
Multiple Targets for Oxysterols in Their Regulation of the Immune System


 , and
, and 
Abstract
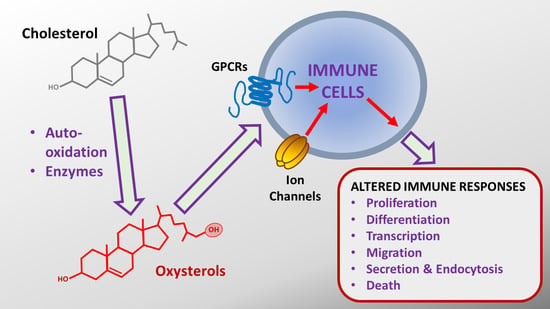
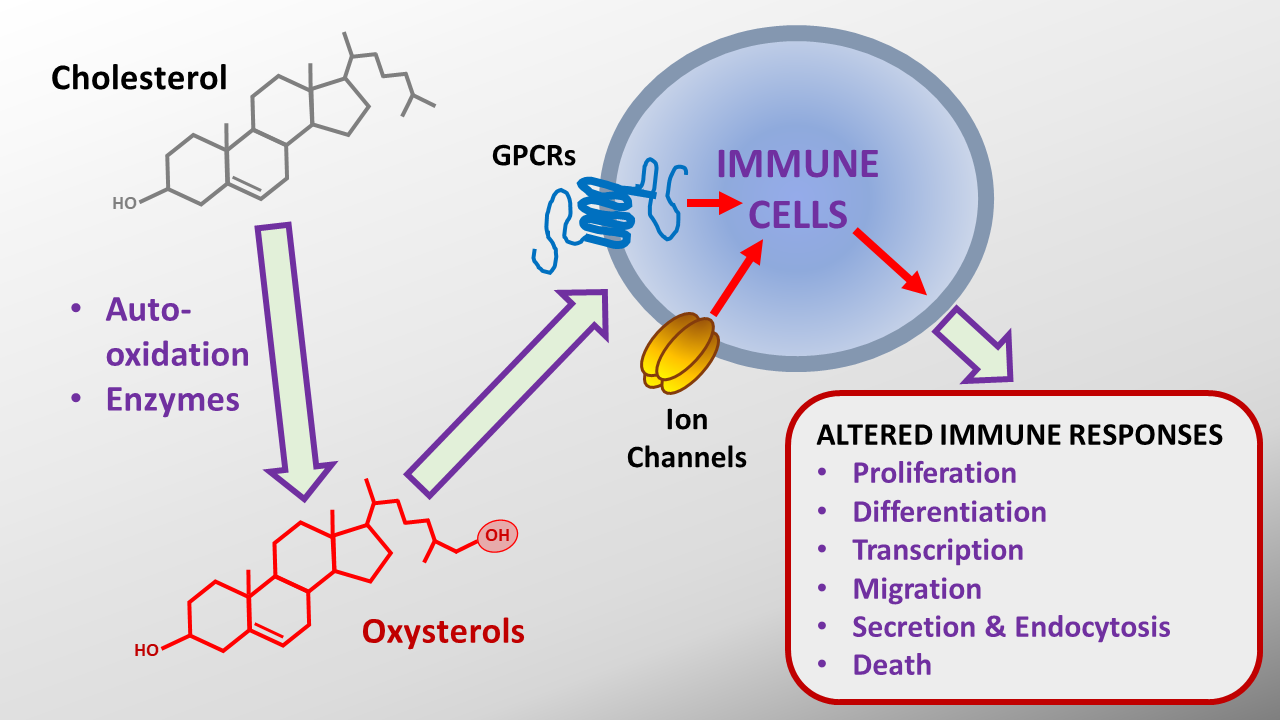
:
{kind=link}
{kind=link}
{kind=link}
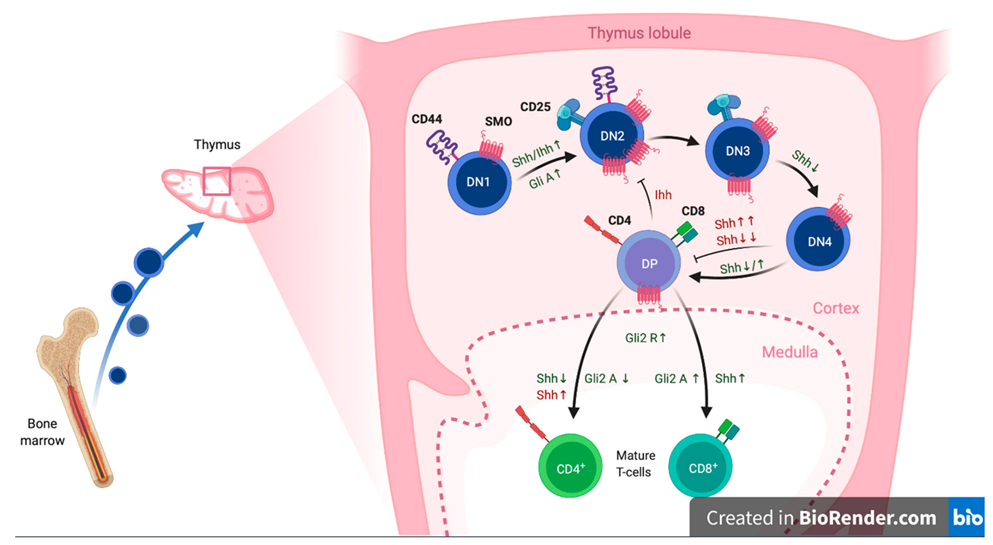{kind=link}
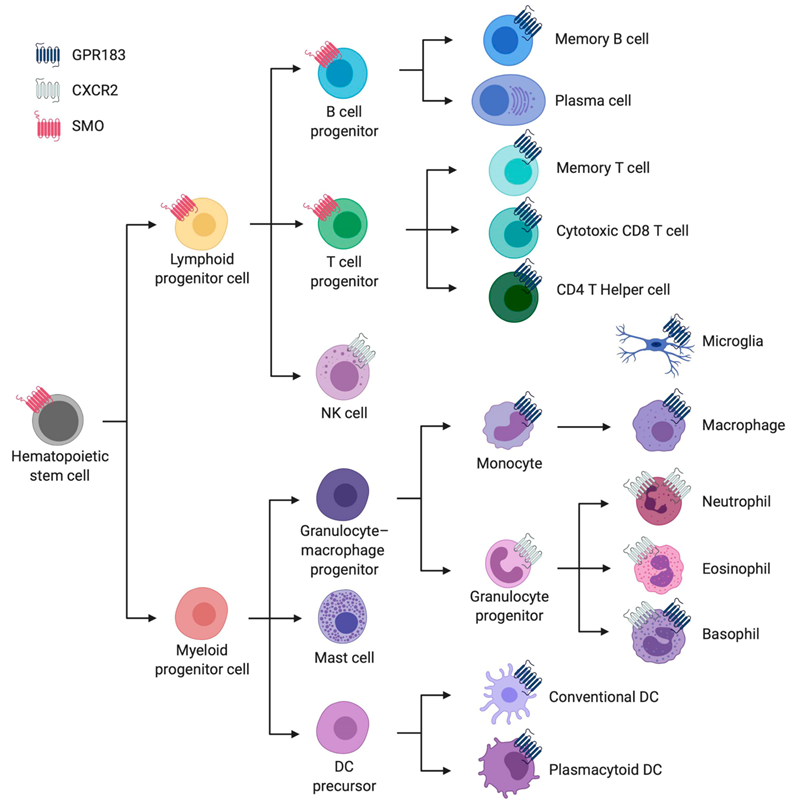
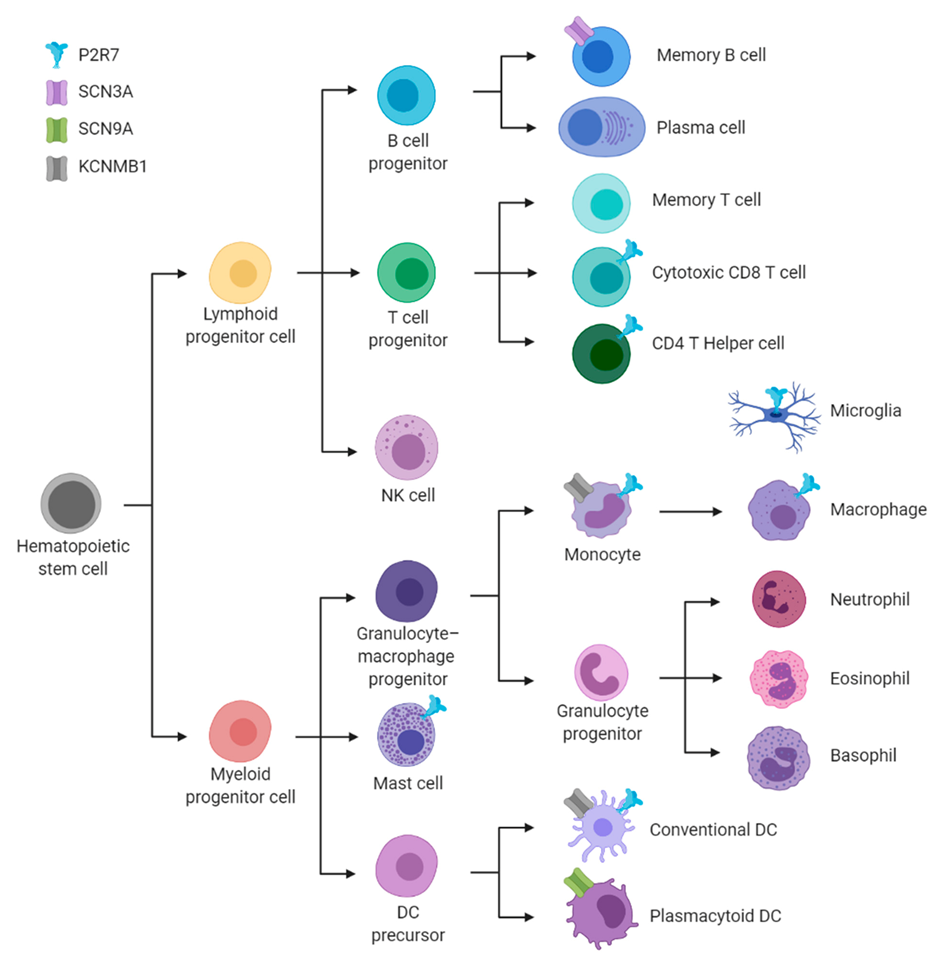{kind=link}
1. Introduction
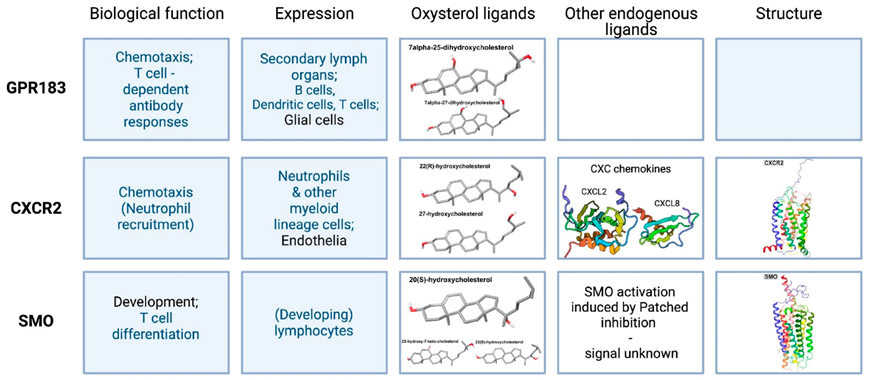
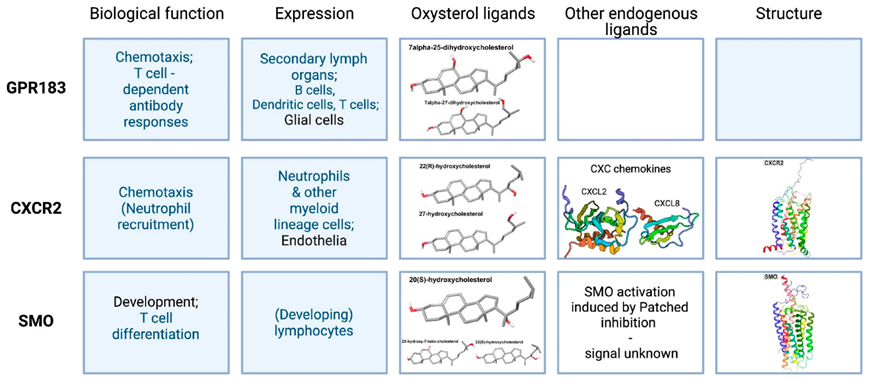
2. G Protein-Coupled Receptors
2.1. Epstein–Barr Virus-Induced Receptor 2, or GPR183
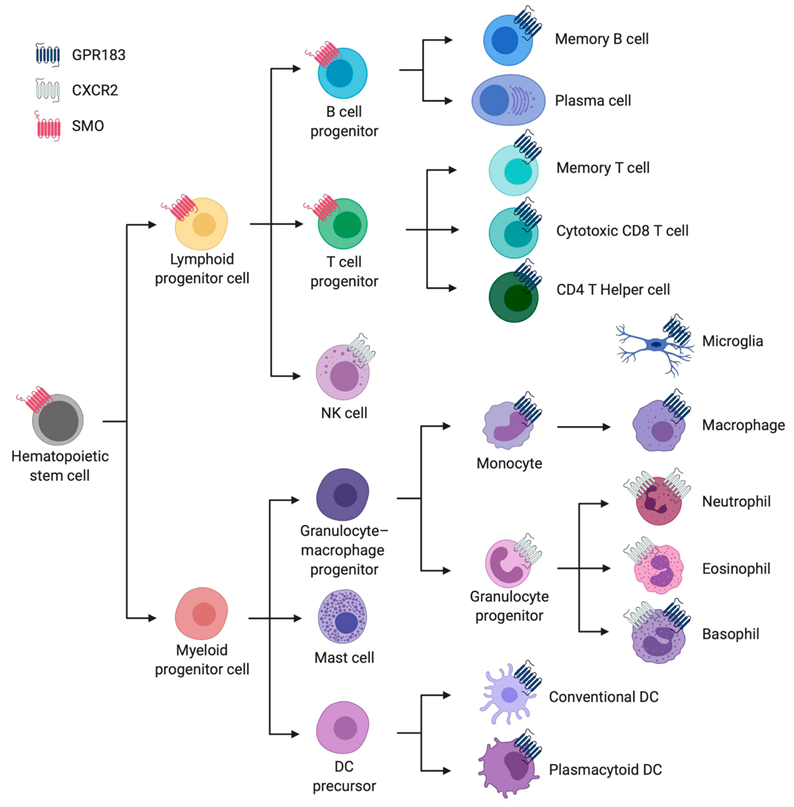
2.1.1. Cellular Expression Pattern and Overall Biological Function
2.1.2. The Chemistry and Production of Endogenous Oxysterols That Bind GPR183
2.1.3. GPR183 Receptor Structure
2.2. CXC Chemokine Receptor 2 (CXCR2)
2.2.1. Cellular Expression Pattern and Overall Biological Function
2.2.2. The Chemistry and Production of Endogenous CXCR2 Ligands
2.2.3. CXCR2 Structure
2.3. Smoothened (SMO)
2.3.1. Cellular Expression Pattern and Overall Biological Function
2.3.2. Chemistry and Production of Endogenous SMO Ligands
2.3.3. SMO Receptor Structure
2.4. Gamma-Amino Butyric Acid (GABA) Type B Receptors
3. Ion Channels
3.1. Ligand-Gated Ion Channels
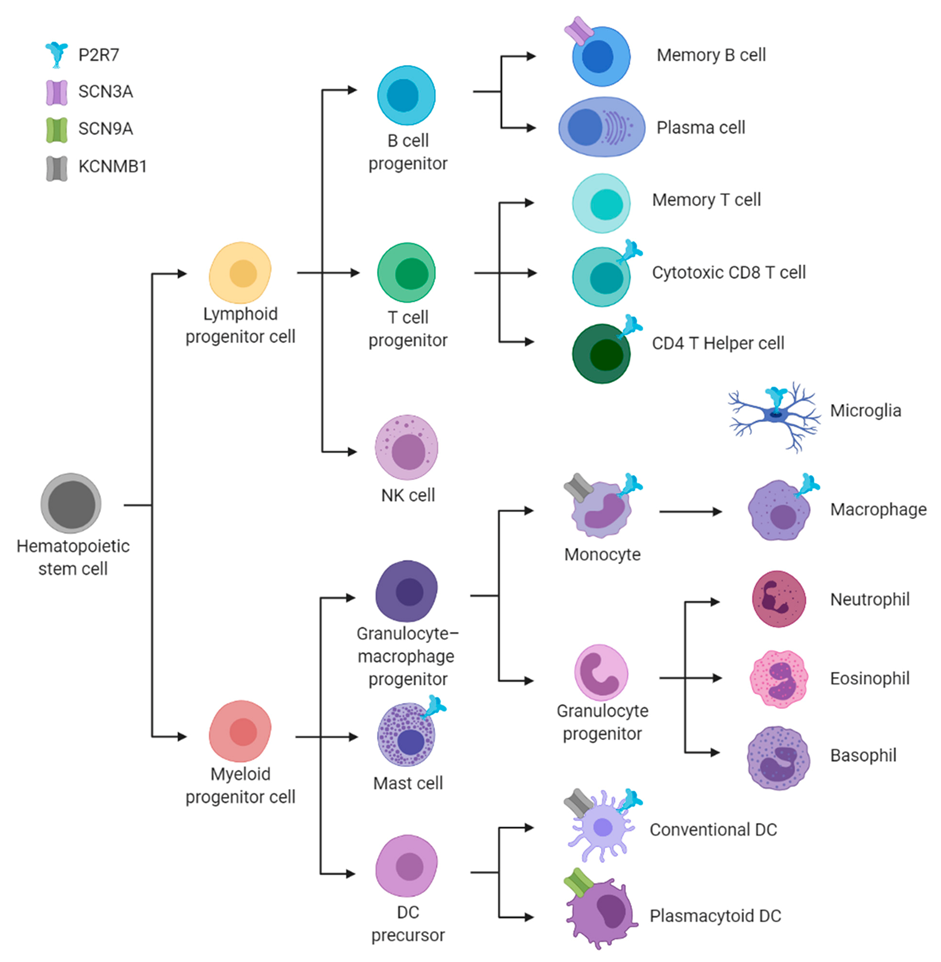
3.1.1. The P2X7 Purinoreceptor (P2X7R)
3.1.2. Ionotropic Glutamate Receptors
3.1.3. Ionotropic GABAARs
3.2. Voltage-Gated Ion Channels
3.2.1. Voltage-Gated Potassium Channels
3.2.1.1. Kv3.1
3.2.2. Voltage-Gated Sodium Channels
3.3. Ion Channels Gated by Multiple Stimuli
3.3.1. Ca2+ and Voltage-Dependent K+ Channels
3.3.2. Transient Receptor Potential Channels
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Luu, W.; Sharpe, L.J.; Capell-Hattam, I.; Gelissen, I.C.; Brown, A.J. Oxysterols: Old Tale, New Twists. Annu. Rev. Pharm. Toxicol. 2016, 56, 447–467. [Google Scholar] [CrossRef]
- Kloudova, A.; Guengerich, F.P.; Soucek, P. The Role of Oxysterols in Human Cancer. Trends Endocrinol. Metab. 2017, 28, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Zmysłowski, A.; Szterk, A. Current knowledge on the mechanism of atherosclerosis and pro-atherosclerotic properties of oxysterols. Lipids Health Dis. 2017, 16, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.; Finlay, D.K. Diverse Immunoregulatory Roles of Oxysterols—The Oxidized Cholesterol Metabolites. Metabolites 2020, 10, 384. [Google Scholar] [CrossRef]
- Duc, D.; Vigne, S.; Pot, C. Oxysterols in Autoimmunity. Int. J. Mol. Sci. 2019, 20, 4522. [Google Scholar] [CrossRef] [Green Version]
- Spann, N.J.; Glass, C.K. Sterols and oxysterols in immune cell function. Nat. Immunol. 2013, 14, 893–900. [Google Scholar] [CrossRef]
- Massey, J.B. Membrane and protein interactions of oxysterols. Curr. Opin. Lipidol. 2006, 17, 296–301. [Google Scholar] [CrossRef] [Green Version]
- Zang, R.; Case, J.B.; Yutuc, E.; Ma, X.; Shen, S.; Castro, M.F.G.; Liu, Z.; Zeng, Q.; Zhao, H.; Son, J.; et al. Cholesterol 25-hydroxylase suppresses SARS-CoV-2 replication by blocking membrane fusion. Proc. Natl. Acad. Sci. USA 2020, 117, 32105–32113. [Google Scholar] [CrossRef] [PubMed]
- Domingues, M.; Gomes, B.; Hollmann, A.; Santos, N. 25-Hydroxycholesterol Effect on Membrane Structure and Mechanical Properties. Int. J. Mol. Sci. 2021, 22, 2574. [Google Scholar] [CrossRef]
- Ghzaiel, I.; Sassi, K.; Zarrouk, A.; Nury, T.; Ksila, M.; Leoni, V.; Bouhaouala-Zahar, B.; Hammami, S.; Hammami, M.; Mackrill, J.J.; et al. 7-Ketocholesterol: Effects on viral infections and hypothetical contribution in COVID-19. J. Steroid Biochem. Mol. Biol. 2021, 212, 105939. [Google Scholar] [CrossRef] [PubMed]
- Abrams, M.E.; Johnson, K.; Perelman, S.S.; Zhang, L.-S.; Endapally, S.; Mar, K.B.; Thompson, B.M.; McDonald, J.G.; Schoggins, J.W.; Radhakrishnan, A.; et al. Oxysterols provide innate immunity to bacterial infection by mobilizing cell surface accessible cholesterol. Nat. Microbiol. 2020, 5, 929–942. [Google Scholar] [CrossRef]
- Antonny, B.; Bigay, J.; Mesmin, B. The Oxysterol-Binding Protein Cycle: Burning Off PI(4)P to Transport Cholesterol. Annu. Rev. Biochem. 2018, 87, 809–837. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A.; Ridgway, N.D. Bridging the molecular and biological functions of the oxysterol-binding protein family. Cell. Mol. Life Sci. 2018, 75, 3079–3098. [Google Scholar] [CrossRef] [PubMed]
- Olkkonen, V.M. OSBP-Related Protein Family in Lipid Transport over Membrane Contact Sites. Lipid Insights 2015, 8s1, LPI-S31726. [Google Scholar] [CrossRef] [Green Version]
- Mackrill, J.J. Oxysterols and calcium signal transduction. Chem. Phys. Lipids 2011, 164, 488–495. [Google Scholar] [CrossRef]
- Zhong, W.; Yi, Q.; Xu, B.; Li, S.; Wang, T.; Liu, F.; Zhu, B.; Hoffmann, P.R.; Ji, G.; Lei, P.; et al. ORP4L is essential for T-cell acute lymphoblastic leukemia cell survival. Nat. Commun. 2016, 7, 12702. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Chen, J.; Li, D.; Xie, P.; Xu, M.; Lin, W.; Li, S.; Pan, G.; Tang, Y.; Xu, J.; et al. ORP4L couples IP3to ITPR1 in control of endoplasmic reticulum calcium release. FASEB J. 2019, 33, 13852–13865. [Google Scholar] [CrossRef]
- Lessmann, E.; Ngo, M.; Leitges, M.; Minguet, S.; Ridgway, N.D.; Huber, M. Oxysterol-binding protein-related protein (ORP) 9 is a PDK-2 substrate and regulates Akt phosphorylation. Cell. Signal. 2007, 19, 384–392. [Google Scholar] [CrossRef]
- Strating, J.; Van Der Linden, L.; Albulescu, L.; Bigay, J.; Arita, M.; Delang, L.; Leyssen, P.; Van Der Schaar, H.M.; Lanke, K.H.; Thibaut, H.J.; et al. Itraconazole Inhibits Enterovirus Replication by Targeting the Oxysterol-Binding Protein. Cell Rep. 2015, 10, 600–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Nelson, E.R. Oxysterols and nuclear receptors. Mol. Cell. Endocrinol. 2019, 484, 42–51. [Google Scholar] [CrossRef]
- Korf, H.; Beken, S.V.; Romano, M.; Steffensen, K.; Stijlemans, B.; Gustafsson, J.; Grooten, J.; Huygen, K. Liver X receptors contribute to the protective immune response against Mycobacterium tuberculosis in mice. J. Clin. Investig. 2009, 119, 1626–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A DeBose-Boyd, R.; Brown, M.S.; Li, W.-P.; Nohturfft, A.; Goldstein, J.L.; Espenshade, P.J. Transport-Dependent Proteolysis of SREBP: Relocation of Site-1 Protease from Golgi to ER Obviates the Need for SREBP Transport to Golgi. Cell 1999, 99, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Lee, J.N.; Brown, M.S.; Goldstein, J.L.; Ye, J. Juxtamembranous aspartic acid in Insig-1 and Insig-2 is required for cholesterol homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 6154–6159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Kumar, N.; Crumbley, C.; Griffin, P.R.; Burris, T.P. A second class of nuclear receptors for oxysterols: Regulation of RORα and RORγ activity by 24S-hydroxycholesterol (cerebrosterol). Mol. Cell Biol. Lipids 2010, 1801, 917–923. [Google Scholar] [CrossRef] [Green Version]
- DuSell, C.D.; Umetani, M.; Shaul, P.W.; Mangelsdorf, D.; McDonnell, D.P. 27-Hydroxycholesterol Is an Endogenous Selective Estrogen Receptor Modulator. Mol. Endocrinol. 2008, 22, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Umetani, M.; Domoto, H.; Gormley, A.K.; Yuhanna, I.S.; Cummins, C.; Javitt, N.B.; Korach, K.; Shaul, P.W.; Mangelsdorf, D. 27-Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen. Nat. Med. 2007, 13, 1185–1192. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, L.; Pandak, W.M.; Heuman, D.; Hylemon, P.B.; Ren, S. High Glucose Induces Lipid Accumulation via 25-Hydroxycholesterol DNA-CpG Methylation. iScience 2020, 23, 101102. [Google Scholar] [CrossRef]
- Bauman, D.R.; Bitmansour, A.D.; McDonald, J.G.; Thompson, B.; Liang, G.; Russell, D.W. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl. Acad. Sci. USA 2009, 106, 16764–16769. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, L.D.; Pontini, L.; Marinozzi, M.; Sanchez-Aranguren, L.C.; Reis, A.; Dias, I.H. Cholesterol and oxysterol sulfates: Pathophysiological roles and analytical challenges. Br. J. Pharm. 2020, 178, 3327–3341. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, W.; Brown, J.E.; Chen, L.; Pandak, W.M.; Hylemon, P.B.; Ren, S. 25-Hydroxycholesterol 3-sulfate is an endogenous ligand of DNA methyltransferases in hepatocytes. J. Lipid Res. 2021, 62, 100063. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, X.; Ren, S. Cholesterol Metabolites 25-Hydroxycholesterol and 25-Hydroxycholesterol 3-Sulfate Are Potent Paired Regulators: From Discovery to Clinical Usage. Metabolites 2020, 11, 9. [Google Scholar] [CrossRef]
- Bottemanne, P.; Paquot, A.; Ameraoui, H.; Guillemot-Legris, O.; Alhouayek, M.; Muccioli, G.G. 25-Hydroxycholesterol metabolism is altered by lung inflammation, and its local administration modulates lung inflammation in mice. FASEB J. 2021, 35, e21514. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Miyamoto, T.; Kakizawa, T.; Nishio, S.-I.; Oiwa, A.; Takeda, T.; Suzuki, S.; Hashizume, K. Inhibition of LXRα signaling by vitamin D receptor: Possible role of VDR in bile acid synthesis. Biochem. Biophys. Res. Commun. 2006, 351, 176–184. [Google Scholar] [CrossRef]
- Wamil, M.; Andrew, R.; Chapman, K.; Street, J.; Morton, N.M.; Seckl, J.R. 7-Oxysterols Modulate Glucocorticoid Activity in Adipocytes through Competition for 11β-Hydroxysteroid Dehydrogenase Type. Endocrinology 2008, 149, 5909–5918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repa, J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.-M.A.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000, 14, 2819–2830. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Yang, J.; Horton, J.D.; Hammer, R.E.; Goldstein, J.L.; Brown, M.S. Diminished Hepatic Response to Fasting/Refeeding and Liver X Receptor Agonists in Mice with Selective Deficiency of Sterol Regulatory Element-binding Protein-1c. J. Biol. Chem. 2002, 277, 9520–9528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maïga, A.; Lemieux, S.; Pabst, C.; Lavallée, V.-P.; Bouvier, M.; Sauvageau, G.; Hébert, J. Transcriptome analysis of G protein-coupled receptors in distinct genetic subgroups of acute myeloid leukemia: Identification of potential disease-specific targets. Blood Cancer J. 2016, 6, e431. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, B.J.; Singh, D.; Madrigal, A.; Valdovino-Gonzalez, A.G.; White, B.M.; Zapardiel-Gonzalo, J.; Ha, B.; Altay, G.; Greenbaum, J.A.; McVicker, G.; et al. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell 2018, 175, 1701–1715. [Google Scholar] [CrossRef] [Green Version]
- Villani, A.-C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef] [Green Version]
- Rosenkilde, M.M.; Benned-Jensen, T.; Andersen, H.; Holst, P.J.; Kledal, T.N.; Luttichau, H.R.; Larsen, J.K.; Christensen, J.; Schwartz, T.W. Molecular Pharmacological Phenotyping of EBI2. J. Biol. Chem. 2006, 281, 13199–13208. [Google Scholar] [CrossRef] [Green Version]
- Yi, T.; Wang, X.; Kelly, L.M.; An, J.; Xu, Y.; Sailer, A.; Gustafsson, J.-A.; Russell, D.; Cyster, J.G. Oxysterol Gradient Generation by Lymphoid Stromal Cells Guides Activated B Cell Movement during Humoral Responses. Immunity 2012, 37, 535–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, L.M.; Pereira, J.; Yi, T.; Xu, Y.; Cyster, J.G. EBI2 Guides Serial Movements of Activated B Cells and Ligand Activity Is Detectable in Lymphoid and Nonlymphoid Tissues. J. Immunol. 2011, 187, 3026–3032. [Google Scholar] [CrossRef]
- Gatto, M.; Wood, K.; Brink, R. EBI2 Operates Independently of but in Cooperation with CXCR5 and CCR7 To Direct B Cell Migration and Organization in Follicles and the Germinal Center. J. Immunol. 2011, 187, 4621–4628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, J.; Kelly, L.M.; Xu, Y.; Cyster, J.G. EBI2 mediates B cell segregation between the outer and centre follicle. Nat. Cell Biol. 2009, 460, 1122–1126. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.T.; Choi, J.-Y.; Lainez, B.; Schulz, V.P.; Karas, D.E.; Baum, E.D.; Setlur, J.; Gallagher, P.G.; Craft, J. Human Extrafollicular CD4+ Th Cells Help Memory B Cells Produce Igs. J. Immunol. 2018, 201, 1359–1372. [Google Scholar] [CrossRef]
- Ki, S.; Thyagarajan, H.M.; Hu, Z.; Lancaster, J.N.; Ehrlich, L.I. EBI2 contributes to the induction of thymic central tolerance in mice by promoting rapid motility of medullary thymocytes. Eur. J. Immunol. 2017, 47, 1906–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baptista, A.P.; Gola, A.; Huang, Y.; Milanez-Almeida, P.; Torabi-Parizi, P.; Urban, J.; Shapiro, V.S.; Gerner, M.Y.; Germain, R.N. The Chemoattractant Receptor Ebi2 Drives Intranodal Naive CD4+ T Cell Peripheralization to Promote Effective Adaptive Immunity. Immunity 2019, 50, 1188–1201. [Google Scholar] [CrossRef]
- Li, J.; Lu, E.; Yi, T.; Cyster, J.G. EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nat. Cell Biol. 2016, 533, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Yi, T.; Cyster, J.G. EBI2-mediated bridging channel positioning supports splenic dendritic cell homeostasis and particulate antigen capture. eLife 2013, 2, e00757. [Google Scholar] [CrossRef]
- Lu, E.; Dang, E.V.; McDonald, J.G.; Cyster, J.G. Distinct oxysterol requirements for positioning naïve and activated dendritic cells in the spleen. Sci. Immunol. 2017, 2, eaal5237. [Google Scholar] [CrossRef] [Green Version]
- Chiang, E.Y.; Johnston, R.J.; Grogan, J.L. EBI2 Is a Negative Regulator of Type I Interferons in Plasmacytoid and Myeloid Dendritic Cells. PLoS ONE 2013, 8, e83457. [Google Scholar] [CrossRef]
- Shen, Z.-J.; Hu, J.; Kashi, V.P.; Kelly, E.; Denlinger, L.C.; Lutchman, K.; McDonald, J.G.; Jarjour, N.N.; Malter, J.S. Epstein-Barr Virus–induced Gene 2 Mediates Allergen-induced Leukocyte Migration into Airways. Am. J. Respir. Crit. Care Med. 2017, 195, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Emgård, J.; Kammoun, H.; García-Cassani, B.; Chesné, J.; Parigi, S.M.; Jacob, J.-M.; Cheng, H.-W.; Evren, E.; Das, S.; Czarnewski, P.; et al. Oxysterol Sensing through the Receptor GPR183 Promotes the Lymphoid-Tissue-Inducing Function of Innate Lymphoid Cells and Colonic Inflammation. Immunity 2018, 48, 120–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyss, A.; Raselli, T.; Perkins, N.; Ruiz, F.; Schmelczer, G.; Klinke, G.; Moncsek, A.; Roth, R.; Spalinger, M.R.; Hering, L.; et al. The EBI2-oxysterol axis promotes the development of intestinal lymphoid structures and colitis. Mucosal Immunol. 2019, 12, 733–745. [Google Scholar] [CrossRef] [Green Version]
- Mutemberezi, V.; Buisseret, B.; Masquelier, J.; Guillemot-Legris, O.; Alhouayek, M.; Muccioli, G.G. Oxysterol levels and metabolism in the course of neuroinflammation: Insights from in vitro and in vivo models. J. Neuroinflamm. 2018, 15, 1–16. [Google Scholar] [CrossRef]
- Rutkowska, A.; O’Sullivan, S.A.; Christen, I.; Zhang, J.; Sailer, A.; Dev, K.K. The EBI2 signalling pathway plays a role in cellular crosstalk between astrocytes and macrophages. Sci. Rep. 2016, 6, 25520. [Google Scholar] [CrossRef] [Green Version]
- Barington, L.; Wanke, F.; Arfelt, K.N.; Holst, P.J.; Kurschus, F.C.; Rosenkilde, M.M. EBI2 in splenic and local immune responses and in autoimmunity. J. Leukoc. Biol. 2018, 104, 313–322. [Google Scholar] [CrossRef]
- Van Der Poel, M.; Ulas, T.; Mizee, M.R.; Hsiao, C.-C.; Miedema, S.; Adelia; Schuurman, K.G.; Helder, B.; Tas, S.W.; Schultze, J.L.; et al. Transcriptional profiling of human microglia reveals grey–white matter heterogeneity and multiple sclerosis-associated changes. Nat. Commun. 2019, 10, 1139. [Google Scholar] [CrossRef] [Green Version]
- Braden, K.; Giancotti, L.A.; Chen, Z.; DeLeon, C.; Latzo, N.; Boehm, T.; D’Cunha, N.; Thompson, B.M.; Doyle, T.M.; McDonald, J.G.; et al. GPR183-Oxysterol Axis in Spinal Cord Contributes to Neuropathic Pain. J. Pharm. Exp. 2020, 375, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Wanke, F.; Moos, S.; Croxford, A.L.; Heinen, A.P.; Gräf, S.; Kalt, B.; Tischner, D.; Zhang, J.; Christen, I.; Bruttger, J.; et al. EBI2 Is Highly Expressed in Multiple Sclerosis Lesions and Promotes Early CNS Migration of Encephalitogenic CD4 T Cells. Cell Rep. 2017, 18, 1270–1284. [Google Scholar] [CrossRef] [Green Version]
- Clottu, A.S.; Mathias, A.; Sailer, A.W.; Schluep, M.; Seebach, J.; Du Pasquier, R.; Pot, C. EBI2 Expression and Function: Robust in Memory Lymphocytes and Increased by Natalizumab in Multiple Sclerosis. Cell Rep. 2017, 18, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Chalmin, F.; Rochemont, V.; Lippens, C.; Clottu, A.; Sailer, A.; Merkler, D.; Hugues, S.; Pot, C. Oxysterols regulate encephalitogenic CD4+ T cell trafficking during central nervous system autoimmunity. J. Autoimmun. 2015, 56, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Nevius, E.; Pinho, F.; Dhodapkar, M.; Jin, H.; Nadrah, K.; Horowitz, M.C.; Kikuta, J.; Ishii, M.; Pereira, J.P. Oxysterols and EBI2 promote osteoclast precursor migration to bone surfaces and regulate bone mass homeostasis. J. Exp. Med. 2015, 212, 1931–1946. [Google Scholar] [CrossRef]
- Hannedouche, S.; Zhang, J.; Yi, T.; Shen, W.; Nguyen, D.; Pereira, J.; Guerini, D.; Baumgarten, B.U.; Roggo, S.; Wen, B.; et al. Oxysterols direct immune cell migration via EBI2. Nat. Cell Biol. 2011, 475, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, X.V.; Wu, J.; Kuei, C.; Mani, N.S.; Zhang, L.; Yu, J.; Sutton, S.W.; Qin, N.; Banie, H.; et al. Oxysterols direct B-cell migration through EBI2. Nat. Cell Biol. 2011, 475, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Miyazaki, T.; Ikegami, T.; Iwamoto, J.; Maeda, T.; Hirayama, T.; Saito, Y.; Teramoto, T.; Matsuzaki, Y. Cholesterol 25-hydroxylation activity of CYP3A. J. Lipid Res. 2011, 52, 1509–1516. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, W.J.; Wang, Y. Oxysterols as lipid mediators: Their biosynthetic genes, enzymes and metabolites. Prostaglandins Other Lipid Mediat. 2019, 147, 106381. [Google Scholar] [CrossRef]
- Park, K.; Scott, A.L. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J. Leukoc. Biol. 2010, 88, 1081–1087. [Google Scholar] [CrossRef] [Green Version]
- Diczfalusy, U.; Olofsson, K.E.; Carlsson, A.-M.; Gong, M.; Golenbock, D.T.; Rooyackers, O.; Fläring, U.; Björkbacka, H. Marked upregulation of cholesterol 25-hydroxylase expression by lipopolysaccharide. J. Lipid Res. 2009, 50, 2258–2264. [Google Scholar] [CrossRef] [Green Version]
- Preuss, I.; Ludwig, M.-G.; Baumgarten, B.; Bassilana, F.; Gessier, F.; Seuwen, K.; Sailer, A.W. Transcriptional regulation and functional characterization of the oxysterol/EBI2 system in primary human macrophages. Biochem. Biophys. Res. Commun. 2014, 446, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Madenspacher, J.H.; Morrell, E.D.; Gowdy, K.M.; McDonald, J.G.; Thompson, B.M.; Muse, G.W.; Martinez, J.; Thomas, S.Y.; Mikacenic, C.; Nick, J.A.; et al. Cholesterol-25-hydroxylase promotes efferocytosis and resolution of lung inflammation. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Jia, J.; Conlon, T.M.; Sarker, R.S.; Taşdemir, D.; Smirnova, N.; Srivastava, B.; Verleden, S.; Güneş, G.; Wu, X.; Prehn, C.; et al. Cholesterol metabolism promotes B-cell positioning during immune pathogenesis of chronic obstructive pulmonary disease. EMBO Mol. Med. 2018, 10, e8349. [Google Scholar] [CrossRef]
- Li-Hawkins, J.; Lund, E.G.; Turley, S.D.; Russell, D.W. Disruption of the Oxysterol 7α-Hydroxylase Gene in Mice. J. Biol. Chem. 2000, 275, 16536–16542. [Google Scholar] [CrossRef] [Green Version]
- Dulos, J.; Van Der Vleuten, M.A.J.; Kavelaars, A.; Heijnen, C.J.; Boots, A.M. CYP7B expression and activity in fibroblast-like synoviocytes from patients with rheumatoid arthritis: Regulation by proinflammatory cytokines. Arthritis Rheum. 2005, 52, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Stoilov, I.; Krueger, W.; Mankowski, D.; Guernsey, L.; Kaur, A.; Glynn, J.; Thrall, R.S. The cytochromes P450 (CYP) response to allergic inflammation of the lung. Arch. Biochem. Biophys. 2006, 456, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Raselli, T.; Hearn, T.; Wyss, A.; Atrott, K.; Peter, A.; Frey-Wagner, I.; Spalinger, M.R.; Maggio, E.M.; Sailer, A.W.; Schmitt, J.; et al. Elevated oxysterol levels in human and mouse livers reflect nonalcoholic steatohepatitis. J. Lipid Res. 2019, 60, 1270–1283. [Google Scholar] [CrossRef] [Green Version]
- Kakiyama, G.; Marques, D.; Martin, R.; Takei, H.; Rodriguez-Agudo, D.; LaSalle, S.A.; Hashiguchi, T.; Liu, X.; Green, R.; Erickson, S.; et al. Insulin resistance dysregulates CYP7B1 leading to oxysterol accumulation: A pathway for NAFL to NASH transition. J. Lipid Res. 2020, 61, 1629–1644. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.-Z.; Lee, E.-J.; Lin, Y.-J.; Chen, L.; Zheng, H.-Y.; He, X.-Q.; Peng, J.-Y.; Noonepalle, S.K.; Shull, A.Y.; Pei, F.C.; et al. Recruitment of monocytes and epigenetic silencing of intratumoral CYP7B1 primarily contribute to the accumulation of 27-hydroxycholesterol in breast cancer. Am. J. Cancer Res. 2019, 9, 2194–2208. [Google Scholar]
- Benned-Jensen, T.; Rosenkilde, M.M. Structural Motifs of Importance for the Constitutive Activity of the Orphan 7TM Receptor EBI2: Analysis of Receptor Activation in the Absence of an Agonist. Mol. Pharm. 2008, 74, 1008–1021. [Google Scholar] [CrossRef] [Green Version]
- Benned-Jensen, T.; Rosenkilde, M.M. Distinct expression and ligand-binding profiles of two constitutively active GPR17 splice variants. Br. J. Pharm. 2010, 159, 1092–1105. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Shih, A.Y.; Yang, X.V.; Kuei, C.; Wu, J.; Deng, X.; Mani, N.S.; Mirzadegan, T.; Sun, S.; Lovenberg, T.W.; et al. Identification of Structural Motifs Critical for Epstein-Barr Virus-Induced Molecule 2 Function and Homology Modeling of the Ligand Docking Site. Mol. Pharm. 2012, 82, 1094–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benned-Jensen, T.; Norn, C.; Laurent, S.; Madsen, C.M.; Larsen, H.M.; Arfelt, K.N.; Wolf, R.M.; Frimurer, T.; Sailer, A.; Rosenkilde, M.M. Molecular Characterization of Oxysterol Binding to the Epstein-Barr Virus-induced Gene 2 (GPR183). J. Biol. Chem. 2012, 287, 35470–35483. [Google Scholar] [CrossRef] [Green Version]
- Sensi, C.; Daniele, S.; Parravicini, C.; Zappelli, E.; Russo, V.; Trincavelli, M.L.; Martini, C.; Abbracchio, M.P.; Eberini, I. Oxysterols act as promiscuous ligands of class-A GPCRs: In silico molecular modeling and in vitro validation. Cell. Signal. 2014, 26, 2614–2620. [Google Scholar] [CrossRef]
- Daugvilaite, V.; Madsen, C.M.; Lückmann, M.; Echeverria, C.C.; Sailer, A.W.; Frimurer, T.M.; Rosenkilde, M.M.; Benned-Jensen, T. Biased agonism and allosteric modulation of G protein-coupled receptor 183—A 7TM receptor also known as Epstein-Barr virus-induced gene 2. Br. J. Pharm. 2017, 174, 2031–2042. [Google Scholar] [CrossRef]
- Thal, D.M.; Glukhova, A.; Sexton, P.; Christopoulos, A. Structural insights into G-protein-coupled receptor allostery. Nat. Cell Biol. 2018, 559, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, Z.; Xiao, W.; Lu, S.; Zhang, J. Allosteric binding sites at the receptor–lipid bilayer interface: Novel targets for GPCR drug discovery. Drug Discov. Today 2020, 26, 690–703. [Google Scholar] [CrossRef]
- Guixà-González, R.; Albasanz, J.L.; Rodriguez-Espigares, I.; Pastor, M.; Sanz, F.; Marti-Solano, M.; Manna, M.; Martinez-Seara, H.; Hildebrand, P.W.; Martín, M.; et al. Membrane cholesterol access into a G-protein-coupled receptor. Nat. Commun. 2017, 8, 14505. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.-S.; Verstegen, N.; Ciampricotti, M.; Hawinkels, L.; Jonkers, J.; et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nat. Cell Biol. 2015, 522, 345–348. [Google Scholar] [CrossRef]
- Baek, A.E.; Yu, Y.-R.A.; He, S.; Wardell, S.E.; Chang, C.-Y.; Kwon, S.; Pillai, R.V.; McDowell, H.B.; Thompson, J.W.; Dubois, L.G.; et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat. Commun. 2017, 8, 864. [Google Scholar] [CrossRef]
- Addison, C.L.; Daniel, T.O.; Burdick, M.D.; Liu, H.; Ehlert, J.E.; Xue, Y.Y.; Buechi, L.; Walz, A.; Richmond, A.; Strieter, R.M. The CXC Chemokine Receptor 2, CXCR2, Is the Putative Receptor for ELR + CXC Chemokine-Induced Angiogenic Activity. J. Immunol. 2000, 165, 5269–5277. [Google Scholar] [CrossRef] [Green Version]
- Khaw, Y.M.; Cunningham, C.; Tierney, A.; Sivaguru, M.; Inoue, M. Neutrophil-selective deletion of Cxcr2 protects against CNS neurodegeneration in a mouse model of multiple sclerosis. J. Neuroinflamm. 2020, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Carlson, T.; Kroenke, M.; Rao, P.; Lane, T.E.; Segal, B. The Th17–ELR + CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J. Exp. Med. 2008, 205, 811–823. [Google Scholar] [CrossRef]
- Steele, C.W.; A Karim, S.; Foth, M.; Rishi, L.; Leach, J.; Porter, R.; Nixon, C.; Evans, T.J.; Carter, C.R.; Nibbs, R.; et al. CXCR2 inhibition suppresses acute and chronic pancreatic inflammation. J. Pathol. 2015, 237, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Farooq, S.M.; Stillie, R.; Svensson, M.; Svanborg, C.; Strieter, R.M.; Stadnyk, A.W. Therapeutic Effect of Blocking CXCR2 on Neutrophil Recruitment and Dextran Sodium Sulfate-Induced Colitis. J. Pharm. Exp. 2009, 329, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.; He, H.; Fan, L.; Ma, C.; Xu, Z.; Xue, Y.; Wang, Y.; Zhang, C.; Zhou, G. Blockade of CXCR2 suppresses proinflammatory activities of neutrophils in ulcerative colitis. Am. J. Transl. Res. 2020, 12, 5237–5251. [Google Scholar] [PubMed]
- Sinclair, A.; Park, L.; Shah, M.; Drotar, M.; Calaminus, S.; Hopcroft, L.E.M.; Kinstrie, R.; Guitart, A.; Dunn, K.; Abraham, S.; et al. CXCR2 and CXCL4 regulate survival and self-renewal of hematopoietic stem/progenitor cells. Blood 2016, 128, 371–383. [Google Scholar] [CrossRef] [Green Version]
- Eash, K.J.; Greenbaum, A.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Investig. 2010, 120, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Shi, H.; Sun, Y.; Shang, C.; Luan, T.; Wang, D.; Ba, X.; Zeng, X. CXCR2 expression on granulocyte and macrophage progenitors under tumor conditions contributes to mo-MDSC generation via SAP18/ERK/STAT3. Cell Death Dis. 2019, 10, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahuja, S.K.; Murphy, P.M. The CXC Chemokines Growth-regulated Oncogene (GRO)α, GROβ, GROγ, Neutrophil-activating Peptide-2, and Epithelial Cell-derived Neutrophil-activating Peptide-78 Are Potent Agonists for the Type B, but Not the Type A, Human Interleukin-8 Receptor. J. Biol. Chem. 1996, 271, 20545–20550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.T.; Rajagopalan, L.; Guerrero-Plata, A.; Sai, J.; Richmond, A.; Garofalo, R.P.; Rajarathnam, K. Monomeric and Dimeric CXCL8 Are Both Essential for In Vivo Neutrophil Recruitment. PLoS ONE 2010, 5, e11754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaik, S.S.; Soltau, T.D.; Chaturvedi, G.; Totapally, B.; Hagood, J.S.; Andrews, W.W.; Athar, M.; Voitenok, N.N.; Killingsworth, C.R.; Patel, R.; et al. Low Intensity Shear Stress Increases Endothelial ELR + CXC Chemokine Production via a Focal Adhesion Kinase-p38β MAPK-NF-κB Pathway. J. Biol. Chem. 2009, 284, 5945–5955. [Google Scholar] [CrossRef] [Green Version]
- Raccosta, L.; Fontana, R.; Traversari, C.; Russo, V. Oxysterols recruit tumor-supporting neutrophils within the tumor microenvironment. OncoImmunology 2013, 2, e26469. [Google Scholar] [CrossRef] [Green Version]
- Raccosta, L.; Fontana, R.; Maggioni, D.; Lanterna, C.; Villablanca, E.; Paniccia, A.; Musumeci, A.; Chiricozzi, E.; Trincavelli, M.L.; Daniele, S.; et al. The oxysterol–CXCR2 axis plays a key role in the recruitment of tumor-promoting neutrophils. J. Exp. Med. 2013, 210, 1711–1728. [Google Scholar] [CrossRef]
- Liu, K.; Wu, L.; Yuan, S.; Wu, M.; Xu, Y.; Sun, Q.; Li, S.; Zhao, S.; Hua, T.; Liu, Z.-J. Structural basis of CXC chemokine receptor 2 activation and signalling. Nat. Cell Biol. 2020, 585, 135–140. [Google Scholar] [CrossRef]
- Catusse, J.; Liotard, A.; Loillier, B.; Pruneau, D.; Paquet, J.-L. Characterization of the molecular interactions of interleukin-8 (CXCL8), growth related oncogen α (CXCL1) and a non-peptide antagonist (SB 225002) with the human CXCR2. Biochem. Pharm. 2003, 65, 813–821. [Google Scholar] [CrossRef]
- Nicholls, D.J.; Tomkinson, N.P.; Wiley, K.E.; Brammall, A.; Bowers, L.; Grahames, C.; Gaw, A.; Meghani, P.; Shelton, P.; Wright, T.J.; et al. Identification of a Putative Intracellular Allosteric Antagonist Binding-Site in the CXC Chemokine Receptors 1 and 2. Mol. Pharm. 2008, 74, 1193–1202. [Google Scholar] [CrossRef] [Green Version]
- De Kruijf, P.; Van Heteren, J.; Lim, H.D.; Conti, P.G.; Van Der Lee, M.M.C.; Bosch, L.; Ho, K.-K.; Auld, U.; Ohlmeyer, M.; Smit, M.J.; et al. Nonpeptidergic Allosteric Antagonists Differentially Bind to the CXCR2 Chemokine Receptor. J. Pharm. Exp. 2009, 329, 783–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salchow, K.; Bond, M.; Evans, S.; Press, N.; Charlton, S.; Hunt, P.; Bradley, M. A common intracellular allosteric binding site for antagonists of the CXCR2 receptor. Br. J. Pharm. 2010, 159, 1429–1439. [Google Scholar] [CrossRef] [Green Version]
- Baugher, P.J.; Richmond, A. The Carboxyl-terminal PDZ Ligand Motif of Chemokine Receptor CXCR2 Modulates Post-endocytic Sorting and Cellular Chemotaxis. J. Biol. Chem. 2008, 283, 30868–30878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Wang, S.; Farooq, S.M.; Castelvetere, M.P.; Hou, Y.; Gao, J.-L.; Navarro, J.; Oupicky, D.; Sun, F.; Li, C. A Chemokine Receptor CXCR2 Macromolecular Complex Regulates Neutrophil Functions in Inflammatory Diseases. J. Biol. Chem. 2012, 287, 5744–5755. [Google Scholar] [CrossRef] [Green Version]
- Legler, D.F.; Matti, C.; Laufer, J.M.; Jakobs, B.D.; Purvanov, V.; Allmen, E.U.-V.; Thelen, M. Modulation of Chemokine Receptor Function by Cholesterol: New Prospects for Pharmacological Intervention. Mol. Pharm. 2017, 91, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Taipale, J.; Young, K.E.; Maiti, T.; Beachy, P.A. Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. USA 2002, 99, 14071–14076. [Google Scholar] [CrossRef] [Green Version]
- Frank-Kamenetsky, M.; Zhang, X.M.; Bottega, S.; Guicherit, O.; Wichterle, H.; Dudek, H.; Bumcrot, D.; Wang, F.Y.; Jones, S.; Shulok, J.; et al. Small-molecule modulators of Hedgehog signaling: Identification and characterization of Smoothened agonists and antagonists. J. Biol. 2002, 1, 10. [Google Scholar] [CrossRef] [Green Version]
- Rohatgi, R.; Milenkovic, L.; Corcoran, R.B.; Scott, M.P. Hedgehog signal transduction by Smoothened: Pharmacologic evidence for a 2-step activation process. Proc. Natl. Acad. Sci. USA 2009, 106, 3196–3201. [Google Scholar] [CrossRef] [Green Version]
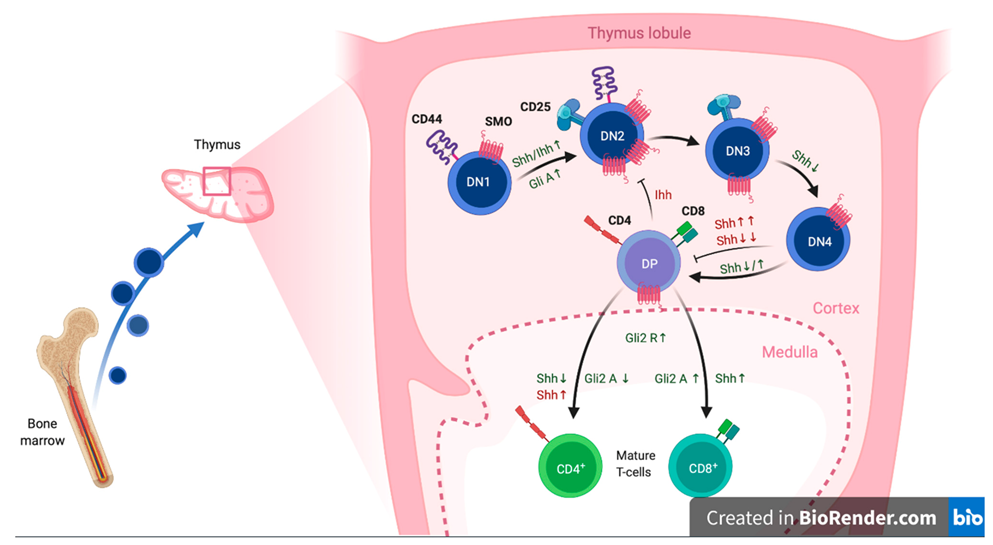
- Outram, S.V.; Varas, A.; Pepicelli, C.V.; Crompton, T. Hedgehog Signaling Regulates Differentiation from Double-Negative to Double-Positive Thymocyte. Immunity 2000, 13, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.K.; Hager-Theodorides, A.; Outram, S.V.; Ross, S.E.; Varas, A.; Crompton, T. Reduced Thymocyte Development in Sonic Hedgehog Knockout Embryos. J. Immunol. 2004, 172, 2296–2306. [Google Scholar] [CrossRef] [Green Version]
- Rowbotham, N.J.; Hager-Theodorides, A.L.; Cebecauer, M.; Shah, D.K.; Drakopoulou, E.; Dyson, J.; Outram, S.V.; Crompton, T. Activation of the Hedgehog signaling pathway in T-lineage cells inhibits TCR repertoire selection in the thymus and peripheral T-cell activation. Blood 2007, 109, 3757–3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowbotham, N.J.; Furmanski, A.L.; Hager-Theodorides, A.L.; Ross, S.E.; Drakopoulou, E.; Koufaris, C.; Outram, S.V.; Crompton, T. Repression of Hedgehog signal transduction in T-lineage cells increases TCR-induced activation and proliferation. Cell Cycle 2008, 7, 904–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mengrelis, K.; Lau, C.-I.; Rowell, J.; Solanki, A.; Norris, S.; Ross, S.; Ono, M.; Outram, S.; Crompton, T. Sonic Hedgehog Is a Determinant of γδ T-Cell Differentiation in the Thymus. Front. Immunol. 2019, 10, 1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldana, J.I.; Solanki, A.; Lau, C.-I.; Sahni, H.; Ross, S.; Furmanski, A.L.; Ono, M.; Holländer, G.; Crompton, T. Sonic Hedgehog regulates thymic epithelial cell differentiation. J. Autoimmun. 2016, 68, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Furmanski, A.L.; Saldana, J.I.; Ono, M.; Sahni, H.; Paschalidis, N.; D’Acquisto, F.; Crompton, T. Tissue-Derived Hedgehog Proteins Modulate Th Differentiation and Disease. J. Immunol. 2013, 190, 2641–2649. [Google Scholar] [CrossRef] [Green Version]
- de la Roche, M.; Ritter, A.T.; Angus, K.L.; Dinsmore, C.; Earnshaw, C.H.; Reiter, J.F.; Griffiths, G.M. Hedgehog Signaling Controls T Cell Killing at the Immunological Synapse. Science 2013, 342, 1247–1250. [Google Scholar] [CrossRef] [Green Version]
- Syn, W.-K.; Witek, R.P.; Curbishley, S.M.; Jung, Y.; Choi, S.S.; Enrich, B.; Omenetti, A.; Agboola, K.M.; Fearing, C.M.; Tilg, H.; et al. Role for hedgehog pathway in regulating growth and function of invariant NKT cells. Eur. J. Immunol. 2009, 39, 1879–1892. [Google Scholar] [CrossRef] [Green Version]
- Solanki, A.; Lau, C.I.; Saldaña, J.I.; Ross, S.; Crompton, T. The transcription factor Gli3 promotes B cell development in fetal liver through repression of Shh. J. Exp. Med. 2017, 214, 2041–2058. [Google Scholar] [CrossRef]
- Solanki, A.; Yanez, D.C.; Ross, S.; Lau, C.I.; Papaioannou, E.; Li, J.; Saldaña, J.I.; Crompton, T. In the fetal thymus, Gli3 in thymic epithelial cells promotes thymocyte positive selection and differentiation by repression of Shh. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Papaioannou, E.; Yánez, D.C.; Ross, S.; Lau, C.-I.; Solanki, A.; Chawda, M.M.; Virasami, A.; Ranz, I.; Ono, M.; O’Shaughnessy, R.F.L.; et al. Sonic Hedgehog signaling limits atopic dermatitis via Gli2-driven immune regulation. J. Clin. Investig. 2019, 129, 3153–3170. [Google Scholar] [CrossRef] [PubMed]
- Myers, B.R.; Sever, N.; Chong, Y.C.; Kim, J.; Belani, J.D.; Rychnovsky, S.; Bazan, J.F.; Beachy, P.A. Hedgehog Pathway Modulation by Multiple Lipid Binding Sites on the Smoothened Effector of Signal Response. Dev. Cell 2013, 26, 346–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, M.K.; Wassif, C.; Krakowiak, P.A.; Taipale, J.; Gong, R.; Kelley, R.I.; Porter, F.D.; Beachy, P.A. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat. Genet. 2003, 33, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Tang, J.-J.; Peng, C.; Wang, Y.; Fu, L.; Qiu, Z.-P.; Xiong, Y.; Yang, L.-F.; Cui, H.-W.; He, X.-L.; et al. Cholesterol Modification of Smoothened Is Required for Hedgehog Signaling. Mol. Cell 2017, 66, 154–162.e10. [Google Scholar] [CrossRef] [Green Version]
- Raleigh, D.R.; Sever, N.; Choksi, P.K.; Sigg, M.; Hines, K.; Thompson, B.; Elnatan, D.; Jaishankar, P.; Bisignano, P.; Garcia-Gonzalo, F.; et al. Cilia-Associated Oxysterols Activate Smoothened. Mol. Cell 2018, 72, 316–327.e5. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176–1187. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhao, F.; Wu, Y.; Yang, J.; Han, G.W.; Zhao, S.; Ishchenko, A.; Ye, L.; Lin, X.; Ding, K.; et al. Crystal structure of a multi-domain human smoothened receptor in complex with a super stabilizing ligand. Nat. Commun. 2017, 8, 15383. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Zheng, S.; Wierbowski, B.; Kim, Y.; Nedelcu, D.; Aravena, L.; Liu, J.; Kruse, A.C.; Salic, A. Structural Basis of Smoothened Activation in Hedgehog Signaling. Cell 2018, 174, 312–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, I.; Liang, J.; Hedeen, D.; Roberts, K.J.; Zhang, Y.; Ha, B.; Latorraca, N.R.; Faust, B.; Dror, R.O.; Beachy, P.A.; et al. Smoothened stimulation by membrane sterols drives Hedgehog pathway activity. Nat. Cell Biol. 2019, 571, 284–288. [Google Scholar] [CrossRef]
- Qi, X.; Friedberg, L.; De Bose-Boyd, R.; Long, T.; Li, X. Sterols in an intramolecular channel of Smoothened mediate Hedgehog signaling. Nat. Chem. Biol. 2020, 16, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Phelan, K.; Mahler, H. Acute Exposure to 25-Hydroxy-cholesterol Selectively Reduces GABAb and Not GABAa Receptor-Mediated Synaptic Inhibition. Biochem. Biophys. Res. Commun. 1997, 237, 68–73. [Google Scholar] [CrossRef]
- A Kuhn, S.; van Landeghem, F.; Zacharias, R.; Färber, K.; Rappert, A.; Pavlovic, S.; Hoffmann, A.; Nolte, C.; Kettenmann, H. Microglia express GABAB receptors to modulate interleukin release. Mol. Cell. Neurosci. 2004, 25, 312–322. [Google Scholar] [CrossRef]
- Huang, S.; Mao, J.; Wei, B.; Pei, G. The anti-spasticity drug baclofen alleviates collagen-induced arthritis and regulates dendritic cells. J. Cell. Physiol. 2015, 230, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Hammoud, Y.; Rice, T.; Mackrill, J.J. Oxysterols modulate calcium signalling in the A7r5 aortic smooth muscle cell-line. Biochimie 2013, 95, 568–577. [Google Scholar] [CrossRef]
- Yan, Z.; Li, S.; Liang, Z.; Tomić, M.; Stojilkovic, S.S. The P2X7 Receptor Channel Pore Dilates under Physiological Ion Conditions. J. Gen. Physiol. 2008, 132, 563–573. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, A.E.; Yoshioka, C.; Mansoor, S.E. Full-Length P2X7 Structures Reveal How Palmitoylation Prevents Channel Desensitization. Cell 2019, 179, 659–670. [Google Scholar] [CrossRef]
- Robinson, L.E.; Shridar, M.; Smith, P.; Murrell-Lagnado, R.D. Plasma Membrane Cholesterol as a Regulator of Human and Rodent P2X7 Receptor Activation and Sensitization. J. Biol. Chem. 2014, 289, 31983–31994. [Google Scholar] [CrossRef] [Green Version]
- Lajdova, I.; Chorvát, D.; Chorvatova, A. Rapid effects of 1α,25(OH)2D3 in resting human peripheral blood mononuclear cells. Eur. J. Pharm. 2008, 586, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Olivier, E.; Dutot, M.; Regazzetti, A.; Laprévote, O.; Rat, P. 25-Hydroxycholesterol induces both P2X7-dependent pyroptosis and caspase-dependent apoptosis in human skin model: New insights into degenerative pathways. Chem. Phys. Lipids 2017, 207, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, E.; Giuliani, A.L.; De Marchi, E.; Pegoraro, A.; Orioli, E.; Di Virgilio, F. The P2X7 receptor: A main player in inflammation. Biochem. Pharm. 2018, 151, 234–244. [Google Scholar] [CrossRef]
- Olivier, E.; Dutot, M.; Regazzetti, A.; Leguillier, T.; Dargère, D.; Auzeil, N.; Laprévote, O.; Rat, P. P2X7-pannexin-1 and amyloid β-induced oxysterol input in human retinal cell: Role in age-related macular degeneration? Biochimie 2016, 127, 70–78. [Google Scholar] [CrossRef] [PubMed]
- A Grahames, C.B.; Michel, A.D.; Chessell, I.P.; A Humphrey, P.P. Pharmacological characterization of ATP- and LPS-induced IL-1β release in human monocytes. Br. J. Pharm. 1999, 127, 1915–1921. [Google Scholar] [CrossRef] [Green Version]
- Martinez-García, J.J.; Banaclocha, H.M.; Angosto, D.; De Torre-Minguela, C.; Baroja-Mazo, A.; Alarcón-Vila, C.; Martinez-Alarcon, L.; Amores-Iniesta, J.; Martín-Sánchez, F.; Ercole, G.A.; et al. P2X7 receptor induces mitochondrial failure in monocytes and compromises NLRP3 inflammasome activation during sepsis. Nat. Commun. 2019, 10, 2711. [Google Scholar] [CrossRef] [Green Version]
- Grassi, F. The P2X7 Receptor as Regulator of T Cell Development and Function. Front. Immunol. 2020, 11, 1179. [Google Scholar] [CrossRef] [PubMed]
- Tsukimoto, M.; Tokunaga, A.; Harada, H.; Kojima, S. Blockade of murine T cell activation by antagonists of P2Y6 and P2X7 receptors. Biochem. Biophys. Res. Commun. 2009, 384, 512–518. [Google Scholar] [CrossRef]
- Schenk, U.; Frascoli, M.; Proietti, M.; Geffers, R.; Traggiai, E.; Buer, J.; Ricordi, C.; Westendorf, A.M.; Grassi, F. ATP Inhibits the Generation and Function of Regulatory T Cells Through the Activation of Purinergic P2X Receptors. Sci. Signal. 2011, 4, ra12. [Google Scholar] [CrossRef]
- Frascoli, M.; Marcandalli, J.; Schenk, U.; Grassi, F. Purinergic P2X7 Receptor Drives T Cell Lineage Choice and Shapes Peripheral γδ Cells. J. Immunol. 2012, 189, 174–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beynon, V.; Quintana, F.J.; Weiner, H.L. Activated Human CD4+CD45RO+ Memory T-Cells Indirectly Inhibit NLRP3 Inflammasome Activation through Downregulation of P2X7R Signalling. PLoS ONE 2012, 7, e39576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Salles, M.; Menezes, M.; Siqueira, R.; Da Silva, H.B.; Amaral, E.P.; Castillo-Méndez, S.I.; Cunha, I.; Cassado, A.D.A.; Vieira, F.S.; Olivieri, D.N.; et al. P2X7 receptor drives Th1 cell differentiation and controls the follicular helper T cell population to protect against Plasmodium chabaudi malaria. Plos Pathog. 2017, 13, e1006595. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, H.B.; Beura, L.K.; Wang, H.; Hanse, E.A.; Gore, R.; Scott, M.C.; Walsh, D.A.; Block, K.E.; Fonseca, R.; Yan, Y.; et al. The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8+ T cells. Nat. Cell Biol. 2018, 559, 264–268. [Google Scholar] [CrossRef]
- Dubyak, G.R. P2X7 receptor regulation of non-classical secretion from immune effector cells. Cell. Microbiol. 2012, 14, 1697–1706. [Google Scholar] [CrossRef] [Green Version]
- Baroja-Mazo, A.; Barberà-Cremades, M.; Pelegrín, P. P2X7 Receptor Activation Impairs Exogenous MHC Class I Oligopeptides Presentation in Antigen Presenting Cells. PLoS ONE 2013, 8, e70577. [Google Scholar] [CrossRef] [Green Version]
- Figliuolo, V.R.; Savio, L.E.; Safya, H.; Nanini, H.F.; Bernardazzi, C.; Abalo, A.; de Souza, H.S.; Kanellopoulos, J.; Bobé, P.; Coutinho, C.M.; et al. P2X7 receptor promotes intestinal inflammation in chemically induced colitis and triggers death of mucosal regulatory T cells. Mol. Basis Dis. 2017, 1863, 1183–1194. [Google Scholar] [CrossRef]
- Salussolia, C.L.; Prodromou, M.L.; Borker, P.; Wollmuth, L.P. Arrangement of Subunits in Functional NMDA Receptors. J. Neurosci. 2011, 31, 11295–11304. [Google Scholar] [CrossRef]
- Zhu, S.; Gouaux, E. Structure and symmetry inform gating principles of ionotropic glutamate receptors. Neuropharmacology 2016, 112, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Otaño, I.; Larsen, R.; Wesseling, I.P.-O.J.F. Emerging roles of GluN3-containing NMDA receptors in the CNS. Nat. Rev. Neurosci. 2016, 17, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Hogan-Cann, A.D.; Anderson, C.M. Physiological Roles of Non-Neuronal NMDA Receptors. Trends Pharm. Sci. 2016, 37, 750–767. [Google Scholar] [CrossRef]
- Ganor, Y.; Levite, M. The neurotransmitter glutamate and human T cells: Glutamate receptors and glutamate-induced direct and potent effects on normal human T cells, cancerous human leukemia and lymphoma T cells, and autoimmune human T cells. J. Neural Transm. 2014, 121, 983–1006. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Doherty, J.J.; Robichaud, A.J.; Belfort, G.M.; Chow, B.Y.; Hammond, R.S.; Crawford, D.; Linsenbardt, A.J.; Shu, H.-J.; Izumi, Y.; et al. The Major Brain Cholesterol Metabolite 24(S)-Hydroxycholesterol Is a Potent Allosteric Modulator of N-Methyl-d-Aspartate Receptors. J. Neurosci. 2013, 33, 17290–17300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emnett, C.M.; Eisenman, L.N.; Mohan, J.; A Taylor, A.; Doherty, J.J.; Paul, S.M.; Zorumski, C.F.; Mennerick, S. Interaction between positive allosteric modulators and trapping blockers of the NMDA receptor channel. Br. J. Pharm. 2015, 172, 1333–1347. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.-Y.; Izumi, Y.; Benz, A.M.; Zorumski, C.F.; Mennerick, S. Endogenous 24S-hydroxycholesterol modulates NMDAR-mediated function in hippocampal slices. J. Neurophysiol. 2016, 115, 1263–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilding, T.J.; Lopez, M.N.; Huettner, J.E. Chimeric Glutamate Receptor Subunits Reveal the Transmembrane Domain Is Sufficient for NMDA Receptor Pore Properties but Some Positive Allosteric Modulators Require Additional Domains. J. Neurosci. 2016, 36, 8815–8825. [Google Scholar] [CrossRef]
- Wei, X.; Nishi, T.; Kondou, S.; Kimura, H.; Mody, I. Preferential enhancement of GluN2B-containing native NMDA receptors by the endogenous modulator 24S-hydroxycholesterol in hippocampal neurons. Neuropharmacology 2018, 148, 11–20. [Google Scholar] [CrossRef]
- Sun, M.-Y.; Taylor, A.; Zorumski, C.F.; Mennerick, S. 24S-hydroxycholesterol and 25-hydroxycholesterol differentially impact hippocampal neuronal survival following oxygen-glucose deprivation. PLoS ONE 2017, 12, e0174416. [Google Scholar] [CrossRef]
- Mateos, L.; Akterin, S.; Gil-Bea, F.; Spulber, S.; Rahman, A.; Björkhem, I.; Schultzberg, M.; Flores-Morales, A.; Cedazo-Mínguez, A. Activity-Regulated Cytoskeleton-Associated Protein in Rodent Brain is Down-Regulated by High Fat Dietin vivoand by 27-Hydroxycholesterolin vitro. Brain Pathol. 2008, 19, 69–80. [Google Scholar] [CrossRef]
- Boldyrev, A.A.; Carpenter, D.O.; Johnson, P. Emerging evidence for a similar role of glutamate receptors in the nervous and immune systems. J. Neurochem. 2005, 95, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Mashkina, A.P.; Tyulina, O.V.; Solovyova, T.I.; Kovalenko, E.; Kanevski, L.M.; Johnson, P.; Boldyrev, A.A. The excitotoxic effect of NMDA on human lymphocyte immune function. Neurochem. Int. 2007, 51, 356–360. [Google Scholar] [CrossRef]
- Kahlfuß, S.; Simma, N.; Mankiewicz, J.; Bose, T.; Lowinus, T.; Klein-Hessling, S.; Sprengel, R.; Schraven, B.; Heine, M.; Bommhardt, U. Immunosuppression by N-Methyl-d-Aspartate Receptor Antagonists Is Mediated through Inhibition of Kv1.3 and KCa3.1 Channels in T Cells. Mol. Cell. Biol. 2013, 34, 820–831. [Google Scholar] [CrossRef] [Green Version]
- Simma, N.; Bose, T.; Kahlfuß, S.; Mankiewicz, J.; Lowinus, T.; Lühder, F.; Schüler, T.; Schraven, B.; Heine, M.; Bommhardt, U. NMDA-receptor antagonists block B-cell function but foster IL-10 production in BCR/CD40-activated B cells. Cell Commun. Signal. 2014, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.S.; Aricescu, A.R. Crystal structure of a human GABAA receptor. Nat. Cell Biol. 2014, 512, 270–275. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, L.D.; Pecci, A.; Estrin, D.A. In Search of GABAA Receptor’s Neurosteroid Binding Sites. J. Med. Chem. 2018, 62, 5250–5260. [Google Scholar] [CrossRef] [PubMed]
- Barragan, A.; Weidner, J.M.; Jin, Z.; Korpi, E.R.; Birnir, B. GABAergic signalling in the immune system. Acta Physiol. 2015, 213, 819–827. [Google Scholar] [CrossRef]
- Rudy, B.; McBain, C.J. Kv3 channels: Voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 2001, 24, 517–526. [Google Scholar] [CrossRef]
- Bezine, M.; Maatoug, S.; Ben Khalifa, R.; Debbabi, M.; Zarrouk, A.; Wang, Y.; Griffiths, W.J.; Nury, T.; Samadi, M.; Vejux, A.; et al. Modulation of Kv3.1b potassium channel level and intracellular potassium concentration in 158N murine oligodendrocytes and BV-2 murine microglial cells treated with 7-ketocholesterol, 24S-hydroxycholesterol or tetracosanoic acid (C24:0). Biochimie 2018, 153, 56–69. [Google Scholar] [CrossRef] [Green Version]
- Grissmer, S.; Ghanshani, S.; Dethlefs, B.; McPherson, J.D.; Wasmuth, J.J.; Gutman, G.A.; Cahalan, M.D.; Chandy, K.G. The Shaw-related potassium channel gene, Kv3.1, on human chromosome 11, encodes the type l K+ channel in T cells. J. Biol. Chem. 1992, 267, 20971–20979. [Google Scholar] [CrossRef]
- Black, J.A.; Waxman, S.G. Noncanonical Roles of Voltage-Gated Sodium Channels. Neuron 2013, 80, 280–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roselli, F.; Livrea, P.; Jirillo, E. Voltage-Gated Sodium Channel Blockers as Immunomodulators. Recent Pat. CNS Drug Discov. 2006, 1, 83–91. [Google Scholar] [CrossRef]
- Bezine, M.; Namsi, A.; Sghaier, R.; Ben Khalifa, R.; Hamdouni, H.; Brahmi, F.; Badreddine, I.; Mihoubi, W.; Nury, T.; Vejux, A.; et al. The effect of oxysterols on nerve impulses. Biochimie 2018, 153, 46–51. [Google Scholar] [CrossRef]
- Pristerà, A.; Baker, M.D.; Okuse, K. Association between Tetrodotoxin Resistant Channels and Lipid Rafts Regulates Sensory Neuron Excitability. PLoS ONE 2012, 7, e40079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.; Wang, Y.; Leng, T.; Sun, H.; Zhou, Y.; Zhu, W.; Qiu, P.; Zhang, J.; Lu, B.; Yan, M.; et al. Cholesterol metabolite cholestane-3β,5α,6β-triol suppresses epileptic seizures by negative modulation of voltage-gated sodium channels. Steroids 2015, 98, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Yan, M.; Leng, T.; Yin, W.; Cai, S.; Duan, S.; Zhu, W.; Lin, S.; Huang, J.; Yan, G.; et al. Cholestane-3β, 5α, 6β-triol suppresses neuronal hyperexcitability via binding to voltage-gated sodium channels. Biochem. Biophys. Res. Commun. 2018, 496, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Mandler, R.N.; Seamer, L.C.; Whitlinger, D.; Lennon, M.; Rosenberg, E.; Bankhurst, A.D. Human natural killer cells express Na+ channels. A pharmacologic flow cytometric study. J. Immunol. 1990, 144, 2365–2370. [Google Scholar]
- Lai, Z.-F.; Chen, Y.-Z.; Nishimura, Y.; Nishi, K. An amiloride-sensitive and voltage-dependent Na+ channel in an HLA-DR-restricted human T cell clone. J. Immunol. 2000, 165, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Lu, C.; Wu, Y.; Ouyang, S.; Chen, Y. Identification and functional characterization of voltage-gated sodium channels in lymphocytes. Biochem. Biophys. Res. Commun. 2015, 458, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Milam, A.A.V.; Bartleson, J.M.; Donermeyer, D.L.; Horvath, S.; Durai, V.; Raju, S.; Yu, H.; Redmann, V.; Zinselmeyer, B.; White, J.M.; et al. Tuning T Cell Signaling Sensitivity Alters the Behavior of CD4+ T Cells during an Immune Response. J. Immunol. 2018, 200, 3429–3437. [Google Scholar] [CrossRef] [Green Version]
- Zsiros, E.; Kis-Toth, K.; Hajdu, P.; Gaspar, R.; Bielanska, J.; Felipe, A.; Rajnavolgyi, E.; Panyi, G. Developmental Switch of the Expression of Ion Channels in Human Dendritic Cells. J. Immunol. 2009, 183, 4483–4492. [Google Scholar] [CrossRef] [Green Version]
- Black, J.A.; Waxman, S.G. Sodium channels and microglial function. Exp. Neurol. 2011, 234, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Carrithers, M.D.; Dib-Hajj, S.; Carrithers, L.M.; Tokmoulina, G.; Pypaert, M.; Jonas, E.A.; Waxman, S.G. Expression of the Voltage-Gated Sodium Channel NaV1.5 in the Macrophage Late Endosome Regulates Endosomal Acidification. J. Immunol. 2007, 178, 7822–7832. [Google Scholar] [CrossRef] [Green Version]
- Carrithers, L.M.; Hulseberg, P.; Sandor, M.; Carrithers, M.D. The human macrophage sodium channel NaV1.5 regulates mycobacteria processing through organelle polarization and localized calcium oscillations. FEMS Immunol. Med. Microbiol. 2011, 63, 319–327. [Google Scholar] [CrossRef]
- Jones, A.; Kainz, D.; Khan, F.; Lee, C.; Carrithers, M.D. Human Macrophage SCN5A Activates an Innate Immune Signaling Pathway for Antiviral Host Defense. J. Biol. Chem. 2014, 289, 35326–35340. [Google Scholar] [CrossRef] [Green Version]
- White, C.R.; Dungan, M.; Carrithers, M.D. Activation of human macrophage sodium channels regulates RNA processing to increase expression of the DNA repair protein PPP1R10. Immunobiology 2018, 224, 80–93. [Google Scholar] [CrossRef]
- Sun, H.; Jiang, J.; Gong, L.; Li, X.; Yang, Y.; Luo, Y.; Guo, Z.; Lu, R.; Li, H.; Li, J.; et al. Voltage-gated sodium channel inhibitor reduces atherosclerosis by modulating monocyte/macrophage subsets and suppressing macrophage proliferation. Biomed. Pharm. 2019, 120, 109352. [Google Scholar] [CrossRef]
- Gonzalez-Perez, V.; Lingle, C.J. Regulation of BK Channels by Beta and Gamma Subunits. Annu. Rev. Physiol. 2019, 81, 113–137. [Google Scholar] [CrossRef]
- Tian, Y.; Heinemann, S.H.; Hoshi, T. Large-conductance Ca2+- and voltage-gated K+channels form and break interactions with membrane lipids during each gating cycle. Proc. Natl. Acad. Sci. USA 2019, 116, 8591–8596. [Google Scholar] [CrossRef] [Green Version]
- Dopico, A.M.; Bukiya, A.N. Regulation of Ca2+-Sensitive K+ Channels by Cholesterol and Bile Acids via Distinct Channel Subunits and Sites. Curr. Top. Membr. 2017, 80, 53–93. [Google Scholar] [CrossRef] [PubMed]
- Tajima, N.; Xiaoyan, L.; Taniguchi, M.; Kato, N. 24S-hydroxycholesterol alters activity of large-conductance Ca2+-dependent K+ (slo1 BK) channel through intercalation into plasma membrane. Mol. Cell Biol. Lipids 2019, 1864, 1525–1535. [Google Scholar] [CrossRef]
- Son, Y.; Chun, W.; Ahn, Y.-T.; Kim, K.; Lee, C.-W.; Kim, J.-M.; Lee, C.; An, W.G. 7-Ketocholesterol induces the reduction of KCNMB1 in atherosclerotic blood vessels. Biochem. Biophys. Res. Commun. 2015, 457, 324–327. [Google Scholar] [CrossRef]
- Erdogan, A.; Schaefer, M.B.; Kuhlmann, C.R.W.; Most, A.; Hartmann, M.; Mayer, K.; Renner, F.C.; Schaefer, C.; Abdallah, Y.; Hoelschermann, H.; et al. Activation of Ca2+-activated potassium channels is involved in lysophosphatidylcholine-induced monocyte adhesion to endothelial cells. Atherosclerosis 2007, 190, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Papavlassopoulos, M.; Stamme, C.; Thon, L.; Adam, D.; Hillemann, D.; Seydel, U.; Schromm, A.B. MaxiK Blockade Selectively Inhibits the Lipopolysaccharide-Induced IκB-α/NF-κB Signaling Pathway in Macrophages. J. Immunol. 2006, 177, 4086–4093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Xu, M.; Cao, Q.; Huang, P.; Zhu, X.; Dong, X.-P. A lysosomal K+ channel regulates large particle phagocytosis by facilitating lysosome Ca2+ release. Sci. Rep. 2020, 10, 1038. [Google Scholar] [CrossRef] [PubMed]
- Selezneva, A.; Yoshida, M.; Gibb, A.; Willis, D. Nuclear BK channels regulate CREB phosphorylation in RAW264.7 macrophages. Pharm. Rep. 2021, 73, 881–890. [Google Scholar] [CrossRef]
- Chen, X.; Sooch, G.; Demaree, I.; White, F.; Obukhov, A. Transient Receptor Potential Canonical (TRPC) Channels: Then and Now. Cells 2020, 9, 1983. [Google Scholar] [CrossRef]
- Berthier, A.; Lemaire-Ewing, S.; Prunet, C.; Monier, S.; Athias, A.; Bessède, G.; De Barros, J.-P.P.; Laubriet, A.; Gambert, P.; Lizard, G.; et al. Involvement of a calcium-dependent dephosphorylation of BAD associated with the localization of Trpc-1 within lipid rafts in 7-ketocholesterol-induced THP-1 cell apoptosis. Cell Death Differ. 2004, 11, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Ingueneau, C.; Huynh-Do, U.; Marcheix, B.; Athias, A.; Gambert, P.; Nègre-Salvayre, A.; Salvayre, R.; Vindis, C. TRPC1 is regulated by caveolin-1 and is involved in oxidized LDL-induced apoptosis of vascular smooth muscle cells. J. Cell. Mol. Med. 2008, 13, 1620–1631. [Google Scholar] [CrossRef]
- Wenning, A.S.; Neblung, K.; Strauß, B.; Wolfs, M.-J.; Sappok, A.; Hoth, M.; Schwarz, E.C. TRP expression pattern and the functional importance of TRPC3 in primary human T-cells. Mol. Cell Res. 2011, 1813, 412–423. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, O.; Strodthoff, C.; Horstmann, M.; Nielsen, N.; Jung, F.; Schimmelpfennig, S.; Heitzmann, M.; Schwab, A. TRPC1 regulates fMLP-stimulated migration and chemotaxis of neutrophil granulocytes. Mol. Cell Res. 2015, 1853, 2122–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, A.; Sun, Y.; Sukumaran, P.; Zangbede, F.O.Q.; Jondle, C.N.; Sharma, A.; Evans, D.L.; Chauhan, P.; Szlabick, R.E.; Aaland, M.O.; et al. M1 Macrophage Polarization Is Dependent on TRPC1-Mediated Calcium Entry. iScience 2018, 8, 85–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Conceicao, V.N.; Sun, Y.; Zboril, E.K.; De la Chapa, J.J.; Singh, B.B. Loss of Ca2+ entry via Orai-TRPC1 induces ER stress that initiates immune activation in macrophage cells. J. Cell Sci. 2019, 133. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chauhan, A.; Sukumaran, P.; Sharma, J.; Singh, B.B.; Mishra, B.B. Inhibition of store-operated calcium entry in microglia by helminth factors: Implications for immune suppression in neurocysticercosis. J. Neuroinflamm. 2014, 11, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reinmuth, L.; Hsiao, C.-C.; Hamann, J.; Rosenkilde, M.; Mackrill, J. Multiple Targets for Oxysterols in Their Regulation of the Immune System. Cells 2021, 10, 2078. https://doi.org/10.3390/cells10082078
Reinmuth L, Hsiao C-C, Hamann J, Rosenkilde M, Mackrill J. Multiple Targets for Oxysterols in Their Regulation of the Immune System. Cells. 2021; 10(8):2078. https://doi.org/10.3390/cells10082078
Chicago/Turabian StyleReinmuth, Lisa, Cheng-Chih Hsiao, Jörg Hamann, Mette Rosenkilde, and John Mackrill. 2021. "Multiple Targets for Oxysterols in Their Regulation of the Immune System" Cells 10, no. 8: 2078. https://doi.org/10.3390/cells10082078
APA StyleReinmuth, L., Hsiao, C.-C., Hamann, J., Rosenkilde, M., & Mackrill, J. (2021). Multiple Targets for Oxysterols in Their Regulation of the Immune System. Cells, 10(8), 2078. https://doi.org/10.3390/cells10082078