoideum, profilin has diverged into multiple isoforms that, in higher organisms, have acquired cell-specific expression patterns and possibly unique functions. In mammals, four profilin isoforms have been found. Profilin 1 is ubiquitously expressed [
6], while profilin 2 is highly expressed in neurons in the entire central and peripheral nervous system [
6,
7], in testis [
8], and to a much lesser extent in kidney, thymus, and spleen [
6,
9]. Profilin 3 and 4 are only found in testis [
10,
11]. Therefore, in the mammalian brain, the profilin paralogs 1 and 2 were found expressed at high levels, while they were shown to participate in different complexes and cellular pathways, most likely through a unique poly-
l-proline-binding domain [
6,
12].
Mouse models lacking one or the other isoform have shed some light on the specific functions of profilin 1 and 2 in vivo. Conventional knockout of
profilin 1 (
Pfn1) has shown its requirement for cell division in early embryos, with the mutant zygote not reaching the four-cell stage [
9]. The absence of profilin 2 expression at early embryonic stages precludes any compensatory function. The conventional knockout of
profilin 2 (
Pfn2), instead, resulted in a fairly normal embryonic development. Despite profilin 2 expression as early as embryonic day (E) 10.5 [
9], the central nervous system (CNS) developed normally in mutant mice. However, we observed striking alterations of the synaptic physiology in mutant neurons, such as hyperexcitability of glutamatergic neurons due to increased neurotransmitter release probability, resulting in behavioral alterations and epileptic-like seizures in the animals [
7]. In this mouse model, profilin 1, although present in the same neuronal compartments [
13], was not able to compensate for the loss of profilin 2, suggesting that the regulation of synaptic activity in higher eukaryotes is a specific function of profilin 2. On the contrary, postnatal deletion of
profilin 1 in mature neurons of the forebrain, using a
Camk2a-Cre mouse line, had no effect on excitatory neurons morphology and physiology [
14]. Instead, deletion of
Pfn1 in neuronal precursor cells (NPCs) of the developing CNS, using a
Nes-Cre mouse line, affected mainly radial migration of cerebellar granule neurons due to deficits in cell–cell adhesion to the radial glia cells [
15,
16] and reduced Purkinje cell survival starting already 2 weeks after birth in a non-cell-autonomous fashion [
17]. Yet, cell proliferation and granule neuron–Purkinje cell physiology in the postnatal cerebellum were not affected. In the same context, nevertheless, defective establishment of the cleavage plane of apical radial glia cells in the ventricular zone of the telencephalon was reported. This did not appear to affect cell division, but mildly altered cortical development [
18]. It is possible that, in these cell types, profilin 2 can compensate for the functions of profilin 1 in cell division that were reported for preimplantation embryonic development.
In order to better understand the specific and redundant functions of profilin 1 and 2 during brain development and in the adult brain, we generated and analyzed two mouse models. To study brain development, we depleted both profilins at mid-gestation by combining the conventional
Pfn2 knockout allele [
7] with the conditional inactivation of
Pfn1, using the
Pfn1-flox allele [
19] and
Nes-Cre-mediated deletion.
Nes-Cre expression was detected in neuronal precursor cells of the CNS starting at about E9 [
20]. To study the profilin requirement in adult neurons, the
Pfn1 allele was inactivated postnatally using a
Camk2a-Cre mouse line in the mentioned
Pfn2 knockout background.
Camk2a-Cre is expressed in forebrain glutamatergic neurons and striatal medium spiny neurons starting around postnatal day (P) 18 [
21]. Our data show that embryos lacking both profilins starting from E9 develop a normal body axis, but completely lack forebrain, midbrain, and hindbrain structures, resulting in an embryonic lethal phenotype. This phenotype is completely rescued by the presence of a single
Pfn1 allele; however, it is only partially rescued by a single
Pfn2 allele. We observed a specific requirement of profilin 1 for proper embryonic cerebral cortex formation, while profilin 2 can support only hind- and midbrain development due to its lower expression level. Inactivation of both profilins in the adult brain resulted in a progressive collapse of dendritic arborizations of cortical and hippocampal pyramidal neurons. A partial rescue of the phenotype by either a single
Pfn2 or
Pfn1 allele could be observed, dependent on the expression level of the respective isoform. This finding suggests similar functions of both profilins in the adult neurons with respect to their structural integrity.
2. Materials and Methods
Mice. The
profilin 2 knockout (
Pfn2tm1(lacZ)Wit, herein denoted as
Pfn2het and
Pfn2ko) mouse model was previously described [
7] (
http://www.informatics.jax.org/allele/key/54529, accessed on 1 September 2021). The
profilin 1 flox mouse line (
Pfn1tm1.1(loxPs)Ref), a generous gift from R. Fässler [
19] (
http://www.informatics.jax.org/allele/key/67261, accessed on 1 September 2021), was crossed with either the
Nes-Cre driver line [
20] (
http://www.informatics.jax.org/allele/key/6206, accessed on 1 September 2021) to generate a mid-embryonic conditional deletion of
profilin 1 in neuroepithelial cells (herein denoted as n-
Pfn1het or n-
Pfn1ko) or the
Camk2a-Cre driver line [
21] (
http://www.informatics.jax.org/allele/key/6312, accessed on 1 September 2021) for conditional deletion of
profilin 1 in postnatal forebrain neurons (herein denoted as n-
Pfn1het or n-
Pfn1ko). The
Pfn2 knockout and the
Pfn1 conditional knockout lines were crossed to generate the double
profilin knockout (herein denoted as n-dko). All genotypes used in the experiments were obtained as littermates according to the mating scheme in
Figure S1A,B. As also indicated in
Figure S1, controls (ctrl) in the embryonic studies were
Pfn1wt/wt;
Pfn2wt/wt or
Pfn1flx/wt;
Pfn2wt/wt, while controls in the adult studies were
Pfn1flx/wt;
Pfn2wt/wt or
Pfn1flx/flx;
Pfn2wt/wt. Since the two Cre mouse lines were obtained by transgenic insertion, the presence of one Cre allele is indicated by the simple line name in the genotype (see
Figure S1A,B). All mouse lines were extensively back-crossed (>10 times) into C57Bl/6NCrl background. Mouse genotyping was performed by PCR as schematically shown in
Figure S1C. Mice were socially housed with a standard 12 h light/dark cycle at 22 °C and 50–55% humidity, with free access to water and food pellets. Line breeding and experiments were performed according to European regulations and local permission (AZ 84-02.04.2013.A233, AZ 84-02.04.2017.A088).
Histology. E11.5, E14.5, and E16.5 embryos were dissected, photographed, and fixed O/N in 4% formaldehyde solution in PBS at 4 °C, then dehydrated in an increasing alcohol concentration series followed by xylene, then impregnated in paraffin O/N at 60 °C, and lastly transferred into plastic molds to solidify according to a classical protocol. E14.5 and E16.5 embryos were decapitated to facilitate paraffin embedding. The embryos were then serially cut sagittally using a microtome with 8 μm thickness. The sections were then rehydrated with an inverse xylene/alcohol series and stained with Meyer’s hemalum (Merck KGaA, Darmstadt, DE, 109249) and eosin Y (Sigma, St. Louis, MO, USA, HT110132).
Immunofluorescence. E11.5 embryos were fixed O/N in 4% formaldehyde at 4 °C and exchanged in sucrose (15% and 30% in PBS), then frozen in Tissue-Tek® in a mold on powdered dry ice, and sagittally cut with 14 μm thickness at a cryostat. Sections were collected on Superfrost slides, dried for 30 min at RT and stored at −80 °C. For the immunofluorescence, the frozen slides were quickly dipped in 4% formaldehyde to postfix the slices and blocked for 2 h at RT in 5% goat serum and 2% DMSO in TBS-T (Tris-buffered saline with 0.05% TritonX-100). Primary antibodies were diluted in blocking solution and incubated O/N in a humidified chamber. Washes were performed with TBS-T, and secondary antibody incubation was again conducted in blocking solution for 2 h at RT. Primary antibodies used were anti-phospho-histone 3 (Ser10) rabbit pcl (Upstate/Merck, 06-570, 1:500) and anti-βIII-tubulin mouse mcl (Promega, Madison, WI, USA, G7121, 1:1000). Alexa 488-conjugated anti-rabbit and Alexa 594-conjugated anti-mouse secondary antibodies (Thermo Fisher Scientific, Waltham, MA, USA, 1:400) were used. Nuclei were stained with DAPI (Sigma, 0.2 μg/mL). Images were taken with a Keyence (Osaka, Japan) BZ-9000 microscope.
P80–90 mice were sacrificed by cervical dislocation, and the brain was quickly dissected on ice, washed in PBS, and fixed in 4% formaldehyde in PBS for 40 h at 4 °C. The brain was then sliced coronally using a vibratome with 40 μm thickness. Slices were stored in PBS at 4 °C until use. Immunofluorescence staining was performed according to the classical protocol for floating sections. In brief, sections were treated with 50 mM NH4Cl for 1 h at RT, then blocked in 5% NGS and 2% DMSO in TBS-T O/N at 4 °C, incubated in primary antibodies diluted in blocking solution O/N at 4 °C, washed in TBS-T, incubated in secondary antibodies diluted in blocking solution for 2 h at RT, washed again, and mounted in 20% Mowiol with 5% N-propylgallate. Primary antibodies used were anti-neurogranin rabbit pcl (Proteintech, Rosemont, IL, USA, 10440-1-AP, 1:500), followed by Alexa 488-conjugated anti-rabbit secondary antibodies. Draq5 (Abcam, Cambridge, UK, 1:1000) was used for nuclear labeling in the red spectrum. Imaging was performed with a Zeiss LSM510 confocal microscope.
Golgi staining. The FD Rapid GolgiStain™ Kit (FD NeuroTechnologies, Columbia, MD, USA) was used, according to the manufacturer’s instructions. Briefly, P80–90 mice were sacrificed by cervical dislocation, and the brain was quickly dissected on ice, washed in PBS, and impregnated in staining solution for 15 days. It was then washed in the provided solution for 3 days, cut coronally with 250 μm thickness, applied to chrom-alum gelatin-coated slides, dried O/N, developed, dehydrated, and mounted with Entellan (Merck). Imaging was performed with a Keyence BZ-9000 microscope taking z-stacks of entire V layer neurons in the motor cortex region. The branching analysis was performed manually and blind of the analyzed genotype, as much as possible given the strong phenotype in some of the genotypes, counting the branches and their length using Image J. The full projection image used for the analysis was always compared with the original stack to avoid false attribution of crossing branches from neighboring stained neurons.
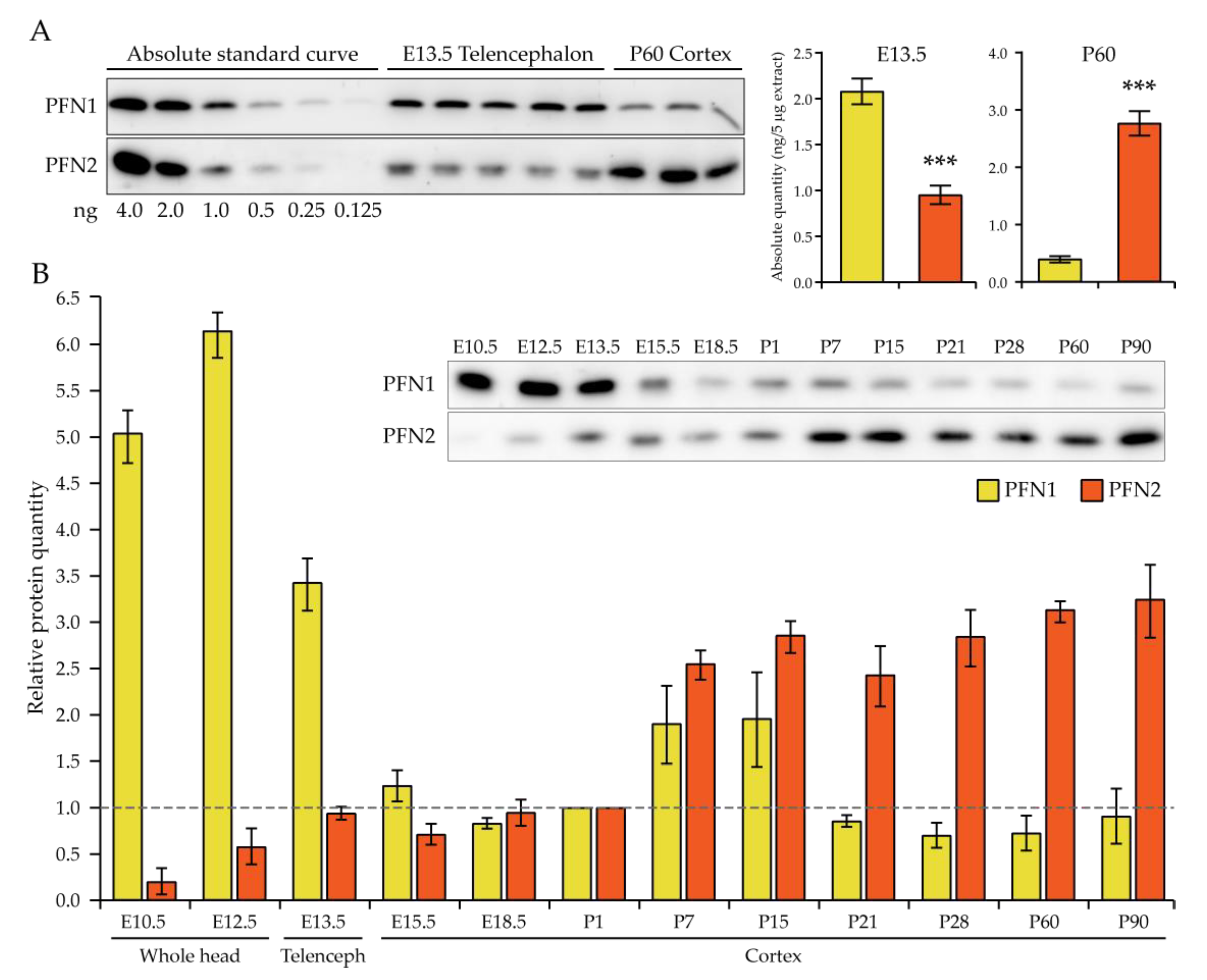
Western blotting. After dissection, embryonic and adult brain tissue was quickly frozen in N
2(l) and stored at −80 °C until lysed. Total tissue lysis was performed in adequate volumes of 2× Laemmli buffer in a glass Teflon tissue homogenizer with an electrically controlled rotation at 600 rpm, to obtain a protein concentration between 2 and 4 mg/mL. The concentration was estimated with the Bradford method using BSA to produce a standard curve. Western blotting was performed according to standard protocols, loading 2.5 to 5 μg of protein for more accurate relative quantification. Submerged blotting was adapted when necessary to the small-molecular-weight proteins, transferring at 160 mA for 30 min. Antibody incubation and washes were performed in TBS-T (0.05% Tween-20) at pH 8.2. Primary antibodies used were homemade anti-PFN2 3003 (1:500) and anti-PFN1 T1 (1:1000) rabbit sera [
6], anti-cofilin 1 KG60 (1:500) rabbit serum [
22], anti-phospho-cofilin 1 (Ser 3) rabbit pcl (Cell Signaling Technology, Danvers, MA, USA, 3313, 1:500), anti-actin mouse mcl (MP Biomedicals, Santa Ana, CA, USA, 08-691002, clone C4, 1:5000), anti phospho-H3 (Ser 10) rabbit pcl (Upstate/Merck 06-570, 1:1000), anti-γ-tubulin mouse mcl (Sigma T6557, 1:2000), anti-phospho-CDC2/CDK1 (Tyr15) rabbit pcl (Cell Signaling Technology 9911, 1:1000), anti-PTTG1 rabbit pcl (Abcam ab26273, 1:1000), anti-vimentin rabbit pcl (Abcam ab92547, 1:500), and anti-GFAP rabbit pcl (Sigma G9269, 1:1000). Chemiluminescence images were acquired with a LAS4000-Mini Imager (FujiFilm/Cytiva, Marlborough, MA, USA). Coomassie staining of the membrane was performed after the antibody detection following a standard protocol. Briefly, the membrane was incubated in 0.2% Coomassie R250 in 50% methanol/10% acetic acid for 10–15 min, then washed 2× in 40% methanol/10% acetic acid for 10 min, and lastly left in 10% methanol/10% acetic acid for 1 h, air-dried, and imaged.
F/G-actin ratio. Dissected cortices from P120 mice were lysed on ice in McRobbie’s PHEM buffer (PIPES-Na 60 mM, HEPES-Na 25 mM, EGTA 10 mM, MgCl2 2 mM at pH 6.9) with 1% TritonX-100 in a glass-Teflon homogenizer at a constant electrically regulated speed of 600 rpm. Lysates were spun at 10,000× g for 10 min at 4 °C, the supernatant was set aside, and the pellet was dried of any supernatant, before fragmenting and resuspending in an equal volume of PHEM buffer with 1% TritonX-100. Both fractions were then complemented with 1× Laemmli buffer and boiled at 100 °C for 10 min. Equal volumes of the pellet (F-actin) and supernatant (G-actin) fractions were loaded on a gel and quantified by Western blotting to calculate the F/G-actin ratio.
Statistics. Statistical evaluation was performed with Microsoft Office Excel, GraphPad, or Minitab software. Applied tests are specified in the figure legends. In general, one-way ANOVA was applied when comparing the four genotypes, followed by Dunnett’s post hoc test to compare the mutant genotypes to the control. Two-sided Welch’s t-test (not assuming equal variance of the samples) was used when comparing the profilin double knockouts to controls, since there is no guarantee that, in the mutant mice, the variance was the same as in the controls. The one-sided t-test was applied only on occasions where, due to the genetic manipulation, it was not possible to have an overlap of the mutant and the control data distributions, mainly to evaluate the significance of the profilin and the actin loss in the profilin double mutants.
4. Discussion
The
profilin gene found already in yeast (PFY1) and up to the fly (
D. melanogaster, chickadee) has undergone several DNA- and RNA-based diversification events during the evolution of the Chordata, and, already in fish (
D. rerio), at least three isoforms have been found. In mammals, until now, four isoforms have been identified, while, in plants, there are even more. Most eukaryotic cells, therefore, simultaneously express at least two profilins; in particular, neuronal cells in mouse have been shown to express both
profilin 1 and
profilin 2 [
7,
13]. The basic properties of profilin isoforms appear to be the same: binding of G-actin in a 1:1 complex, catalytically inducing ADP to ATP exchange on the actin monomer, and, in general, positively regulating actin polymerization in concert with other polymerization factors [
33]. The difference in their biological function when they are present in the same cell has largely remained elusive. Three aspects should be considered: (1) their absolute expression levels in a given cell; (2) their unique interaction and/or affinity with ligands involved in specific cellular processes [
6,
34]; (3) specific post-translational regulatory mechanisms (e.g., phosphorylation) that can regulate the different isoforms in response to stimuli [
35,
36,
37,
38,
39]. In this work, we attempted to address the specific and redundant functions of profilin 1 and 2 in the nervous system of the mouse in two different in vivo settings: (i) cell expansion and differentiation at the beginning of mouse brain development, and (ii) morphology of postmitotic neurons in the mouse adult brain. For this purpose, we employed two separate time- and cell-specific conditional knockout approaches for
profilin 1 in conjunction with a
profilin 2 conventional knockout background.
Our study in the embryonic system supports a mostly redundant function of profilin 1 and 2 in the cell division of neural progenitor cells. Broadly speaking, it is the absolute level of profilins which is critical at this early stage. In fact, we show that, in E13.5 telencephalon, the earliest timepoint where we addressed profilin expression specifically in the neural tissue, the absolute amount of profilin 2 appears to be only about half that of profilin 1, remaining constant until birth. In previous studies, this novel finding was masked by the use of whole embryo or whole head extracts [
9,
23]. The double
profilin knockout in the neuroepithelial stem cells (obtained using the
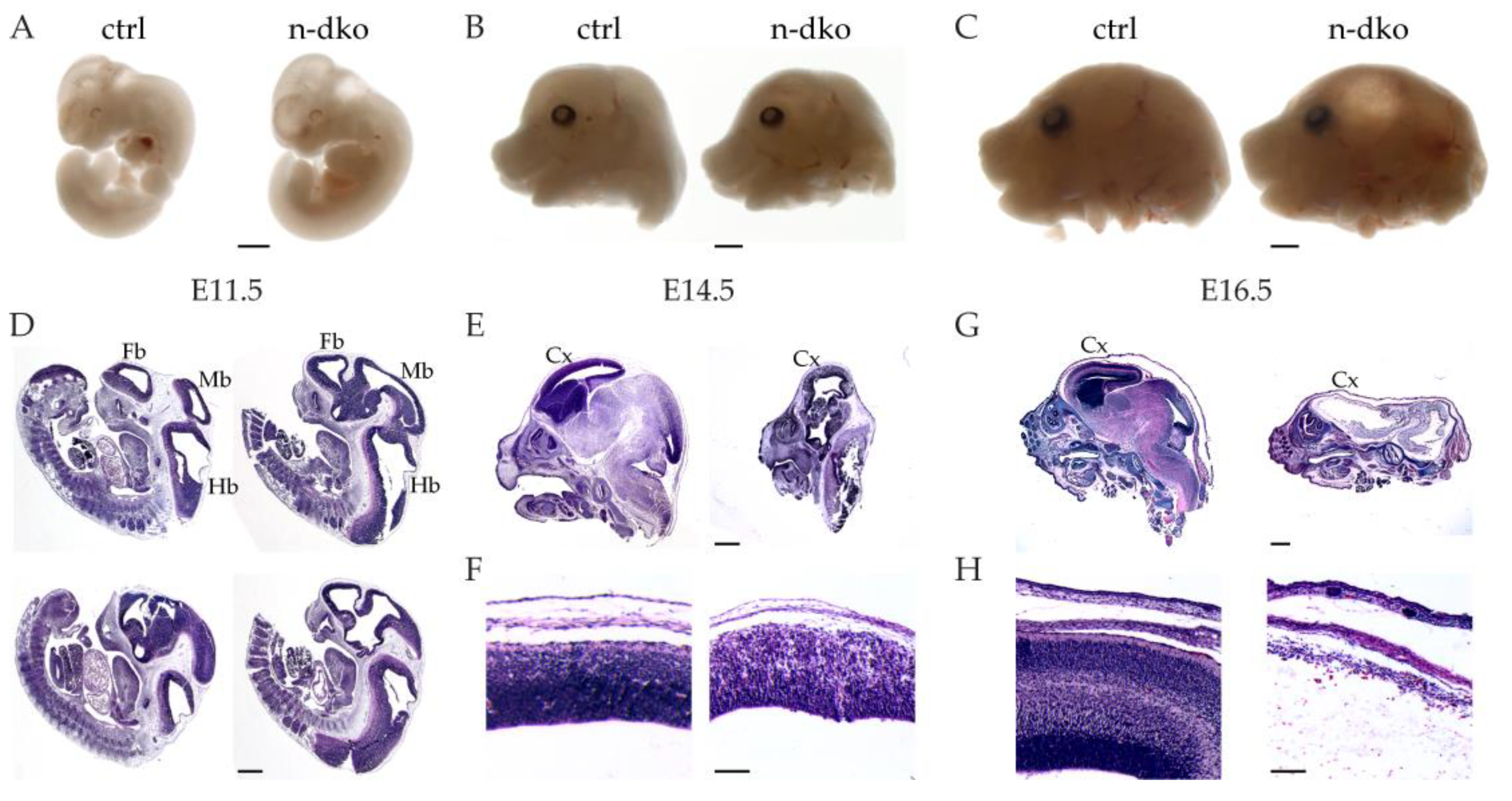
Nes-Cre driver line) completely arrests telencephalic development between E11.5 and E14.5 and significantly affects mid-/hindbrain development. Corticogenesis in mouse embryos occurs more or less between E11 and E18 and is regulated by the cycles of cell division of the neural precursor cells (also known as radial glia) that can divide both symmetrically to maintain their pool and asymmetrically to implement neurogenesis, as well as astrogenesis [
40]. We show that, in our mouse model, profilin 1 protein depletion from the neuroepithelial cells is seemingly complete at E11.5, therefore it is not surprising that the expansion of the neuroepithelial stem-cell pool, occurring before E11, is not significantly affected in
profilin double knockout embryos or
Pfn2 haploinsufficient embryos, which look quite normal following histological analysis at E11.5. The cell division arrest, therefore, starts in apical radial glia cells in this mouse model, in all three brain vesicles, affecting both the maintenance of the NPCs pool and the generation of the majority of the neurons. This results at E14.5–16.5 in the complete absence of the cortex, where proliferation is known to be most important [
41], and in the reduced size of the mid-/hindbrain region. Brain size depends on the NPC cell division rate, with increased proliferation of neural stem cells resulting in an enlarged brain [
42], and defective proliferation resulting in microcephaly [
43,
44].
While a single
profilin 1 allele is able to rescue the
profilin double knockout phenotype and allow viable mice at birth, a single
profilin 2 allele only succeeds in a very partial rescue that does not abolish the embryonic lethal phenotype. Previous work has shown that two
profilin 2 alleles in a
profilin 1;
Nes-Cre knockout mouse model [
15,
18] are sufficient to substantially rescue the cell division phenotype and obtain viable mice, which is in agreement with a redundant role of profilin 1 and 2 in cell division and an allele requirement based on their individual expression level at this developmental stage. Nevertheless, cell adhesion and neuronal migration deficits in the
profilin 1;
Nes-Cre mouse model were not sufficiently compensated for by profilin 2 [
16], suggesting a more specific role of profilin 1 in migration- and adhesion-dependent cell functions.
The role of profilin in cell division was first uncovered in fission yeast (
S. pombe) [
45], but the strongest evidence has come from the
profilin 1 knockout mouse model [
9], where embryonic development was largely arrested between the two- and four-cell stage, a developmental stage where
profilin 2 is not yet expressed. In the present work, we show that the arrest of cell division in NPCs depleted of both profilins occurs in G2 or at the G2/M transition point of mitosis, as suggested by the accumulation of several markers that should be catabolized in M-phase, such as CDK1 tyrosine 15 phosphorylation (inhibitory for cell division progression) and PTTG1 (that blocks chromatid segregation), and supported by the doubling of p-H3-labeled neural precursor cells and the 50% decrease in dividing NPCs with segregated chromatids. Therefore, it is plausible to conclude that profilin-dependent actin dynamics are required in NPCs for the progression of cell division into the M-phase and finally cytokinesis. A minimal required amount of profilin can be provided by a single
profilin 1 allele or by two
profilin 2 alleles, due to their differential embryonic expression levels. Nevertheless, in the presence of both
profilin 2 alleles, in the conditional
profilin 1 knockout model with the
Nes-Cre driver, the precise regulation of the orientation of the cleavage plane of apical radial glia cells was disrupted [
18], showing fine differences even within the basic common functions of profilin 1 and 2 in cell division.
The fact that the different profilin isoforms can compensate for each other in basic cellular functions is not totally surprising. Profilin is present in multiple isoforms already in early evolutionary organisms, such as
D. discoideum, and the sequence homology between the many different profilin paralogs and orthologs is never particularly high; for example, mouse profilins 1 and 2 share only a 63% identity at the amino-acid level (NCBI blastp), while the identity between chickadee (
D. melanogaster) and mouse profilin 1 is as low as 29% (NCBI blastp). What typically characterizes all profilins, nevertheless, is their secondary and tertiary structure (for a review, see [
46]). All the crystal structures determined until now are highly superimposable. This is due to the fact that all profilins possess a seven-strand flat beta sheet core, on one side of which, together with two alpha helices, the actin-binding domain is formed, while, on the opposite side, the N- and C-terminal helices come close together to form the SH3-like poly-
l-proline-binding domain. Lastly, all profilin ligand interactions are negatively regulated by the binding of phosphatidylinositol phosphates. The structural identity of the profilin orthologs from yeast to man has preserved profilin’s fundamental role in actin dynamics for basic functions such as cell division, cell adhesion, and cell migration to the point that null phenotypes can be rescued by evolutionarily very distant profilins [
47]. It is, therefore, not unreasonable to also expect partial functional overlap among paralogs. Of course, the amino-acid sequence diversity in the paralogs can introduce fine functional differences, such as what has been observed between profilin 1 and 2 in the orientation of the radial glia cleavage plane discussed above, or add specific functions.
Why the complete loss of profilin, accompanied by a consistent loss of actin and a general reduction in actin dynamics, results in a cell-cycle arrest in the G2-phase or G2/M transition, could have at least two explanations. First, the genetic ablation of both
profilin genes in neuroepithelial cells might activate a mitotic actin checkpoint. There has been some evidence for such a mechanism, first described 20 years ago in fission yeast [
48], and later shown in different cell culture systems [
49,
50], using actin-depolymerizing drugs. Only in one report has the mechanism been proposed in vivo, in
filamin A knockout mice [
43], which nevertheless displayed only a mild microcephaly phenotype due to a delay of cell division. The double
profilin knockout mouse model shows a severe mitosis arrest, with lack of disinhibition of the CDK1 kinase by Tyr15 dephosphorylation, a common signature found in all the abovementioned systems where actin dynamics were disrupted. A second explanation for the severe mitotic arrest of NPCs in double
profilin knockout embryos is that a minimum profilin level is required for interkinetic nuclear migration (INM), a characteristic process occurring in many neuroepithelial and epithelial thin proliferating layers to increase the cell density and the efficiency of cell division [
41]. In radial glia cells, mitosis can occur only at the apical surface, close to the ventricle, since the centrosome, necessary for spindle formation, is located apically in these cells to support cilia extension into the ventricle. As soon as cell division is complete, the nucleus is translocated toward the basal side, creating a pseudostratification of the cells that increases compactness and leaves space at the apical surface for other cells to continue dividing [
41]. Furthermore, the S-phase occurs at the basal side, but then the nucleus before nuclear membrane breakdown must be moved to the apical surface. Translocation of the nucleus from the basal to the apical region in NPCs must occur quickly and has been shown to depend on microtubules, as well as actin dynamics [
44,
51,
52,
53,
54]. It is, therefore, plausible that, in the absence of profilins, this translocation cannot occur or is severely slowed down, physically impeding cell division. A similar phenotype was observed in conditional
cofilin 1 knockout embryos [
44], albeit with a less severe phenotype due to partial functional compensation by the
cofilin 1 paralog, actin-depolymerizing factor (
Adf). The apparent thickening of the p-H3-stained layer in
profilin double knockout E11.5 embryos supports this view. The ectopic p-H3 staining in the basal region of the cortical primordium is also in agreement with this explanation. It indicates a defoliation process of the ventricular zone cell layer due to crowding-induced mechanical stress [
55,
56], which can be caused by impaired INM, improper pseudostratification of the cells, and increased number of cells in G2 phase, when they occupy the highest volume. This defoliation process results in the complete loss of dividing cells in the VZ and eventually the absence of the cortex in E16.5
profilin double knockout embryos.
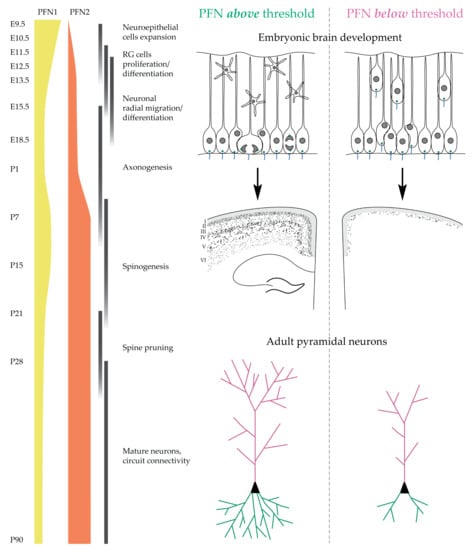
Changing perspective from embryonic development to adult brain morphology, another novel and fascinating aspect of versatile profilin function emerged. In early development and cell-cycle control, profilin 1 is the driving isoform in terms of expression and activity, with profilin 2 behaving more as a bystander that can support the same functions if expressed at a sufficient level. After birth, in mature postmitotic neurons, the two isoforms profilin 1 and profilin 2 exchange roles (or importance), with profilin 2 now becoming the limiting factor in the context of cell morphology and function and profilin 1 retaining a partially compensating activity due to a much lower expression level. Structural defects occur in the
profilin knockout neurons at 3 months of age, such as shortening of dendritic branches and loss of dendritic arborization complexity, with a profilin dosage-dependent severity, while no specific defects seem to occur depending on the deleted
profilin isoform. Therefore, profilin in postmitotic neurons appears to be necessary for the cytoskeletal support of the complex neuronal structure. In adult mice, profilin 2 levels are markedly higher than profilin 1 levels [
6]; in particular, in the cortex, profilin 2 appears to be about sevenfold higher, in good agreement with the previous data. Accordingly, the neuronal phenotype is less severe when a single
profilin 2 allele that can express more profilin is present, more severe when a single
profilin 1 allele, able to express much less profilin, is still present, and finally the most severe when both profilins are depleted. The phenotype dependence on the specific profilin isoform is the opposite of that in embryonic stages, but conceptually identical, due to the reversed quantity ratio. A function of profilin 2 in supporting neuronal morphology has been previously shown using a knockdown approach in hippocampal cultured neurons [
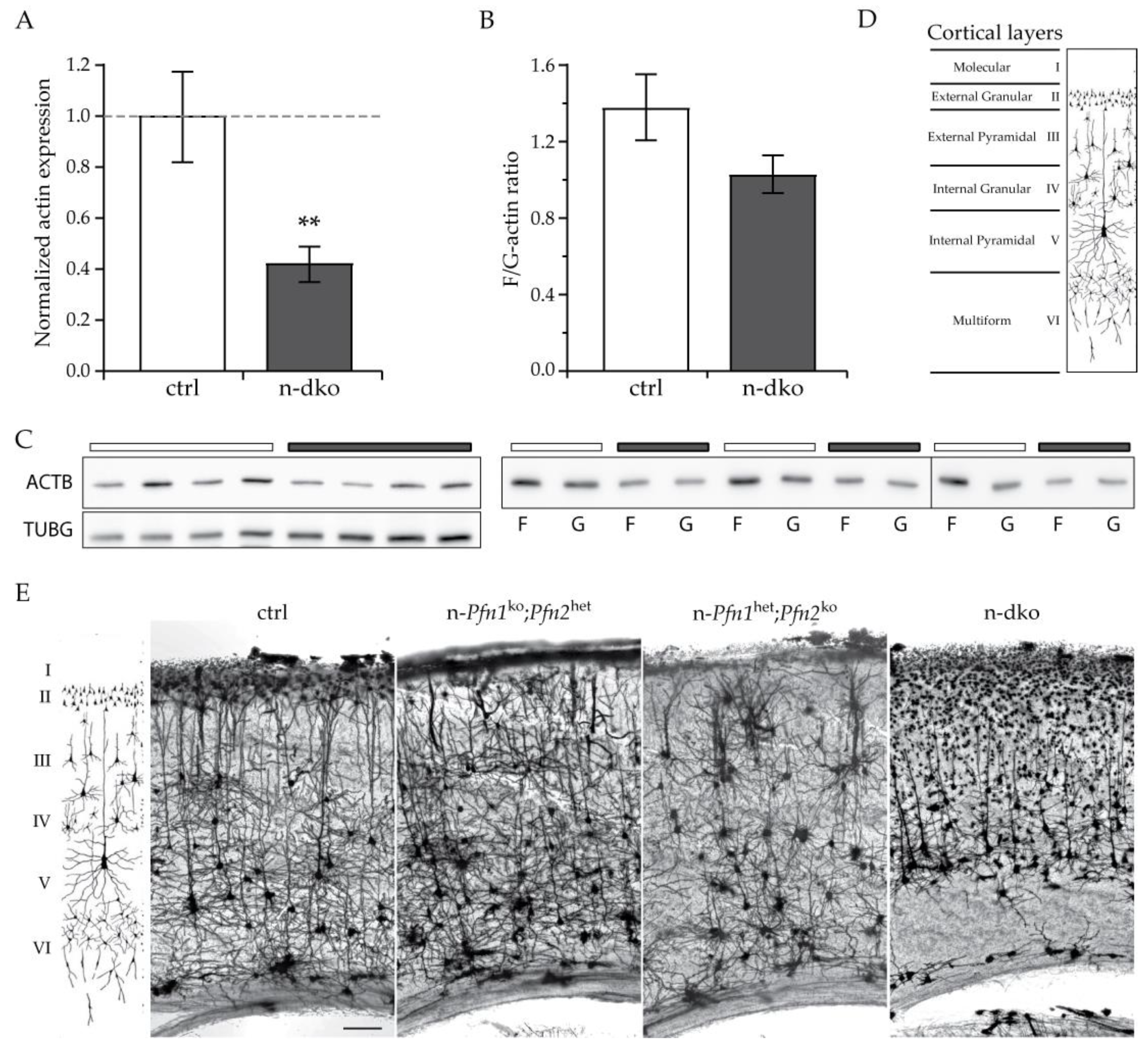
57], but the redundant function of profilin 1 in supporting dendritic complexity was not recognized. One possibility could be an insufficient ectopic overexpression of profilin 1, difficult to evaluate in single cells; alternatively, it is possible that, in isolated cultured neurons, profilin isoforms are not equivalent. In our model, the collapse of the dendritic arbor in the absence of both profilins appears to be due to the considerable loss of actin dynamics, as reflected by the large reduction in actin levels and the alteration of F/G-actin ratios. Considering that neurons in the cortex represent only 25–30% of the cells [
31], the 40% actin reduction calculated on a total cortical extract suggests a significant loss of actin in
profilin double knockout neurons. In addition to the loss of total actin, the filamentous to monomeric actin ratio was decreased, accounting for a substantial loss of structural F-actin in
profilin double knockout neurons. The model shows the importance and the necessity for structural actin in the arborization of postmitotic neurons, in addition to the well-recognized role of actin dynamics in synaptic plasticity. In the Golgi-stained images, it is possible to observe that, in the genotypes lacking
profilin 2, the density of the stained neurons is decreased. Neuronal death following collapse of the dendritic arborization is plausible and could proceed according to mechanisms similar to transneuronal degeneration [
58], since the collapsing dendritic arbors would lose connectivity, and it is a general principle that nerve cells need to be integrated into a functional network and receive trophic signals from other neurons in order to remain viable.
In conclusion, we show here that basic cellular functions in mouse NPCs and neurons can be supported by both profilin 1 and 2, and the main requirement is a threshold profilin level. By no means does the work presented here exclude specific functions in which one profilin isoform is favored or uniquely required. Here, we simply provide two aspects of profilin function—cell proliferation and pyramidal neuron morphogenesis—where the two profilin isoforms can, in principle, compensate for each other. There have already been described activities which suggest additional and rather specific functions of profilins. One example might be cell adhesion and neuronal migration that appear to selectively require profilin 1 [
15,
16]. Similarly, synaptic transmission appears to mainly depend on profilin 2, according to the hyperexcitability phenotype of the
profilin 2 knockout mouse model and the specific interaction of profilin 2 with synaptic proteins [
6,
7,
59]. We would like to predict that the ligand interaction network and regulatory post-translational mechanisms of the different profilin isoforms will shed more light on the very (cell) specific aspects of profilin isoform function.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






