
α-Synuclein Decreases the Abundance of Proteasome Subunits and Alters Ubiquitin Conjugates in Yeast

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Yeast Transformation and Growth Conditions
2.2. Spotting Assays
2.3. Western Blot Analysis
2.4. Fluorescence Microscopy and Quantifications
2.5. Flow Cytometry
2.6. Quantification and Statistical Analysis
2.7. Sample Preparation for LC-MS Proteome Analysis
2.8. LC-MS Analysis
2.9. BioID-SILAC Analysis
3. Results
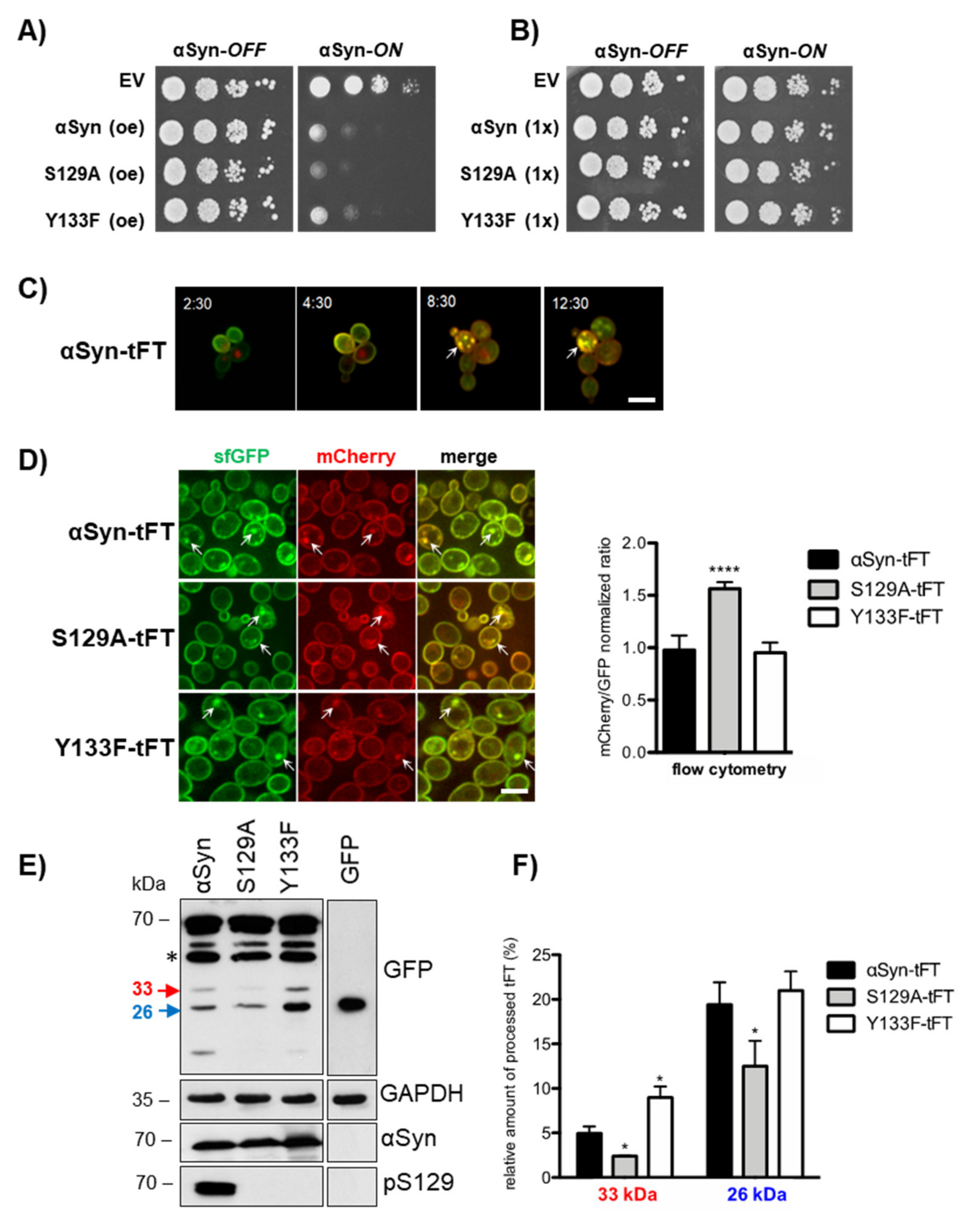
3.1. Tandem Fluorescent Protein Timer Monitoring Reveals That a Y133F Substitution Compensates the Deficiency in S129 Phosphorylation, which Normally Promotes Soluble αSyn Turnover
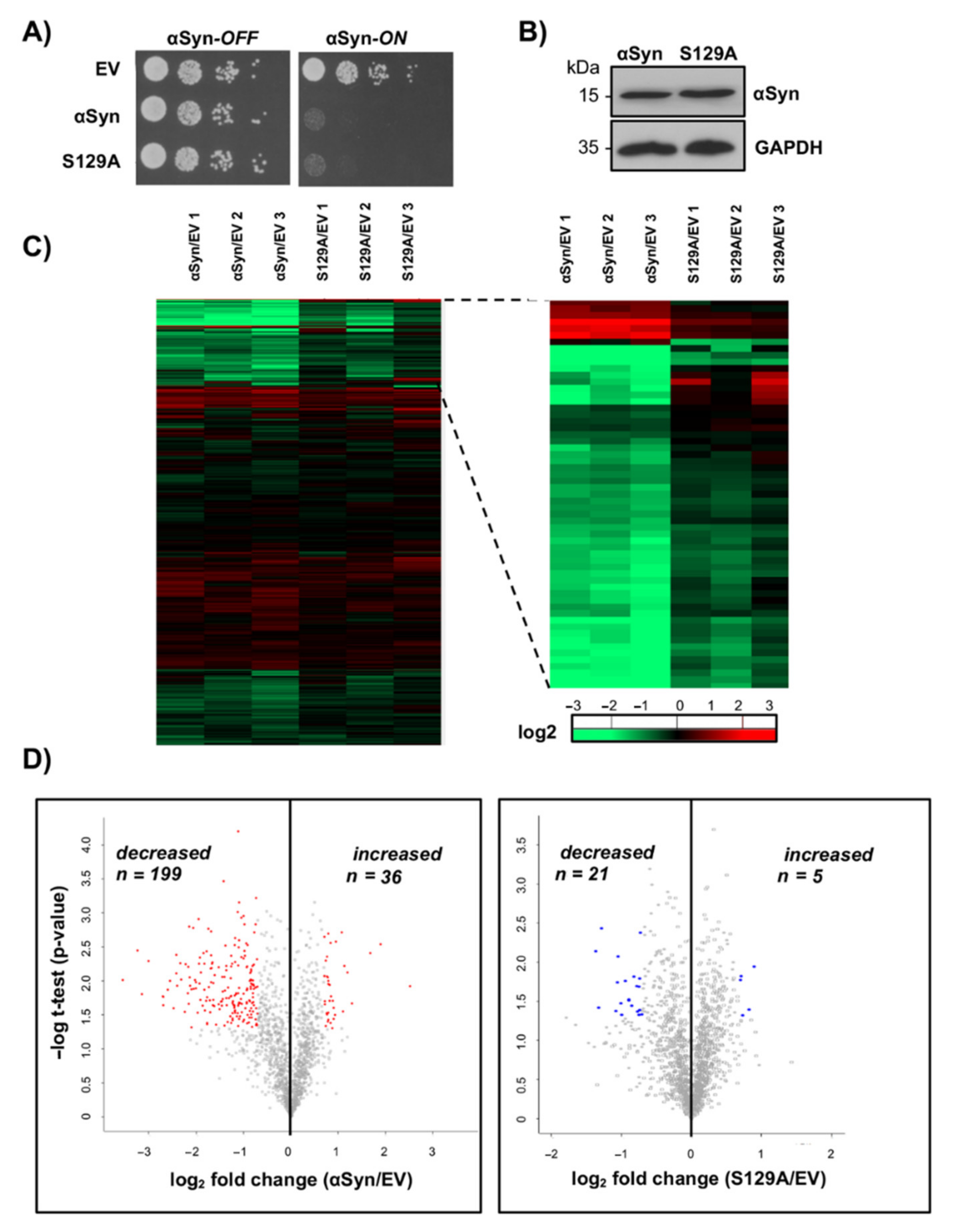
3.2. αSyn Expression Changes the Yeast Proteome and Reduces the Proteasome Subunit Levels
3.3. Downregulation of Genes for Proteasome Subunits Enhances αSyn Toxicity
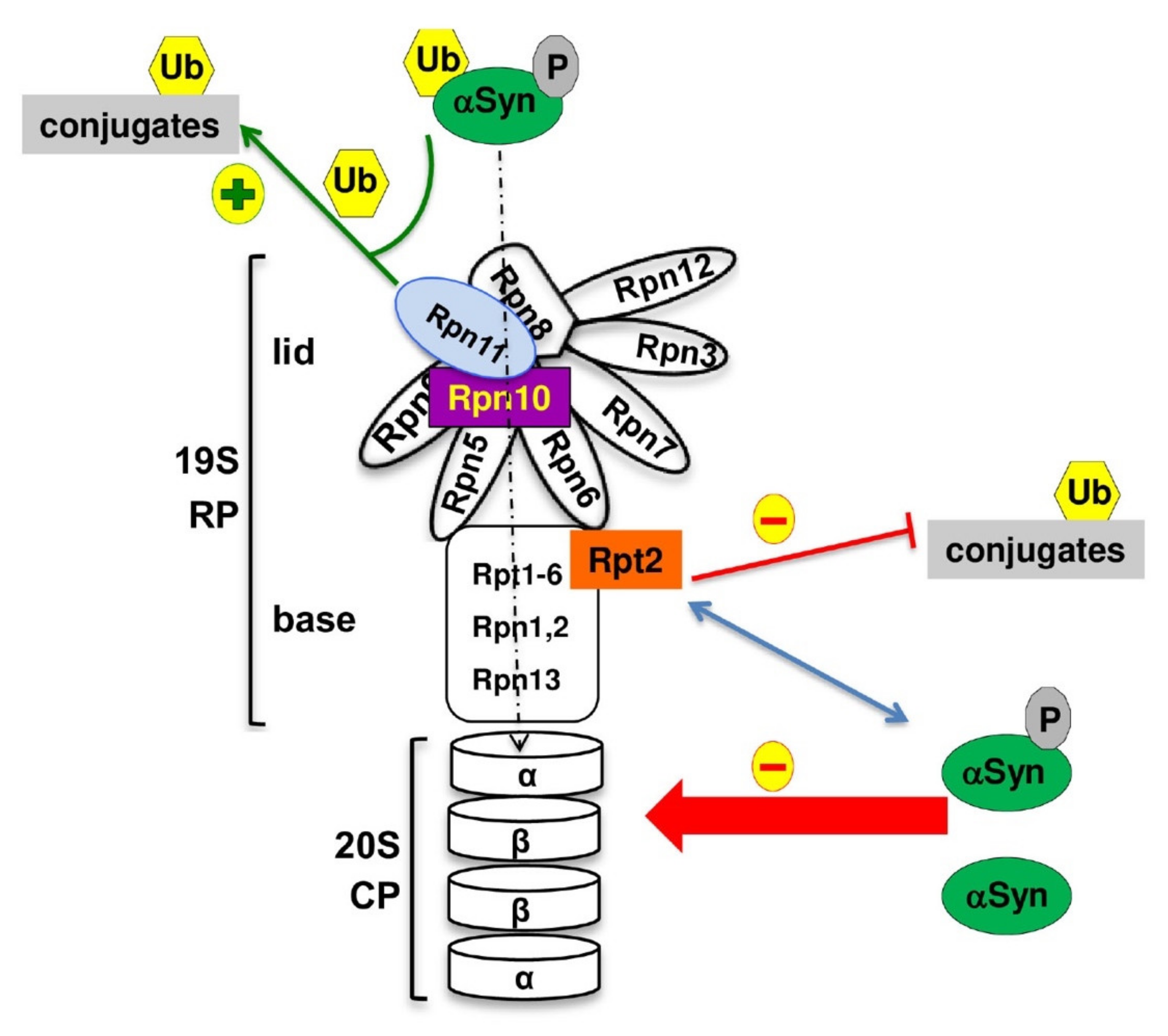
3.4. αSyn Interacts with the Base Subunit Rpt2
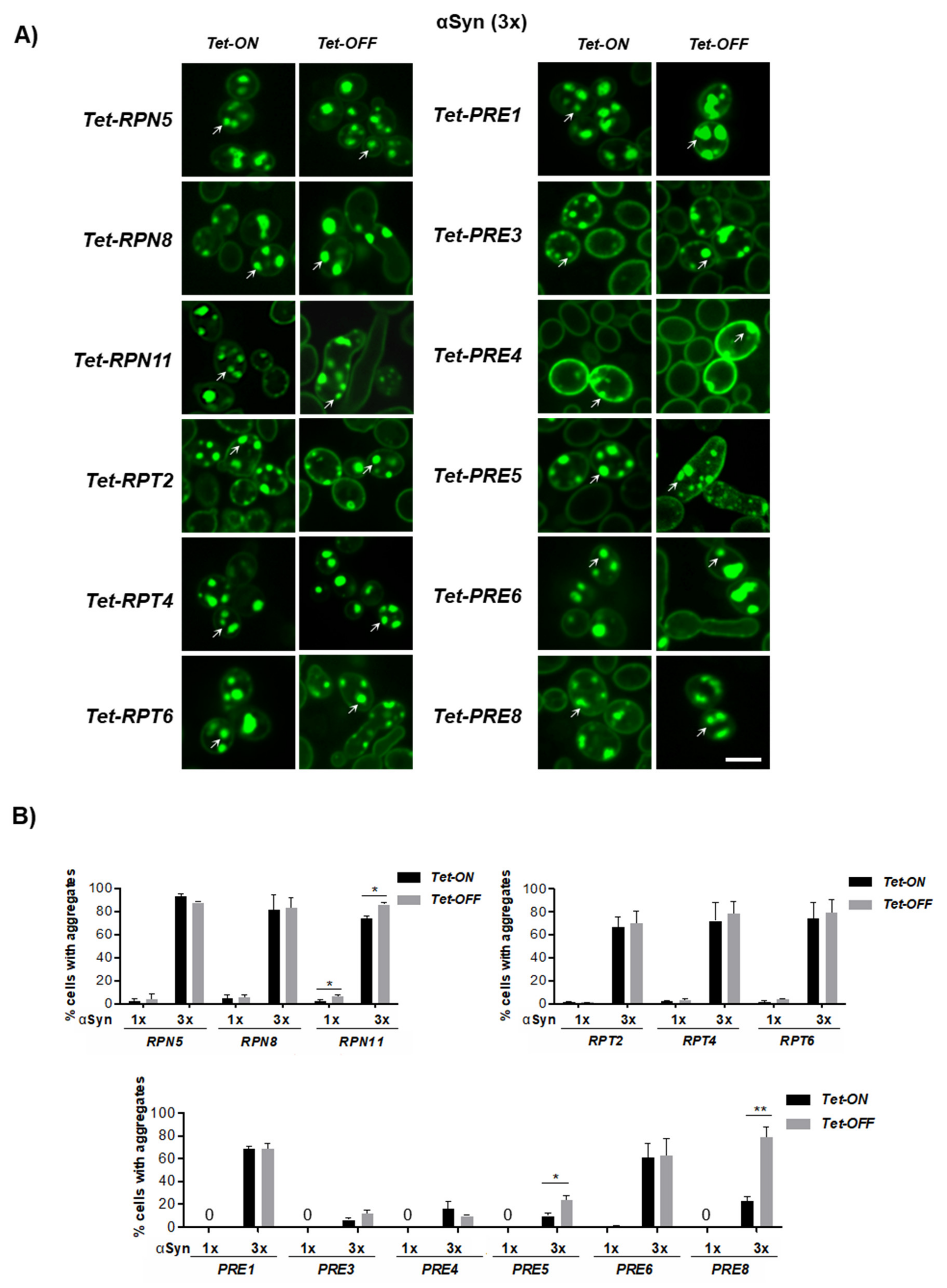
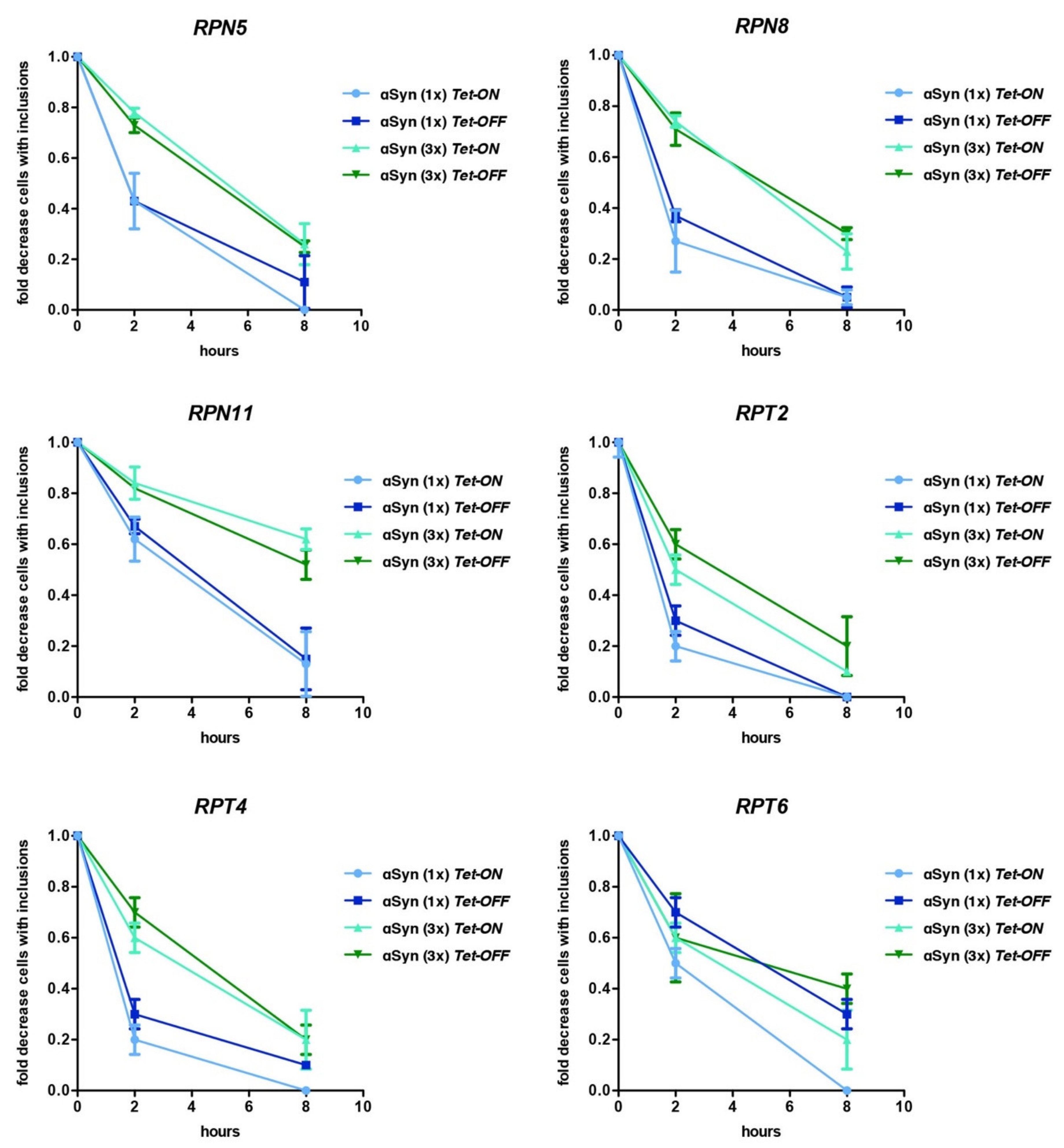
3.5. Downregulation of Genes for Proteasome Subunits Results in Different Outcomes of αSyn Inclusion Formation
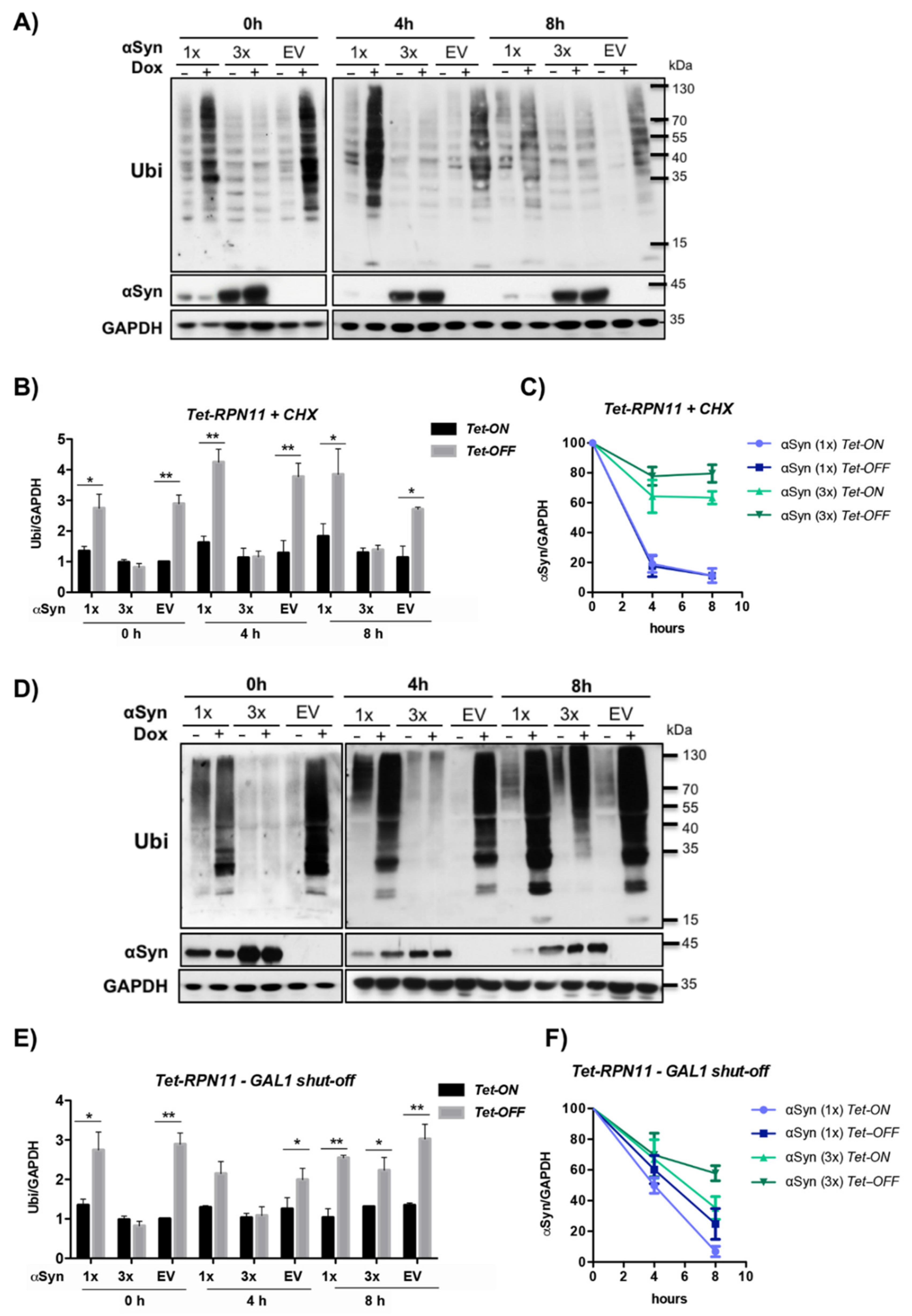
3.6. αSyn Diminishes the Pool of Ubiquitin Conjugates upon Downregulation of Tet-RPN11
3.7. αSyn Increases the Pool of Ubiquitin Conjugates upon Downregulation of Tet-RPT2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dexter, D.T.; Jenner, P. Parkinson disease: From pathology to molecular disease mechanisms. Free. Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Mercuri, N.B.; Venneri, A.; Faustini, G.; Longhena, F.; Pizzi, M.; Missale, C.; Spano, P.; Bellucci, A.; Mercuri, N.B.; et al. Review: Parkinson’s disease: From synaptic loss to connectome dysfunction. Neuropathol. Appl. Neurobiol. 2016, 42, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Sulzer, D.; Edwards, R.H. The physiological role of α-synuclein and its relationship to Parkinson’s disease. J. Neurochem. 2019, 150, 475–486. [Google Scholar] [CrossRef]
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. α-Synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Pinho, R.; Paiva, I.; Jerčić, K.G.; Fonseca-Ornelas, L.; Gerhardt, E.; Fahlbusch, C.; Esparcia, P.G.; Kerimoglu, C.; Pavlou, M.A.S.; Villar-Piqué, A.; et al. Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum. Mol. Genet. 2018, 28, 31–50. [Google Scholar] [CrossRef]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Bsc, C.T.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2003, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-Synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Benskey, M.J.; Perez, R.G.; Manfredsson, F.P. The contribution of alpha synuclein to neuronal survival and function—Implications for Parkinson’s disease. J. Neurochem. 2016, 137, 331–359. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2012, 14, 38–48. [Google Scholar] [CrossRef]
- Karpinar, D.P.; Balija, M.B.; Kugler, S.; Opazo, F.; Rezaei-Ghaleh, N.; Wender, N.; Kim, H.Y.; Taschenberger, G.; Falkenburger, B.H.; Heise, H.; et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. EMBO J. 2009, 28, 3256–3268. [Google Scholar] [CrossRef]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Lázaro, D.F.; Rodrigues, E.F.; Langohr, R.; Shahpasandzadeh, H.; Ribeiro, T.; Guerreiro, P.; Gerhardt, E.; Kröhnert, K.; Klucken, J.; Pereira, M.D.; et al. Systematic comparison of the effects of alpha-synuclein mutations on its oligomerization and aggregation. PLoS Genet. 2014, 10, e1004741. [Google Scholar] [CrossRef]
- Stefanis, L. Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 2, a009399. [Google Scholar] [CrossRef]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Autophagy and Alpha-Synuclein: Relevance to Parkinson’s disease and related synucleopathies. Mov. Disord. 2016, 31, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Petroi, D.; Popova, B.; Taheri-Talesh, N.; Irniger, S.; Shahpasandzadeh, H.; Zweckstetter, M.; Outeiro, T.F.; Braus, G.H. Aggregate clearance of α-synuclein in saccharomyces cerevisiae depends more on autophagosome and vacuole function than on the proteasome. J. Biol. Chem. 2012, 287, 27567–27579. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Alpha-synuclein and protein degradation systems: A reciprocal relationship. Mol. Neurobiol. 2012, 47, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Popova, B.; Kleinknecht, A.; Braus, G.H. Posttranslational modifications and clearing of α-synuclein aggregates in yeast. Biomolecules 2015, 5, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Shahpasandzadeh, H.; Popova, B.; Kleinknecht, A.; Fraser, P.E.; Outeiro, T.F.; Braus, G.H. Interplay between sumoylation and phosphorylation for protection against α-synuclein inclusions. J. Biol. Chem. 2014, 289, 31224–31240. [Google Scholar] [CrossRef] [PubMed]
- Kleinknecht, A.; Popova, B.; Lázaro, D.F.; Pinho, R.; Valerius, O.; Outeiro, T.F.; Braus, G.H. C-terminal tyrosine residue modifications modulate the protective phosphorylation of serine 129 of α-synuclein in a yeast model of Parkinson’s disease. PLoS Genet. 2016, 12, e1006098. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2000, 297, 191–194. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Mytilineou, C.; JnoBaptiste, R.; Yabut, J.; Shashidharan, P.; Jennert, P.; Olanow, C.W. Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J. Neurochem. 2002, 81, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.; Razzaq, A.; Ghetti, B.; Lilley, K.S.; Spillantini, M.G. Ubiquitination of α-Synuclein in lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 2003, 278, 44405–44411. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Jackson, T.; JnoBaptiste, R.; Kapustin, A.; Olanow, C.W. Proteasomal dysfunction in sporadic Parkinson’s disease. Neurology 2006, 66, S37–S49. [Google Scholar] [CrossRef] [PubMed]
- Bentea, E.; Verbruggen, L.; Massie, A. The proteasome inhibition model of Parkinson’s disease. J. Park. Dis. 2017, 7, 31–63. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Belizaire, R.; Jenner, P.; Olanow, C.; Isacson, O. Selective loss of 20S proteasome α-subunits in the substantia nigra pars compacta in Parkinson’s disease. Neurosci. Lett. 2002, 326, 155–158. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Vigouroux, S.; Wong, H.; Guttman, M.; Rajput, A.H.; Ang, L.; Kish, S.J.; Briand, Y.; Buldanlioglu, U. Brain proteasomal function in sporadic Parkinson’s disease and related disorders. Ann. Neurol. 2002, 51, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 2003, 302, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Thorpe, J.; Keller, J. α-Synuclein alters proteasome function, protein synthesis, and stationary phase viability. J. Biol. Chem. 2005, 280, 30009–30017. [Google Scholar] [CrossRef]
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-produced α-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 2010, 31, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.-Y.; Tang, Z.; Liu, C.-W. α-Synuclein protofibrils inhibit 26 s proteasome-mediated protein degradation. J. Biol. Chem. 2008, 283, 20288–20298. [Google Scholar] [CrossRef]
- Lindersson, E.; Beedholm, R.; Højrup, P.; Moos, T.; Gai, W.; Hendil, K.B.; Jensen, P.H. Proteasomal inhibition by α-synuclein filaments and oligomers. J. Biol. Chem. 2004, 279, 12924–12934. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Franssens, V.; Winderickx, J.; Outeiro, T.F. Yeast models of Parkinson’s disease-associated molecular pathologies. Curr. Opin. Genet. Dev. 2017, 44, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Sirati, N.; Popova, B.; Molenaar, M.R.; Verhoek, I.C.; Braus, G.H.; Kaloyanova, D.V.; Helms, J.B. Dynamic and reversible aggregation of the human cap superfamily Member GAPR-1 in protein inclusions in saccharomyces cerevisiae. J. Mol. Biol. 2021, 433, 167162. [Google Scholar] [CrossRef]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef]
- Yeger-Lotem, E.; Riva, L.; Su, L.J.; Gitler, A.D.; Cashikar, A.; King, O.D.; Auluck, P.K.; Geddie, M.L.; Valastyan, J.S.; Karger, D.R.; et al. Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nat. Genet. 2009, 41, 316–323. [Google Scholar] [CrossRef]
- Willingham, S.; Outeiro, T.F.; DeVit, M.J.; Lindquist, S.L.; Muchowski, P.J. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or synuclein. Science 2003, 302, 1769–1772. [Google Scholar] [CrossRef] [PubMed]
- Popova, B.; Wang, D.; Pätz, C.; Akkermann, D.; Lázaro, D.F.; Galka, D.; Gulko, M.K.; Bohnsack, M.T.; Möbius, W.; Bohnsack, K.E.; et al. DEAD-box RNA helicase Dbp4/DDX10 is an enhancer of α-synuclein toxicity and oligomerization. PLoS Genet. 2021, 17, e1009407. [Google Scholar] [CrossRef]
- Gietz, R.D.; Woods, R.A. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002, 350, 87–96. [Google Scholar] [CrossRef]
- Kurtzman, C.P.; Guthrie, C.; Fink, G.R. Guide to yeast genetics and molecular biology. Mycologia 1993, 85, 714. [Google Scholar] [CrossRef]
- Schmitt, K.; Smolinski, N.; Neumann, P.; Schmaul, S.; Hofer-Pretz, V.; Braus, G.H.; Valerius, O. Asc1p/RACK1 connects ribosomes to Eukaryotic Phosphosignaling. Mol. Cell. Biol. 2016, 37, e00279-16. [Google Scholar] [CrossRef]
- Mumberg, D.; Müller, R.; Funk, M. Regulatable promoters of Saccharomyces cerevisiae: Comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 1994, 22, 5767–5768. [Google Scholar] [CrossRef] [PubMed]
- Opitz, N.; Schmitt, K.; Hofer-Pretz, V.; Neumann, B.; Krebber, H.; Braus, G.H.; Valerius, O. Capturing the Asc1p/receptor for activated C kinase 1 (RACK1) microenvironment at the head region of the 40S Ribosome with quantitative BioID in yeast. Mol. Cell. Proteom. 2017, 16, 2199–2218. [Google Scholar] [CrossRef]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, S.T. The perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Wessel, D.; Flügge, U. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Tenreiro, S.; Pinto, M.M.R.; Antas, P.; Rino, J.; Wawrzycka, D.; Macedo, D.; Rosado-Ramos, R.; Amen, T.; Waiss, M.; Magalhães, F.; et al. Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson’s disease. PLoS Genet. 2014, 10, e1004302. [Google Scholar] [CrossRef] [PubMed]
- Gross, L.A.; Baird, G.S.; Hoffman, R.C.; Baldridge, K.K.; Tsien, R.Y. The structure of the chromophore within DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA 2000, 97, 11990–11995. [Google Scholar] [CrossRef]
- Khmelinskii, A.; Keller, P.J.; Bartosik, A.; Meurer, M.; Barry, J.D.; Mardin, B.R.; Kaufmann, A.; Trautmann, S.; Wachsmuth, M.; Pereira, G.; et al. Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nat. Biotechnol. 2012, 30, 708–714. [Google Scholar] [CrossRef]
- Khmelinskii, A.; Meurer, M.; Ho, C.-T.; Besenbeck, B.; Fuller, J.; Lemberg, M.K.; Bukau, B.; Mogk, A.; Knop, M. Incomplete proteasomal degradation of green fluorescent proteins in the context of tandem fluorescent protein timers. Mol. Biol. Cell 2016, 27, 360–370. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Ji, C.H.; Kwon, A.Y.T. Crosstalk and interplay between the ubiquitin-proteasome system and autophagy. Mol. Cells 2017, 40, 441–449. [Google Scholar] [CrossRef]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Rochet, J.-C.; McCaffery, J.M.; Barlowe, C.; et al. The Parkinson’s disease protein synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2007, 105, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J.; Caldwell, K.; Caldwell, G.; Cooper, A.; Rochet, J.-C.; et al. α-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009, 41, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Zabrocki, P.; Bastiaens, I.; Delay, C.; Bammens, T.; Ghillebert, R.; Pellens, K.; De Virgilio, C.; Van Leuven, F.; Winderickx, J. Phosphorylation, lipid raft interaction and traffic of α-synuclein in a yeast model for Parkinson. Biochim. Biophys. Acta Bioenerg. 2008, 1783, 1767–1780. [Google Scholar] [CrossRef]
- Büttner, S.; Bitto, A.; Ring, J.; Augsten, M.; Zabrocki, P.; Eisenberg, T.; Jungwirth, H.; Hutter, S.; Carmona-Gutierrez, D.; Kroemer, G.; et al. Functional mitochondria are required for α-synuclein toxicity in aging yeast. J. Biol. Chem. 2008, 283, 7554–7560. [Google Scholar] [CrossRef] [PubMed]
- Grassi, D.; Howard, S.; Zhou, M.; Diaz-Perez, N.; Urban, N.T.; Guerrero-Given, D.; Kamasawa, N.; Volpicelli-Daley, L.A.; LoGrasso, P.; Lasmézas, C.I. Identification of a highly neurotoxic α-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E2634–E2643. [Google Scholar] [CrossRef]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and rescue of synuclein toxicity in Parkinson patient-derived neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef]
- Koch, Y.; Helferich, A.M.; Steinacker, P.; Oeckl, P.; Walther, P.; Weishaupt, J.H.; Danzer, K.M.; Otto, M. Aggregated α-Synuclein increases SOD1 oligomerization in a mouse model of amyotrophic lateral sclerosis. Am. J. Pathol. 2016, 186, 2152–2161. [Google Scholar] [CrossRef]
- Finley, D.; Ulrich, H.D.; Sommer, T.; Kaiser, P. The ubiquitin-proteasome system of saccharomyces cerevisiae. Genetics 2012, 192, 319–360. [Google Scholar] [CrossRef]
- Tomko, R.J.; Hochstrasser, M. Molecular architecture and assembly of the Eukaryotic Proteasome. Annu. Rev. Biochem. 2013, 82, 415–445. [Google Scholar] [CrossRef]
- Mnaimneh, S.; Davierwala, A.P.; Haynes, J.; Moffat, J.; Peng, W.-T.; Zhang, W.; Yang, X.; Pootoolal, J.; Chua, G.; Lopez, A.; et al. Exploration of essential gene functions via titratable promoter alleles. Cell 2004, 118, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; da Fonseca, T.L.; Eisbach, S.E.; Anduaga, A.M.; Breda, C.; Orcellet, M.L.; Szegő, M.; Guerreiro, P.; Lázaro, D.F.; Braus, G.H.; et al. α-Synuclein interacts with the switch region of Rab8a in a Ser129 phosphorylation-dependent manner. Neurobiol. Dis. 2014, 70, 149–161. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sampaio-Marques, B.; Guedes, A.; Vasilevskiy, I.; Gonçalves, S.; Outeiro, T.F.; Winderickx, J.; Burhans, W.C.; Ludovico, P. α-Synuclein toxicity in yeast and human cells is caused by cell cycle re-entry and autophagy degradation of ribonucleotide reductase 1. Aging Cell 2019, 18, e12922. [Google Scholar] [CrossRef]
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates, J.R.; Koonin, E.V.; Deshaies, R.J. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26s proteasome. Science 2002, 298, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Cohen, R.E. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 2002, 419, 403–407. [Google Scholar] [CrossRef]
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 2016, 73, 3497–3506. [Google Scholar] [CrossRef]
- Ghislain, M.; Udvardy, A.; Mann, C.S. cerevisiae 26S protease mutants arrest cell division in G2/metaphase. Nature 1993, 366, 358–362. [Google Scholar] [CrossRef]
- Hanna, J.; Leggett, D.S.; Finley, D. Ubiquitin depletion as a key mediator of toxicity by translational inhibitors. Mol. Cell. Biol. 2003, 23, 9251–9261. [Google Scholar] [CrossRef]
- Park, C.-W.; Ryu, K.-Y. Cellular ubiquitin pool dynamics and homeostasis. BMB Rep. 2014, 47, 475–482. [Google Scholar] [CrossRef]
- Köhler, A.; Cascio, P.; Leggett, D.S.; Woo, K.M.; Goldberg, A.L.; Finley, D. The axial channel of the proteasome core particle is gated by the Rpt2 ATPase and controls both substrate entry and product release. Mol. Cell 2001, 7, 1143–1152. [Google Scholar] [CrossRef]
- Sakata, E.; Eisele, M.R.; Baumeister, W. Molecular and cellular dynamics of the 26S proteasome. Biochim. Biophys. Acta Proteins Proteom. 2020, 1869, 140583. [Google Scholar] [CrossRef]
- Marshall, R.S.; Vierstra, R.D. Dynamic regulation of the 26s proteasome: From synthesis to degradation. Front. Mol. Biosci. 2019, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.S.; Vierstra, R.D. Proteasome storage granules protect proteasomes from autophagic degradation upon carbon starvation. eLife 2018, 7, e34532. [Google Scholar] [CrossRef]
- Marshall, R.S.; McLoughlin, F.; Vierstra, R.D. Autophagic turnover of inactive 26s proteasomes in yeast is directed by the ubiquitin receptor cue5 and the hsp42 chaperone. Cell Rep. 2016, 16, 1717–1732. [Google Scholar] [CrossRef]
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Fabre, B.; Ziv, T.; Kwon, Y.T.; Ciechanover, A. p62 and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, E7490–E7499. [Google Scholar] [CrossRef]
- Peters, L.Z.; Karmon, O.; David-Kadoch, G.; Hazan, R.; Yu, Z.; Glickman, M.H.; Ben-Aroya, S. The protein quality control machinery regulates its misassembled proteasome subunits. PLoS Genet. 2015, 11, e1005178. [Google Scholar] [CrossRef] [PubMed]
- Gulko, M.K.; Heinrich, G.; Gross, C.; Popova, B.; Valerius, O.; Neumann, P.; Ficner, R.; Braus, G.H. Sem1 links proteasome stability and specificity to multicellular development. PLoS Genet. 2018, 14, e1007141. [Google Scholar] [CrossRef]
- Stefanis, L.; Larsen, K.E.; Rideout, H.J.; Sulzer, D.; Greene, L.A. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 2001, 21, 9549–9560. [Google Scholar] [CrossRef]
- Tanaka, Y.; Engelender, S.; Igarashi, S.; Rao, R.K.; Wanner, T.; Tanzi, R.E.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 2001, 10, 919–926. [Google Scholar] [CrossRef]
- Snyder, H.; Mensah, K.; Theisler, C.; Lee, J.; Matouschek, A.; Wolozin, B. Aggregated and monomeric α-synuclein bind to the s6’ proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 2003, 278, 11753–11759. [Google Scholar] [CrossRef]
- Lander, G.C.; Estrin, E.; Matyskiela, M.E.; Bashore, C.; Nogales, E.; Martin, A. Complete subunit architecture of the proteasome regulatory particle. Nature 2012, 482, 186–191. [Google Scholar] [CrossRef]
- Tonoki, A.; Kuranaga, E.; Tomioka, T.; Hamazaki, J.; Murata, S.; Tanaka, K.; Miura, M. Genetic evidence linking age-dependent attenuation of the 26s proteasome with the aging process. Mol. Cell. Biol. 2009, 29, 1095–1106. [Google Scholar] [CrossRef]
- Bedford, L.; Hay, D.; Devoy, A.; Paine, S.; Powe, D.G.; Seth, R.; Gray, T.; Topham, I.; Fone, K.; Rezvani, N.; et al. Depletion of 26s proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J. Neurosci. 2008, 28, 8189–8198. [Google Scholar] [CrossRef]
- Fernández-Cruz, I.; Sánchez-Díaz, I.; Narváez-Padilla, V.; Reynaud, E. Rpt2 proteasome subunit reduction causes Parkinson’s disease like symptoms in Drosophila. IBRO Rep. 2020, 9, 65–77. [Google Scholar] [CrossRef]
- Gallardo, G.; Schlüter, O.M.; Südhof, T.C. A molecular pathway of neurodegeneration linking α-synuclein to ApoE and Aβ peptides. Nat. Neurosci. 2008, 11, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Meides, A.; Zhang, D.P.; Finley, D. A ubiquitin stress response induces altered proteasome composition. Cell 2007, 129, 747–759. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.J.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Shimura, H.; Schlossmacher, M.G.; Hattori, N.; Frosch, M.P.; Trockenbacher, A.; Schneider, R.; Mizuno, Y.; Kosik, K.S.; Selkoe, D.J. Ubiquitination of a new form of alpha synuclein by Parkin from human brain: Implications for Parkinson’s disease. Science 2001, 293, 263–269. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nature 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.-Y.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated α-synuclein is ubiquitinated in α-synucleinopathy lesions. J. Biol. Chem. 2002, 277, 49071–49076. [Google Scholar] [CrossRef]
- Dorval, V.; Fraser, P.E. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and α-synuclein. J. Biol. Chem. 2006, 281, 9919–9924. [Google Scholar] [CrossRef]
- De Oliveira, R.M.; Miranda, H.V.; Francelle, L.; Pinho, R.; Szegö, M.; Martinho, R.; Munari, F.; Lázaro, D.F.; Moniot, S.; Guerreiro, P.; et al. The mechanism of sirtuin 2–mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLoS Biol. 2017, 15, e2000374. [Google Scholar] [CrossRef]
- Stefanis, L.; Emmanouilidou, E.; Pantazopoulou, M.; Kirik, D.; Vekrellis, K.; Tofaris, G. How is alpha-synuclein cleared from the cell? J. Neurochem. 2019, 150, 577–590. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Genotype | Source |
|---|---|---|
| W303-1A | MATa; ura3-1; trp1D2; leu2-3_112; his3-11; ade2-1; can1-100 | EUROSCARF |
| RH3851 | W303-1A: ura3-1::GAL1-SNCA-mCherry-sfGFP-ADH1-kanMX (one genomic copy) | This Study |
| RH3852 | W303-1A: ura3-1::GAL1-SNCA(S129A)-mCherry-sfGFP-ADH1-kanMX (one genomic copy) | This Study |
| RH3853 | W303-1A: ura3-1::GAL1-SNCA(Y133F)-mCherry-sfGFP-ADH1-kanMX (one genomic copy) | This Study |
| RH3493 | MATα, ura3-52, trp1::hisG, Δarg4::loxP, Δlys1::loxP | [50] |
| R1158 | MATa URA3::CMV-tTA his3-1 leu2-0 met15-0 (kanMX4:G418R) | yTHC collection, Horizon Discovery, UK |
| yTHC-916 | Tet-RPN5 in R1158 | yTHC collection, Horizon Discovery, UK |
| yTHC-481 | Tet-RPN8 in R1158 | yTHC collection, Horizon Discovery, UK |
| yTHC-719 | Tet-RPN11 in R1158 | yTHC collection, Horizon Discovery, UK |
| yTHC-76 | Tet-RPT2 in R1158 | yTHC collection, Horizon Discovery, UK |
| yTHC-24 | Tet-RPT4 in R1158 | yTHC collection, Horizon Discovery, UK |
| yTHC-681 | Tet-RPT6 in R1158 | yTHC collection, Horizon Discovery, UK |
| yTHC-761 | Tet-PRE1 in R1158 | yTHC collection, Horizon Discovery, UK |
| yTHC-345 | Tet-PRE3 in R1158 | yTHC collection, Horizon Discovery, UK |
| yTHC-1012 | Tet-PRE4 in R1158 | yTHC collection, Horizon Discovery, UK |
| yTHC-564 | Tet-PRE5 in R1158 | yTHC collection, Horizon Discovery, UK |
| yTHC-657 | Tet-PRE6 in R1158 | yTHC collection, Horizon Discovery, UK |
| yTHC-545 | Tet-PRE8 in R1158 | yTHC collection, Horizon Discovery, UK |
| RH3854 | Tet-RPN5 in R1158; 1 genomic copy of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3855 | Tet-RPN5 in R1158; 2 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3856 | Tet-RPN5 in R1158; 3 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3857 | Tet-RPN5 in R1158; pME5037 (EV) in trp1 locus | This Study |
| RH3858 | Tet-RPN8 in R1158; 1 genomic copy of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3859 | Tet-RPN8 in R1158; 2 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3860 | Tet-RPN8 in R1158; 3 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3861 | Tet-RPN8 in R1158; pME5037 (EV) in trp1 locus | This Study |
| RH3862 | Tet-RPN11 in R1158; 1 genomic copy of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3863 | Tet-RPN11 in R1158; 2 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3864 | Tet-RPN11 in R1158; 3 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3865 | Tet-RPN11 in R1158; pME5037 (EV) in trp1 locus | This Study |
| RH3866 | Tet-RPT2 in R1158; 1 genomic copy of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3867 | Tet-RPT2 in R1158; 2 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3868 | Tet-RPT2 in R1158; 3 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3869 | Tet-RPT2 in R1158; pME5037 (EV) in trp1 locus | This Study |
| RH3870 | Tet-RPT4 in R1158; 1 genomic copy of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3871 | Tet-RPT4 in R1158; 2 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3872 | Tet-RPT4 in R1158; 3 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3873 | Tet-RPT4 in R1158; pME5037 (EV) in trp1 locus | This Study |
| RH3874 | Tet-RPT6 in R1158; 1 genomic copy of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3875 | Tet-RPT6 in R1158; 2 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3876 | Tet-RPT6 in R1158; 3 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3877 | Tet-RPT6 in R1158; pME5037 (EV) in trp1 locus | This Study |
| RH3878 | Tet-PRE1 in R1158; 1 genomic copy of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3879 | Tet-PRE1 in R1158; 2 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3880 | Tet-PRE1 in R1158; 3 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3881 | Tet-PRE1 in R1158; pME5037 (EV) in trp1 locus | This Study |
| RH3882 | Tet-PRE3 in R1158; 1 genomic copy of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3883 | Tet-PRE3 in R1158; 2 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3884 | Tet-PRE3 in R1158; 3 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3885 | Tet-PRE3 in R1158; pME5037 (EV) in trp1 locus | This Study |
| RH3886 | Tet-PRE4 in R1158; 1 genomic copy of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3887 | Tet-PRE4 in R1158; 2 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3888 | Tet-PRE4 in R1158; 3 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3889 | Tet-PRE4 in R1158; pME503 (EV) in trp1 locus | This Study |
| RH3890 | Tet-PRE5 in R1158; 1 genomic copy of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3891 | Tet-PRE5 in R1158; 2 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3892 | Tet-PRE5 in R1158; 3 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3893 | Tet-PRE5 in R1158; pME5037 (EV) in trp1 locus | This Study |
| RH3894 | Tet-PRE6 in R1158; 1 genomic copy of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3895 | Tet-PRE6 in R1158; 2 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3896 | Tet-PRE6 in R1158; 3 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3897 | Tet-PRE6 in R1158; pME5037 (EV) in trp1 locus | This Study |
| RH3898 | Tet-PRE8 in R1158; 1 genomic copy of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3899 | Tet-PRE8 in R1158; 2 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3900 | Tet-PRE8 in R1158; 3 genomic copies of GAL1-SNCA-GFP in trp1 locus | This Study |
| RH3901 | Tet-PRE8 in R1158; pME5037 (EV) in trp1 locus | This Study |
| Name | Description | Source |
|---|---|---|
| p426 | 2µm, URA3, GAL1, CYC1, AmpR | [51] |
| pME5037 | pRS305 (LEU2, GAL1, CYC1, AmpR) with TRP1 | [47] |
| pME5038 | pME5037 with GAL1-SNCA-GFP | [47] |
| pME5039 | p425 (2µm, LEU2, CYC1, AmpR) with GAL1-SNCA-GFP | [47] |
| pME3760 | p426-GAL1-SNCA | [23] |
| pME3759 | p426-GAL1-GFP | [23] |
| pME5320 | p426-GAL1-SNCA(S129A) | This study |
| pME5321 | pFA6a-GAL1-SNCA-mCherry-sfGFP-kanMX | This study |
| pME5322 | pFA6a-GAL1-SNCA(S129A)-mCherry-sfGFP-kanMX | This study |
| pME5322 | pFA6a-GAL1-SNCA(Y133F)-mCherry-sfGFP-kanMX | This study |
| pME4480 | pME2787-MET25-BirA* | [52] |
| pME5324 | p426-GAL1-αSyn-BirA* | This study |
| pME5325 | p426-GAL1-SNCA(S129A)-BirA* | This study |
| Proteasome Subunit | Description | αSyn/EV (log2) | S129A/EV (log2) |
|---|---|---|---|
| 20S Core: | |||
| Pre9 | Proteasome subunit alpha type-3 | −1.48 | −0.50 |
| Pre7 | Proteasome subunit beta type-6 | −1.26 | −0.44 |
| Pre8 | Proteasome subunit alpha type-2 | −1.26 | −0.49 |
| Pre10 | Proteasome subunit alpha type-7 | −1.12 | −0.27 |
| Pre3 | Proteasome subunit beta type-1 | −0.93 | −0.46 |
| Pre5 | Proteasome subunit alpha type-6 | −0.84 | −0.25 |
| 19S Regulatory particle—LID: | |||
| Rpn12 | 26S proteasome regulatory subunit Rpn12 | −1.11 | −0.34 |
| Rpn8 | 26S proteasome regulatory subunit Rpn8 | −0.77 | −0.30 |
| 19S Regulatory particle—BASE: | |||
| Rpn13 | 26S proteasome regulatory subunit Rpn13 | −0.92 | −0.29 |
| 19S Regulatory particle—ubiquitin receptor: | |||
| Rpn10 | 26S proteasome regulatory subunit Rpn10 | −0.62 | −0.24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popova, B.; Galka, D.; Häffner, N.; Wang, D.; Schmitt, K.; Valerius, O.; Knop, M.; Braus, G.H. α-Synuclein Decreases the Abundance of Proteasome Subunits and Alters Ubiquitin Conjugates in Yeast. Cells 2021, 10, 2229. https://doi.org/10.3390/cells10092229
Popova B, Galka D, Häffner N, Wang D, Schmitt K, Valerius O, Knop M, Braus GH. α-Synuclein Decreases the Abundance of Proteasome Subunits and Alters Ubiquitin Conjugates in Yeast. Cells. 2021; 10(9):2229. https://doi.org/10.3390/cells10092229
Chicago/Turabian StylePopova, Blagovesta, Dajana Galka, Nicola Häffner, Dan Wang, Kerstin Schmitt, Oliver Valerius, Michael Knop, and Gerhard H. Braus. 2021. "α-Synuclein Decreases the Abundance of Proteasome Subunits and Alters Ubiquitin Conjugates in Yeast" Cells 10, no. 9: 2229. https://doi.org/10.3390/cells10092229
APA StylePopova, B., Galka, D., Häffner, N., Wang, D., Schmitt, K., Valerius, O., Knop, M., & Braus, G. H. (2021). α-Synuclein Decreases the Abundance of Proteasome Subunits and Alters Ubiquitin Conjugates in Yeast. Cells, 10(9), 2229. https://doi.org/10.3390/cells10092229