
Immune Responses in the Glaucomatous Retina: Regulation and Dynamics


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
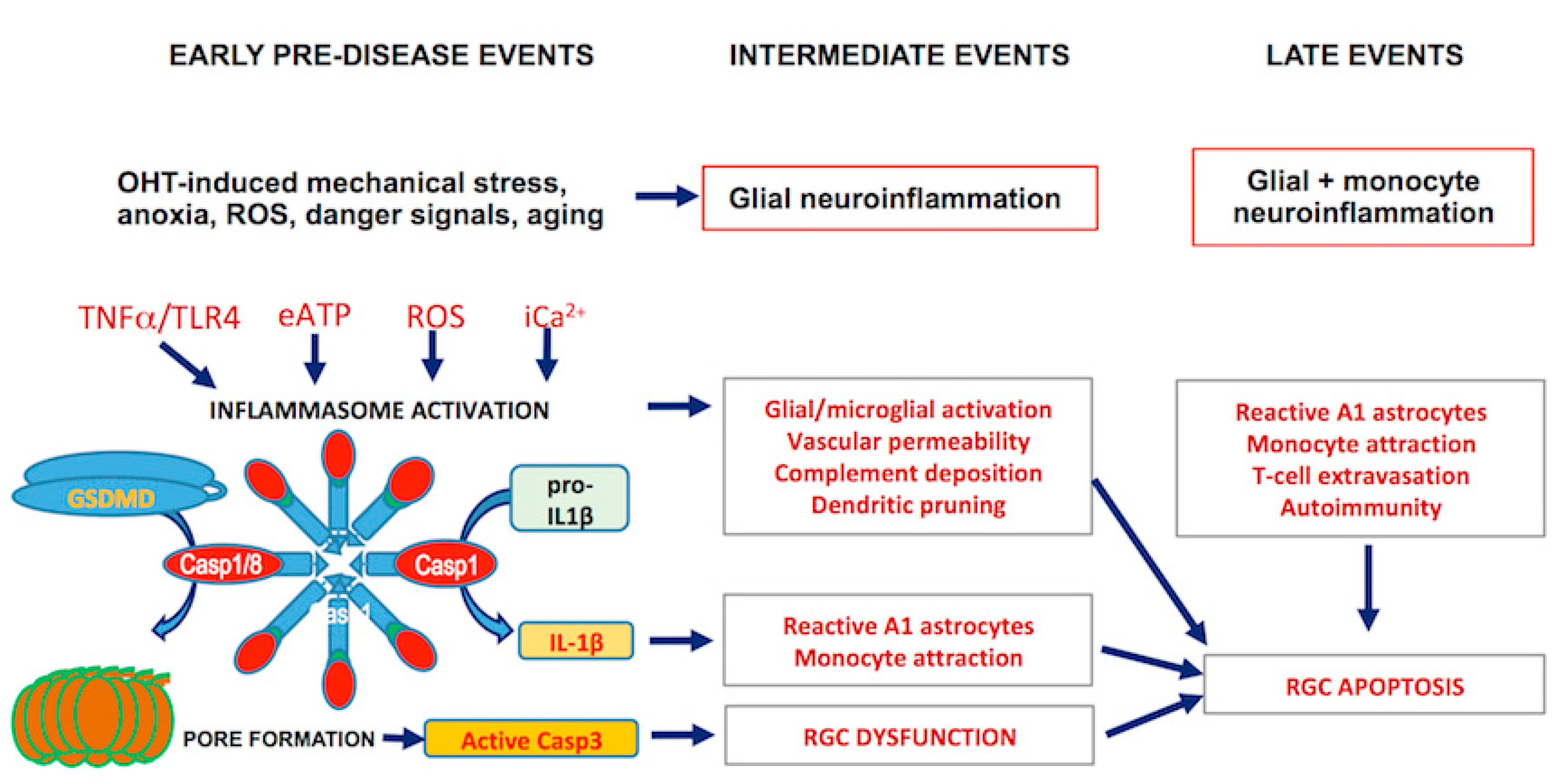
2. Early Events
2.1. Metabolic and Mitochondrial RGC Stress
2.2. Release of Extracellular ATP
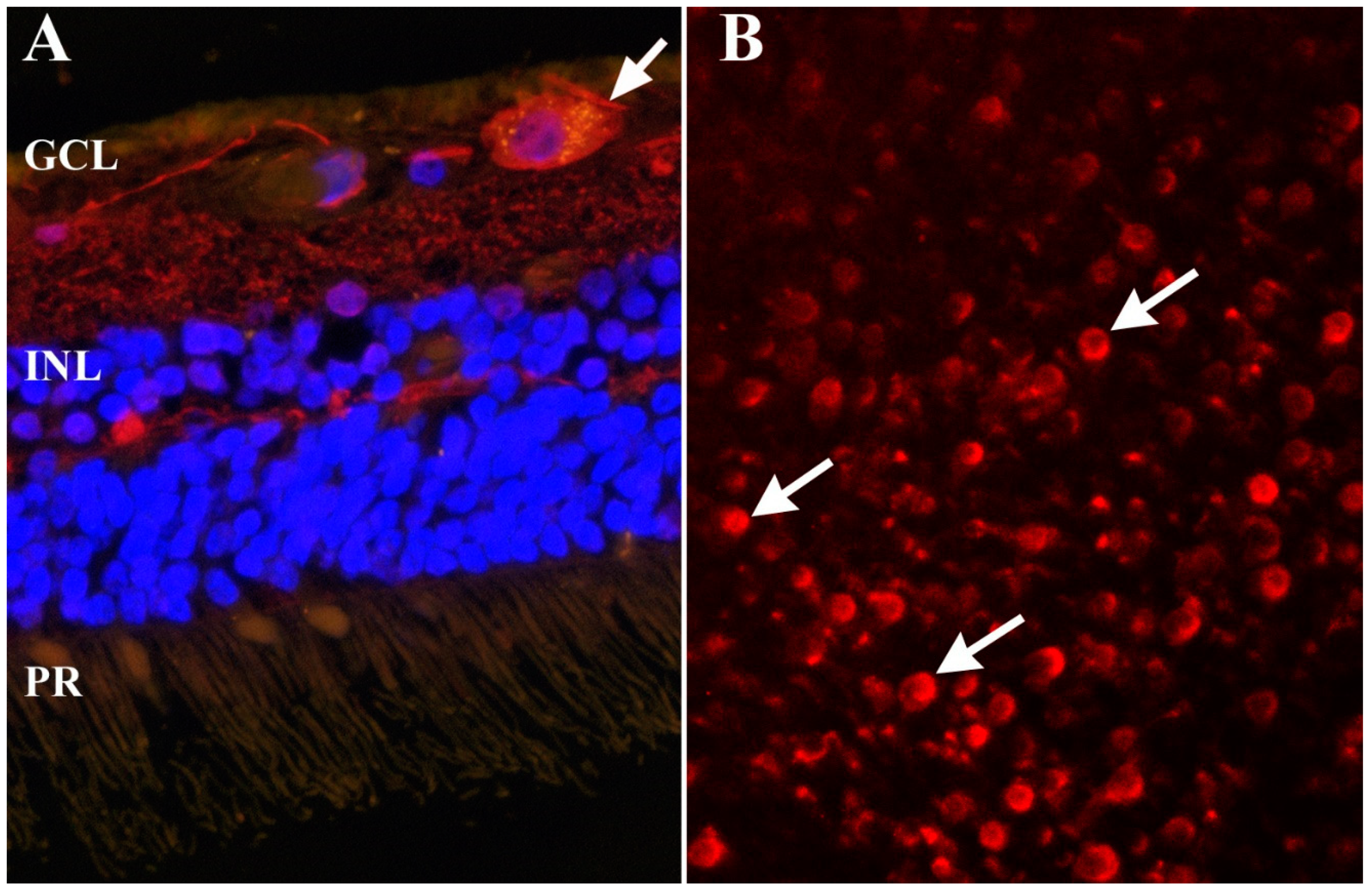
2.3. Inflammasome Formation and Signaling
3. Intermediate Events
3.1. Neuroinflammation
3.2. Complement Cascade Activation
3.3. Inflammasomes Mediate Crosstalk between the Innate and Adaptive Immune Systems
4. Late Events
4.1. Inflammasome Mediated RGC Death
4.2. Autoimmune Responses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Osborne, N.N.; Ugarte, M.; Chao, M.; Chidlow, G.; Bae, J.H.; Wood, J.P.; Nash, M.S. Neuroprotection in relation to retinal ischemia and relevance to glaucoma. Surv. Ophthalmol. 1999, 43, S102–S128. [Google Scholar] [CrossRef]
- Quigley, H.A.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Esen, F.; Eraslan, M.; Cerman, E.; Celiker, H.; Kazokoglu, H. Diurnal Spikes of Intraocular Pressure in Uveitic Glaucoma: A 24-hour Intraocular Pressure Monitoring Study. Semin. Ophthalmol. 2020, 35, 246–251. [Google Scholar] [CrossRef]
- Wilensky, J.T. The role of diurnal pressure measurements in the management of open angle glaucoma. Curr. Opin. Ophthalmol. 2004, 15, 90–92. [Google Scholar] [CrossRef]
- Resta, V.; Novelli, E.; Vozzi, G.; Scarpa, C.; Caleo, M.; Ahluwalia, A.; Solini, A.; Santini, E.; Parisi, V.; Di Virgilio, F.; et al. Acute retinal ganglion cell injury caused by intraocular pressure spikes is mediated by endogenous extracellular ATP. Eur. J. Neurosci. 2007, 25, 2741–2754. [Google Scholar] [CrossRef]
- Gramlich, O.W.; Teister, J.; Neumann, M.; Tao, X.; Beck, S.; Von Pein, H.D.; Pfeiffer, N.; Grus, F.H. Immune response after intermittent minimally invasive intraocular pressure elevations in an experimental animal model of glaucoma. J. Neuroinflamm. 2016, 13, 1–16. [Google Scholar] [CrossRef]
- McMonnies, C.W. Intraocular Pressure Spikes in Keratectasia, Axial Myopia, and Glaucoma. Optom. Vis. Sci. 2008, 85, 1018–1026. [Google Scholar] [CrossRef]
- Ren, R.; Zhang, X.; Wang, N.; Li, B.; Tian, G.; Jonas, J.B. Cerebrospinal fluid pressure in ocular hypertension. Acta Ophthalmol. 2011, 89, e142–e148. [Google Scholar] [CrossRef] [PubMed]
- Nusbaum, D.M.; Wu, S.M.; Frankfort, B.J. Elevated intracranial pressure causes optic nerve and retinal ganglion cell degeneration in mice. Exp. Eye Res. 2015, 136, 38–44. [Google Scholar] [CrossRef]
- Heijl, A.; Leske, M.C.; Bengtsson, B.; Hyman, L.; Bengtsson, B.; Hussein, M.; Early Manifest Glaucoma Trial, G. Reduction of intraocular pressure and glaucoma progression: Results from the Early Manifest Glaucoma Trial. Arch. Ophthalmol. 2002, 120, 1268–1279. [Google Scholar] [CrossRef]
- Caprioli, J. Glaucoma: A Disease of Early Cellular Senescence. Investig. Ophthalmol. Vis. Sci. 2013, 54, ORSF60–ORSF67. [Google Scholar] [CrossRef] [PubMed]
- Ernest, P.J.; Schouten, J.S.; Beckers, H.J.; Hendrikse, F.; Prins, M.H.; Webers, C.A. An Evidence-Based Review of Prognostic Factors for Glaucomatous Visual Field Progression. Ophthalmology 2013, 120, 512–519. [Google Scholar] [CrossRef]
- Kass, M.A.; Gordon, M.O.; Gao, F.; Heuer, D.K.; Higginbotham, E.J.; Johnson, C.A.; Keltner, J.K.; Miller, J.P.; Parrish, R.K.; Wilson, M.R.; et al. Delaying treatment of ocular hypertension: The ocular hypertension treatment study. Arch. Ophthalmol. 2010, 128, 276–287. [Google Scholar] [CrossRef]
- Lee, J.M.; Caprioli, J.; Nouri-Mahdavi, K.; Afifi, A.A.; Morales, E.; Ramanathan, M.; Yu, F.; Coleman, A.L. Baseline prognostic factors predict rapid visual field deterioration in glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2228–2236. [Google Scholar] [CrossRef]
- Leske, M.C.; Heijl, A.; Hussein, M.; Bengtsson, B.; Hyman, L.; Komaroff, E.; Early Manifest Glaucoma Trial, G. Factors for glaucoma progression and the effect of treatment: The early manifest glaucoma trial. Arch. Ophthalmol. 2003, 121, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, S.K.; Johnson, C.A.; Demirel, S. Factors predicting the rate of functional progression in early and suspected glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3598–3604. [Google Scholar] [CrossRef] [PubMed]
- Siesky, B.; Wentz, S.M.; Januleviciene, I.; Kim, D.H.; Burgett, K.M.; Verticchio Vercellin, A.C.; Rowe, L.W.; Eckert, G.J.; Harris, A. Baseline structural characteristics of the optic nerve head and retinal nerve fiber layer are associated with progressive visual field loss in patients with open-angle glaucoma. PLoS ONE 2020, 15, e0236819. [Google Scholar] [CrossRef]
- Dvoriantchikova, G.; Ivanov, D.; Barakat, D.; Grinberg, A.; Wen, R.; Slepak, V.Z.; Shestopalov, V.I. Genetic ablation of Pannexin1 protects retinal neurons from ischemic injury. PLoS ONE 2012, 7, e31991. [Google Scholar] [CrossRef]
- Chi, W.; Chen, H.; Li, F.; Zhu, Y.; Yin, W.; Zhuo, Y. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-kappaB pathway in acute glaucoma. J. Neuroinflamm. 2015, 12, 137. [Google Scholar] [CrossRef]
- Shestopalov, V.I.; Slepak, V.Z. Molecular pathways of pannexin1-mediated neurotoxicity. Front. Physiol. 2014, 5, 23. [Google Scholar] [CrossRef][Green Version]
- Chi, W.; Li, F.; Chen, H.; Wang, Y.; Zhu, Y.; Yang, X.; Zhu, J.; Wu, F.; Ouyang, H.; Ge, J.; et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1beta production in acute glaucoma. Proc. Natl. Acad. Sci. USA 2014, 111, 11181–11186. [Google Scholar] [CrossRef] [PubMed]
- Gramlich, O.W.; Godwin, C.R.; Wadkins, D.; Elwood, B.W.; Kuehn, M.H. Early Functional Impairment in Experimental Glaucoma Is Accompanied by Disruption of the GABAergic System and Inceptive Neuroinflammation. Int. J. Mol. Sci. 2021, 22, 7581. [Google Scholar] [CrossRef]
- Chen, H.; Cho, K.S.; Vu, T.H.K.; Shen, C.H.; Kaur, M.; Chen, G.; Mathew, R.; McHam, M.L.; Fazelat, A.; Lashkari, K.; et al. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat. Commun. 2018, 9, 3209. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Braine, C.E.; Kizhatil, K.; Foxworth, N.E.; Tolman, N.G.; Harder, J.M.; Scott, R.A.; Sousa, G.L.; Panitch, A.; Howell, G.R.; et al. Inhibition of monocyte-like cell extravasation protects from neurodegeneration in DBA/2J glaucoma. Mol. Neurodegener. 2019, 14, 6. [Google Scholar] [CrossRef]
- Gramlich, O.W.; Ding, Q.J.; Zhu, W.; Cook, A.; Anderson, M.G.; Kuehn, M.H. Adoptive transfer of immune cells from glaucomatous mice provokes retinal ganglion cell loss in recipients. Acta Neuropathol. Commun. 2015, 3, 56. [Google Scholar] [CrossRef]
- Barron, M.J.; Griffiths, P.; Turnbull, D.M.; Bates, D.; Nichols, P. The distributions of mitochondria and sodium channels reflect the specific energy requirements and conduction properties of the human optic nerve head. Br. J. Ophthalmol. 2004, 88, 286–290. [Google Scholar] [CrossRef]
- Casson, R.J.; Chidlow, G.; Crowston, J.G.; Williams, P.A.; Wood, J.P.M. Retinal energy metabolism in health and glaucoma. Prog. Retin. Eye Res. 2021, 81, 100881. [Google Scholar] [CrossRef]
- Osborne, N.N. Mitochondria: Their role in ganglion cell death and survival in primary open angle glaucoma. Exp. Eye Res. 2010, 90, 750–757. [Google Scholar] [CrossRef]
- Osborne, N.N.; Kamalden, T.A.; Majid, A.S.; del Olmo-Aguado, S.; Manso, A.G.; Ji, D. Light effects on mitochondrial photosensitizers in relation to retinal degeneration. Neurochem. Res. 2010, 35, 2027–2034. [Google Scholar] [CrossRef]
- Nunez-Alvarez, C.; Osborne, N.N. Blue light exacerbates and red light counteracts negative insults to retinal ganglion cells in situ and R28 cells in vitro. Neurochem. Int. 2019, 125, 187–196. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Morales, J.; Bosley, T.M. Mitochondrial abnormalities in patients with primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.N. Neuroinflammatory Mechanisms of Mitochondrial Dysfunction and Neurodegeneration in Glaucoma. J. Ophthalmol. 2021, 2021, 4581909. [Google Scholar] [CrossRef]
- Garhofer, G.; Zawinka, C.; Resch, H.; Huemer, K.H.; Schmetterer, L.; Dorner, G.T. Response of retinal vessel diameters to flicker stimulation in patients with early open angle glaucoma. J. Glaucoma 2004, 13, 340–344. [Google Scholar] [CrossRef]
- Flammer, J.; Orgul, S.; Costa, V.P.; Orzalesi, N.; Krieglstein, G.K.; Serra, L.M.; Renard, J.P.; Stefansson, E. The impact of ocular blood flow in glaucoma. Prog. Retin. Eye Res. 2002, 21, 359–393. [Google Scholar] [CrossRef]
- Hayreh, S.S. The 1994 Von Sallman Lecture. The optic nerve head circulation in health and disease. Exp. Eye Res. 1995, 61, 259–272. [Google Scholar] [CrossRef]
- Jassim, A.H.; Inman, D.M. Evidence of Hypoxic Glial Cells in a Model of Ocular Hypertension. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Wax, M.B. Hypoxia-inducible factor 1alpha in the glaucomatous retina and optic nerve head. Arch. Ophthalmol. 2004, 122, 1348–1356. [Google Scholar] [CrossRef]
- Hughes, J.M.; Groot, A.J.; van der Groep, P.; Sersansie, R.; Vooijs, M.; van Diest, P.J.; Van Noorden, C.J.; Schlingemann, R.O.; Klaassen, I. Active HIF-1 in the normal human retina. J. Histochem. Cytochem. 2010, 58, 247–254. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Cardozo, B.H.; Foxworth, N.E.; John, S.W.M. Nicotinamide treatment robustly protects from inherited mouse glaucoma. Commun. Integr. Biol. 2018, 11, e1356956. [Google Scholar] [CrossRef]
- Harun-Or-Rashid, M.; Pappenhagen, N.; Zubricky, R.; Coughlin, L.; Jassim, A.H.; Inman, D.M. MCT2 overexpression rescues metabolic vulnerability and protects retinal ganglion cells in two models of glaucoma. Neurobiol. Dis. 2020, 141, 104944. [Google Scholar] [CrossRef]
- Hui, F.; Tang, J.; Williams, P.A.; McGuinness, M.B.; Hadoux, X.; Casson, R.J.; Coote, M.; Trounce, I.A.; Martin, K.R.; van Wijngaarden, P.; et al. Improvement in inner retinal function in glaucoma with nicotinamide (vitamin B3) supplementation: A crossover randomized clinical trial. Clin. Exp. Ophthalmol. 2020, 48, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Bader, V.; Winklhofer, K.F. Mitochondria at the interface between neurodegeneration and neuroinflammation. Semin. Cell Dev. Biol. 2020, 99, 163–171. [Google Scholar] [CrossRef]
- Pronin, A.; Pham, D.; An, W.; Dvoriantchikova, G.; Reshetnikova, G.; Qiao, J.; Kozhekbaeva, Z.; Reiser, A.E.; Slepak, V.Z.; Shestopalov, V.I. Inflammasome Activation Induces Pyroptosis in the Retina Exposed to Ocular Hypertension Injury. Front. Mol. Neurosci. 2019, 12, 36. [Google Scholar] [CrossRef]
- Lu, W.; Hu, H.; Sevigny, J.; Gabelt, B.T.; Kaufman, P.L.; Johnson, E.C.; Morrison, J.C.; Zode, G.S.; Sheffield, V.C.; Zhang, X.; et al. Rat, mouse, and primate models of chronic glaucoma show sustained elevation of extracellular ATP and altered purinergic signaling in the posterior eye. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3075–3083. [Google Scholar] [CrossRef]
- Perez de Lara, M.J.; Guzman-Aranguez, A.; de la Villa, P.; Diaz-Hernandez, J.I.; Miras-Portugal, M.T.; Pintor, J. Increased levels of extracellular ATP in glaucomatous retinas: Possible role of the vesicular nucleotide transporter during the development of the pathology. Mol. Vis. 2015, 21, 1060–1070. [Google Scholar]
- Li, A.; Zhang, X.; Zheng, D.; Ge, J.; Laties, A.M.; Mitchell, C.H. Sustained elevation of extracellular ATP in aqueous humor from humans with primary chronic angle-closure glaucoma. Exp. Eye Res. 2011, 93, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Lim, J.C.; Lu, W.; Beckel, J.M.; Macarak, E.J.; Laties, A.M.; Mitchell, C.H. Neurons respond directly to mechanical deformation with pannexin-mediated ATP release and autostimulation of P2X7 receptors. J. Physiol. 2012, 590, 2285–2304. [Google Scholar] [CrossRef] [PubMed]
- Beckel, J.M.; Argall, A.J.; Lim, J.C.; Xia, J.; Lu, W.; Coffey, E.E.; Macarak, E.J.; Shahidullah, M.; Delamere, N.A.; Zode, G.S.; et al. Mechanosensitive release of adenosine 5’-triphosphate through pannexin channels and mechanosensitive upregulation of pannexin channels in optic nerve head astrocytes: A mechanism for purinergic involvement in chronic strain. Glia 2014, 62, 1486–1501. [Google Scholar] [CrossRef] [PubMed]
- Reigada, D.; Lu, W.; Zhang, M.; Mitchell, C.H. Elevated pressure triggers a physiological release of ATP from the retina: Possible role for pannexin hemichannels. Neuroscience 2008, 157, 396–404. [Google Scholar] [CrossRef][Green Version]
- Beckel, J.M.; Gomez, N.M.; Lu, W.; Campagno, K.E.; Nabet, B.; Albalawi, F.; Lim, J.C.; Boesze-Battaglia, K.; Mitchell, C.H. Stimulation of TLR3 triggers release of lysosomal ATP in astrocytes and epithelial cells that requires TRPML1 channels. Sci. Rep. 2018, 8, 5726. [Google Scholar] [CrossRef]
- Yang, X.; Luo, C.; Cai, J.; Powell, D.W.; Yu, D.; Kuehn, M.H.; Tezel, G. Neurodegenerative and inflammatory pathway components linked to TNF-alpha/TNFR1 signaling in the glaucomatous human retina. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8442–8454. [Google Scholar] [CrossRef]
- Mac Nair, C.E.; Schlamp, C.L.; Montgomery, A.D.; Shestopalov, V.I.; Nickells, R.W. Retinal glial responses to optic nerve crush are attenuated in Bax-deficient mice and modulated by purinergic signaling pathways. J. Neuroinflamm. 2016, 13, 93. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.; Sun, Q.; Xue, S.; Guan, H.; Ji, M. Activation of P2X7R- NLRP3 pathway in Retinal microglia contribute to Retinal Ganglion Cells death in chronic ocular hypertension (COH). Exp. Eye Res. 2019, 188, 107771. [Google Scholar] [CrossRef]
- Albalawi, F.; Lu, W.; Beckel, J.M.; Lim, J.C.; McCaughey, S.A.; Mitchell, C.H. The P2X7 Receptor Primes IL-1beta and the NLRP3 Inflammasome in Astrocytes Exposed to Mechanical Strain. Front. Cell. Neurosci. 2017, 11, 227. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Yang, X.; Luo, C.; Cai, J.; Powell, D.W. An astrocyte-specific proteomic approach to inflammatory responses in experimental rat glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4220–4233. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Albalawi, F.; Beckel, J.M.; Lim, J.C.; Laties, A.M.; Mitchell, C.H. The P2X7 receptor links mechanical strain to cytokine IL-6 up-regulation and release in neurons and astrocytes. J. Neurochem. 2017, 141, 436–448. [Google Scholar] [CrossRef]
- Pelegrin, P.; Barroso-Gutierrez, C.; Surprenant, A. P2X7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage. J. Immunol. 2008, 180, 7147–7157. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; He, Y.; Munoz-Planillo, R.; Liu, Q.; Nunez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef]
- Chakraborty, S.; Kaushik, D.K.; Gupta, M.; Basu, A. Inflammasome signaling at the heart of central nervous system pathology. J. Neurosci. Res. 2010, 88, 1615–1631. [Google Scholar] [CrossRef]
- Dvoriantchikova, G.; Pronin, A.; Kurtenbach, S.; Toychiev, A.; Chou, T.H.; Yee, C.W.; Prindeville, B.; Tayou, J.; Porciatti, V.; Sagdullaev, B.T.; et al. Pannexin 1 sustains the electrophysiological responsiveness of retinal ganglion cells. Sci. Rep. 2018, 8, 5797. [Google Scholar] [CrossRef]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, L.; Pytel, D.; Mucha, B.; Szymanek, K.; Szaflik, J.; Szaflik, J.P.; Majsterek, I. Altered Expression Levels of MMP1, MMP9, MMP12, TIMP1, and IL-1beta as a Risk Factor for the Elevated IOP and Optic Nerve Head Damage in the Primary Open-Angle Glaucoma Patients. Biomed. Res. Int. 2015, 2015, 812503. [Google Scholar] [CrossRef]
- Devi, T.S.; Lee, I.; Huttemann, M.; Kumar, A.; Nantwi, K.D.; Singh, L.P. TXNIP links innate host defense mechanisms to oxidative stress and inflammation in retinal Muller glia under chronic hyperglycemia: Implications for diabetic retinopathy. Exp. Diabetes Res. 2012, 2012, 438238. [Google Scholar] [CrossRef]
- Dick, A.D. Doyne lecture 2016: Intraocular health and the many faces of inflammation. Eye 2016, 31, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Mawhinney, L.J.; de Rivero Vaccari, J.P.; Dale, G.A.; Keane, R.W.; Bramlett, H.M. Heightened inflammasome activation is linked to age-related cognitive impairment in Fischer 344 rats. BMC Neurosci. 2011, 12, 123. [Google Scholar] [CrossRef] [PubMed]
- Mejias, N.H.; Martinez, C.C.; Stephens, M.E.; de Rivero Vaccari, J.P. Contribution of the inflammasome to inflammaging. J. Inflamm. 2018, 15, 23. [Google Scholar] [CrossRef]
- Berger, S.; Savitz, S.I.; Nijhawan, S.; Singh, M.; David, J.; Rosenbaum, P.S.; Rosenbaum, D.M. Deleterious role of TNF-alpha in retinal ischemia-reperfusion injury. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3605–3610. [Google Scholar] [CrossRef]
- McGeough, M.D.; Wree, A.; Inzaugarat, M.E.; Haimovich, A.; Johnson, C.D.; Pena, C.A.; Goldbach-Mansky, R.; Broderick, L.; Feldstein, A.E.; Hoffman, H.M. TNF regulates transcription of NLRP3 inflammasome components and inflammatory molecules in cryopyrinopathies. J. Clin. Investig. 2017, 127, 4488–4497. [Google Scholar] [CrossRef] [PubMed]
- Dvoriantchikova, G.; Barakat, D.; Brambilla, R.; Agudelo, C.; Hernandez, E.; Bethea, J.R.; Shestopalov, V.I.; Ivanov, D. Inactivation of astroglial NF-kappa B promotes survival of retinal neurons following ischemic injury. Eur. J. Neurosci. 2009, 30, 175–185. [Google Scholar] [CrossRef]
- Dvoriantchikova, G.; Santos, A.R.; Danek, D.; Dvoriantchikova, X.; Ivanov, D. The TIR-domain-containing adapter inducing interferon-beta-dependent signaling cascade plays a crucial role in ischemia-reperfusion-induced retinal injury, whereas the contribution of the myeloid differentiation primary response 88-dependent signaling cascade is not as pivotal. Eur. J. Neurosci. 2014, 40, 2502–2512. [Google Scholar] [CrossRef]
- Hara, H.; Tsuchiya, K.; Kawamura, I.; Fang, R.; Hernandez-Cuellar, E.; Shen, Y.; Mizuguchi, J.; Schweighoffer, E.; Tybulewicz, V.; Mitsuyama, M. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 2013, 14, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Huang, Q.; Ye, C.; Wu, R.; Lei, G.; Jiang, J.; Chen, T.; Peng, Y.; Fang, R. Syk and JNK signaling pathways are involved in inflammasome activation in macrophages infected with Streptococcus pneumoniae. Biochem. Biophys. Res. Commun. 2018, 507, 217–222. [Google Scholar] [CrossRef]
- Niyadurupola, N.; Sidaway, P.; Ma, N.; Rhodes, J.D.; Broadway, D.C.; Sanderson, J. P2X7 receptor activation mediates retinal ganglion cell death in a human retina model of ischemic neurodegeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2163–2170. [Google Scholar] [CrossRef] [PubMed]
- Krizaj, D.; Ryskamp, D.A.; Tian, N.; Tezel, G.; Mitchell, C.H.; Slepak, V.Z.; Shestopalov, V.I. From mechanosensitivity to inflammatory responses: New players in the pathology of glaucoma. Curr. Eye Res. 2014, 39, 105–119. [Google Scholar] [CrossRef]
- Sappington, R.M.; Sidorova, T.; Long, D.J.; Calkins, D.J. TRPV1: Contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Investig. Ophthalmol. Vis. Sci. 2009, 50, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, S.; Tanihara, H.; Kido, N.; Honda, Y.; Goto, W.; Hara, H.; Miyawaki, N. Interleukin-1beta mediates ischemic injury in the rat retina. Exp. Eye Res. 2001, 73, 661–667. [Google Scholar] [CrossRef]
- Zhang, X.; Chintala, S.K. Influence of interleukin-1 beta induction and mitogen-activated protein kinase phosphorylation on optic nerve ligation-induced matrix metalloproteinase-9 activation in the retina. Exp. Eye Res. 2004, 78, 849–860. [Google Scholar] [CrossRef]
- Arai, J.; Katai, N.; Kuida, K.; Kikuchi, T.; Yoshimura, N. Decreased retinal neuronal cell death in caspase-1 knockout mice. Jpn. J. Ophthalmol. 2006, 50, 417–425. [Google Scholar] [CrossRef]
- Seki, M.; Soussou, W.; Manabe, S.; Lipton, S.A. Protection of retinal ganglion cells by caspase substrate-binding peptide IQACRG from N-methyl-D-aspartate receptor-mediated excitotoxicity. Investig. Ophthalmol. Vis. Sci. 2009, 51, 1198–1207. [Google Scholar] [CrossRef]
- Puyang, Z.; Feng, L.; Chen, H.; Liang, P.; Troy, J.B.; Liu, X. Retinal Ganglion Cell Loss is Delayed Following Optic Nerve Crush in NLRP3 Knockout Mice. Sci. Rep. 2016, 6, 20998. [Google Scholar] [CrossRef]
- Tribble, J.R.; Harder, J.M.; Williams, P.A.; John, S.W.M. Ocular hypertension suppresses homeostatic gene expression in optic nerve head microglia of DBA/2 J mice. Mol. Brain 2020, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Nikolskaya, T.; Nikolsky, Y.; Serebryiskaya, T.; Zvereva, S.; Sviridov, E.; Dezso, Z.; Rahkmatulin, E.; Brennan, R.J.; Yankovsky, N.; Bhattacharya, S.K.; et al. Network analysis of human glaucomatous optic nerve head astrocytes. BMC Med. Genom. 2009, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Gramlich, O.W.; Godwin, C.R.; Heuss, N.D.; Gregerson, D.S.; Kuehn, M.H. T and B Lymphocyte Deficiency in Rag1−/− Mice Reduces Retinal Ganglion Cell Loss in Experimental Glaucoma. Investig. Ophthalmol. Vis. Sci. 2020, 61, 18. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Tang, Y.; Yi, I.; Chen, D.F. The role of commensal microflora-induced T cell responses in glaucoma neurodegeneration. Prog. Brain Res. 2020, 256, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Dvoriantchikova, G.; Hernandez, E.; Grant, J.; Santos, A.R.; Yang, H.; Ivanov, D. The high-mobility group box-1 nuclear factor mediates retinal injury after ischemia-reperfusion. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7187–7194. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Hernandez, R.; Wax, M.B. Immunostaining of heat shock proteins in the retina and optic nerve head of normal and glaucomatous eyes. Arch. Ophthalmol. 2000, 118, 511–518. [Google Scholar] [CrossRef]
- Luo, C.; Yang, X.; Kain, A.D.; Powell, D.W.; Kuehn, M.H.; Tezel, G. Glaucomatous tissue stress and the regulation of immune response through glial Toll-like receptor signaling. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5697–5707. [Google Scholar] [CrossRef]
- Lim, J.C.; Lu, W.; Beckel, J.M.; Mitchell, C.H. Neuronal Release of Cytokine IL-3 Triggered by Mechanosensitive Autostimulation of the P2X7 Receptor Is Neuroprotective. Front. Cell. Neurosci. 2016, 10, 270. [Google Scholar] [CrossRef]
- Williams, P.A.; Tribble, J.R.; Pepper, K.W.; Cross, S.D.; Morgan, B.P.; Morgan, J.E.; John, S.W.; Howell, G.R. Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Mol. Neurodegener. 2016, 11, 26. [Google Scholar] [CrossRef]
- Kuehn, M.H.; Kim, C.Y.; Ostojic, J.; Bellin, M.; Alward, W.L.; Stone, E.M.; Sakaguchi, D.S.; Grozdanic, S.D.; Kwon, Y.H. Retinal synthesis and deposition of complement components induced by ocular hypertension. Exp. Eye Res. 2006, 83, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; von Bernhardi, R.; Giaume, C.; Saez, J.C. Glial hemichannels and their involvement in aging and neurodegenerative diseases. Rev. Neurosci. 2012, 23, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, M.H.; Kim, C.Y.; Jiang, B.; Dumitrescu, A.V.; Kwon, Y.H. Disruption of the complement cascade delays retinal ganglion cell death following retinal ischemia-reperfusion. Exp. Eye Res. 2008, 87, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.R.; MacNicoll, K.H.; Braine, C.E.; Soto, I.; Macalinao, D.G.; Sousa, G.L.; John, S.W. Combinatorial targeting of early pathways profoundly inhibits neurodegeneration in a mouse model of glaucoma. Neurobiol. Dis. 2014, 71, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Madeira, M.H.; Elvas, F.; Boia, R.; Goncalves, F.Q.; Cunha, R.A.; Ambrosio, A.F.; Santiago, A.R. Adenosine A2AR blockade prevents neuroinflammation-induced death of retinal ganglion cells caused by elevated pressure. J. Neuroinflamm. 2015, 12, 115. [Google Scholar] [CrossRef]
- Silverman, S.M.; Kim, B.J.; Howell, G.R.; Miller, J.; John, S.W.; Wordinger, R.J.; Clark, A.F. C1q propagates microglial activation and neurodegeneration in the visual axis following retinal ischemia/reperfusion injury. Mol. Neurodegener. 2016, 11, 24. [Google Scholar] [CrossRef]
- Ivanov, D.; Dvoriantchikova, G.; Nathanson, L.; McKinnon, S.J.; Shestopalov, V.I. Microarray analysis of gene expression in adult retinal ganglion cells. FEBS Lett. 2006, 580, 331–335. [Google Scholar] [CrossRef]
- Ivanov, D.; Dvoriantchikova, G.; Barakat, D.J.; Nathanson, L.; Shestopalov, V.I. Differential gene expression profiling of large and small retinal ganglion cells. J. Neurosci. Methods 2008, 174, 10–17. [Google Scholar] [CrossRef]
- Namekata, K.; Harada, C.; Guo, X.; Kikushima, K.; Kimura, A.; Fuse, N.; Mitamura, Y.; Kohyama, K.; Matsumoto, Y.; Tanaka, K.; et al. Interleukin-1 attenuates normal tension glaucoma-like retinal degeneration in EAAC1-deficient mice. Neurosci. Lett. 2009, 465, 160–164. [Google Scholar] [CrossRef]
- Qi, Y.; Zhao, M.; Bai, Y.; Huang, L.; Yu, W.; Bian, Z.; Zhao, M.; Li, X. Retinal ischemia/reperfusion injury is mediated by Toll-like receptor 4 activation of NLRP3 inflammasomes. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5466–5475. [Google Scholar] [CrossRef] [PubMed]
- Barakat, D.J.; Dvoriantchikova, G.; Ivanov, D.; Shestopalov, V.I. Astroglial NF-kappaB mediates oxidative stress by regulation of NADPH oxidase in a model of retinal ischemia reperfusion injury. J. Neurochem. 2012, 120, 586–597. [Google Scholar] [CrossRef]
- Schneider, M.; Fuchshofer, R. The role of astrocytes in optic nerve head fibrosis in glaucoma. Exp. Eye Res. 2016, 142, 49–55. [Google Scholar] [CrossRef]
- Crabb, J.W.; Yuan, X.; Dvoriantchikova, G.; Ivanov, D.; Crabb, J.S.; Shestopalov, V.I. Preliminary quantitative proteomic characterization of glaucomatous rat retinal ganglion cells. Exp. Eye Res. 2010, 91, 107–110. [Google Scholar] [CrossRef][Green Version]
- Ren, L.; Danias, J. A role for complement in glaucoma? Adv. Exp. Med. Biol. 2010, 703, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Yang, X.; Luo, C.; Kain, A.D.; Powell, D.W.; Kuehn, M.H.; Kaplan, H.J. Oxidative stress and the regulation of complement activation in human glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5071–5082. [Google Scholar] [CrossRef]
- Hubens, W.H.G.; Mohren, R.J.C.; Liesenborghs, I.; Eijssen, L.M.T.; Ramdas, W.D.; Webers, C.A.B.; Gorgels, T. The aqueous humor proteome of primary open angle glaucoma: An extensive review. Exp. Eye Res. 2020, 197, 108077. [Google Scholar] [CrossRef]
- Adav, S.S.; Wei, J.; Terence, Y.; Ang, B.C.H.; Yip, L.W.L.; Sze, S.K. Proteomic Analysis of Aqueous Humor from Primary Open Angle Glaucoma Patients on Drug Treatment Revealed Altered Complement Activation Cascade. J. Proteome Res. 2018, 17, 2499–2510. [Google Scholar] [CrossRef] [PubMed]
- Kodeboyina, S.K.; Lee, T.J.; Bollinger, K.; Ulrich, L.; Bogorad, D.; Estes, A.; Zhi, W.; Sharma, S.; Sharma, A. Aqueous Humor Proteomic Alterations Associated with Visual Field Index Parameters in Glaucoma Patients: A Pilot Study. J. Clin. Med. 2021, 10, 1180. [Google Scholar] [CrossRef]
- Mirzaei, M.; Gupta, V.B.; Chick, J.M.; Greco, T.M.; Wu, Y.; Chitranshi, N.; Wall, R.V.; Hone, E.; Deng, L.; Dheer, Y.; et al. Age-related neurodegenerative disease associated pathways identified in retinal and vitreous proteome from human glaucoma eyes. Sci. Rep. 2017, 7, 12685. [Google Scholar] [CrossRef]
- Stasi, K.; Nagel, D.; Yang, X.; Wang, R.F.; Ren, L.; Podos, S.M.; Mittag, T.; Danias, J. Complement component 1Q (C1Q) upregulation in retina of murine, primate, and human glaucomatous eyes. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1024–1029. [Google Scholar] [CrossRef]
- Kong, X.; Liu, X.; Huang, X.; Mao, Z.; Zhong, Y.; Chi, W. Damage to the blood-aqueous barrier in eyes with primary angle closure glaucoma. Mol. Vis. 2010, 16, 2026–2032. [Google Scholar]
- Grieshaber, M.C.; Flammer, J. Does the blood-brain barrier play a role in Glaucoma? Surv. Ophthalmol. 2007, 52, S115–S121. [Google Scholar] [CrossRef]
- Alexander, J.J. Blood-brain barrier (BBB) and the complement landscape. Mol. Immunol. 2018, 102, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C.; Alexopoulos, H.; Spaeth, P.J. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat. Rev. Neurol. 2020, 16, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Conway, R.M.; Schlotzer-Schrehardt, U.; Kuchle, M.; Naumann, G.O. Pseudoexfoliation syndrome: Pathological manifestations of relevance to intraocular surgery. Clin. Exp. Ophthalmol. 2004, 32, 199–210. [Google Scholar] [CrossRef]
- Doudevski, I.; Rostagno, A.; Cowman, M.; Liebmann, J.; Ritch, R.; Ghiso, J. Clusterin and complement activation in exfoliation glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2491–2499. [Google Scholar] [CrossRef] [PubMed]
- Boehm, N.; Wolters, D.; Thiel, U.; Lossbrand, U.; Wiegel, N.; Pfeiffer, N.; Grus, F.H. New insights into autoantibody profiles from immune privileged sites in the eye: A glaucoma study. Brain Behav. Immun. 2012, 26, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Bishop, P.N. The eye as a complement dysregulation hotspot. Semin. Immunopathol. 2018, 40, 65–74. [Google Scholar] [CrossRef]
- Mohlin, C.; Sandholm, K.; Ekdahl, K.N.; Nilsson, B. The link between morphology and complement in ocular disease. Mol. Immunol. 2017, 89, 84–99. [Google Scholar] [CrossRef]
- Howell, G.R.; Soto, I.; Ryan, M.; Graham, L.C.; Smith, R.S.; John, S.W. Deficiency of complement component 5 ameliorates glaucoma in DBA/2J mice. J. Neuroinflamm. 2013, 10, 76. [Google Scholar] [CrossRef]
- Bosco, A.; Anderson, S.R.; Breen, K.T.; Romero, C.O.; Steele, M.R.; Chiodo, V.A.; Boye, S.L.; Hauswirth, W.W.; Tomlinson, S.; Vetter, M.L. Complement C3-Targeted Gene Therapy Restricts Onset and Progression of Neurodegeneration in Chronic Mouse Glaucoma. Mol. Ther. 2018, 26, 2379–2396. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, S.; Gomes, S.C.; Gassel, C.J.; Asaad, M.A.; Stute, G.; Schargus, M.; Dick, H.B.; Joachim, S.C. Intravitreal Therapy Against the Complement Factor C5 Prevents Retinal Degeneration in an Experimental Autoimmune Glaucoma Model. Front. Pharmacol. 2019, 10, 1381. [Google Scholar] [CrossRef] [PubMed]
- Jha, P.; Banda, H.; Tytarenko, R.; Bora, P.S.; Bora, N.S. Complement mediated apoptosis leads to the loss of retinal ganglion cells in animal model of glaucoma. Mol. Immunol. 2011, 48, 2151–2158. [Google Scholar] [CrossRef]
- Martin-Sanchez, F.; Diamond, C.; Zeitler, M.; Gomez, A.I.; Baroja-Mazo, A.; Bagnall, J.; Spiller, D.; White, M.; Daniels, M.J.; Mortellaro, A.; et al. Inflammasome-dependent IL-1beta release depends upon membrane permeabilisation. Cell Death Differ. 2016, 23, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Heilig, R.; Dick, M.S.; Sborgi, L.; Meunier, E.; Hiller, S.; Broz, P. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur. J. Immunol. 2018, 48, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.S.; Tan, L.; Jiang, T.; Zhu, X.C.; Wang, H.F.; Jia, C.D.; Yu, J.T. Amyloid-beta induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1382. [Google Scholar] [CrossRef]
- Gan, J.; Huang, M.; Lan, G.; Liu, L.; Xu, F. High Glucose Induces the Loss of Retinal Pericytes Partly via NLRP3-Caspase-1-GSDMD-Mediated Pyroptosis. Biomed. Res. Int. 2020, 2020, 4510628. [Google Scholar] [CrossRef]
- Li, J.; Hao, J.H.; Yao, D.; Li, R.; Li, X.F.; Yu, Z.Y.; Luo, X.; Liu, X.H.; Wang, M.H.; Wang, W. Caspase-1 inhibition prevents neuronal death by targeting the canonical inflammasome pathway of pyroptosis in a murine model of cerebral ischemia. CNS Neurosci. Ther. 2020, 26, 925–939. [Google Scholar] [CrossRef]
- Wang, J.; Yao, J.; Liu, Y.; Huang, L. Targeting the gasdermin D as a strategy for ischemic stroke therapy. Biochem. Pharmacol. 2021, 188, 114585. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Mei, S.; Luo, Y.; Wu, H.; Zhang, J.; Zhu, J. Gasdermin Family: A Promising Therapeutic Target for Stroke. Transl. Stroke Res. 2018, 9, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Qian, J.; Zhang, P.; Li, H.; Shen, H.; Li, X.; Chen, G. Gasdermin D serves as a key executioner of pyroptosis in experimental cerebral ischemia and reperfusion model both in vivo and in vitro. J. Neurosci. Res. 2019, 97, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Adornetto, A.; Russo, R.; Parisi, V. Neuroinflammation as a target for glaucoma therapy. Neural Regen. Res. 2019, 14, 391–394. [Google Scholar] [CrossRef]
- Russo, H.M.; Rathkey, J.; Boyd-Tressler, A.; Katsnelson, M.A.; Abbott, D.W.; Dubyak, G.R. Active Caspase-1 Induces Plasma Membrane Pores That Precede Pyroptotic Lysis and Are Blocked by Lanthanides. J. Immunol. 2016, 197, 1353–1367. [Google Scholar] [CrossRef]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018, 48, 35–44.e6. [Google Scholar] [CrossRef]
- Rogers, C.; Alnemri, E.S. Gasdermins: Novel mitochondrial pore-forming proteins. Mol. Cell. Oncol. 2019, 6, e1621501. [Google Scholar] [CrossRef]
- Wang, J.; Feng, Y.; Huo, H.; Zhang, X.; Yue, J.; Zhang, W.; Yan, Z.; Jiao, X. NLRP3 inflammasome mediates angiotensin II-induced islet beta cell apoptosis. Acta Biochim. Biophys. Sin. 2019, 51, 501–508. [Google Scholar] [CrossRef]
- Lin, C.; Wu, F.; Zheng, T.; Wang, X.; Chen, Y.; Wu, X. Kaempferol attenuates retinal ganglion cell death by suppressing NLRP1/NLRP3 inflammasomes and caspase-8 via JNK and NF-kappaB pathways in acute glaucoma. Eye 2019, 33, 777–784. [Google Scholar] [CrossRef]
- Gupta, K.K.; Khan, M.A.; Singh, S.K. Constitutive Inflammatory Cytokine Storm: A Major Threat to Human Health. J. Interferon Cytokine Res. 2020, 40, 19–23. [Google Scholar] [CrossRef]
- Kerrigan, L.A.; Zack, D.J.; Quigley, H.A.; Smith, S.D.; Pease, M.E. TUNEL-positive ganglion cells in human primary open-angle glaucoma. Arch. Ophthalmol. 1997, 115, 1031–1035. [Google Scholar] [CrossRef]
- Howell, G.R.; Libby, R.T.; Jakobs, T.C.; Smith, R.S.; Phalan, F.C.; Barter, J.W.; Barbay, J.M.; Marchant, J.K.; Mahesh, N.; Porciatti, V.; et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J. Cell Biol. 2007, 179, 1523–1537. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Cui, J.Z.; To, E.; Cao, S.; Matsubara, J.A. Evidence for the activation of pyroptotic and apoptotic pathways in RPE cells associated with NLRP3 inflammasome in the rodent eye. J. Neuroinflamm. 2018, 15, 15. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef]
- Vince, J.E.; Silke, J. The intersection of cell death and inflammasome activation. Cell. Mol. Life Sci. 2016, 73, 2349–2367. [Google Scholar] [CrossRef]
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 2019, 10, 1689. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Li, L.Y.; Patil, R.V.; Wax, M.B. TNF-alpha and TNF-alpha receptor-1 in the retina of normal and glaucomatous eyes. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1787–1794. [Google Scholar]
- Dvoriantchikova, G.; Ivanov, D. Tumor necrosis factor-alpha mediates activation of NF-kappaB and JNK signaling cascades in retinal ganglion cells and astrocytes in opposite ways. Eur. J. Neurosci. 2014, 40, 3171–3178. [Google Scholar] [CrossRef]
- Sanderson, J.; Dartt, D.A.; Trinkaus-Randall, V.; Pintor, J.; Civan, M.M.; Delamere, N.A.; Fletcher, E.L.; Salt, T.E.; Grosche, A.; Mitchell, C.H. Purines in the eye: Recent evidence for the physiological and pathological role of purines in the RPE, retinal neurons, astrocytes, Muller cells, lens, trabecular meshwork, cornea and lacrimal gland. Exp. Eye Res. 2014, 127, 270–279. [Google Scholar] [CrossRef]
- Dvoriantchikova, G.; Barakat, D.J.; Hernandez, E.; Shestopalov, V.I.; Ivanov, D. Toll-like receptor 4 contributes to retinal ischemia/reperfusion injury. Mol. Vis. 2010, 16, 1907–1912. [Google Scholar]
- Del Olmo-Aguado, S.; Nunez-Alvarez, C.; Osborne, N.N. Blue Light Action on Mitochondria Leads to Cell Death by Necroptosis. Neurochem. Res. 2016, 41, 2324–2335. [Google Scholar] [CrossRef]
- Harder, J.M.; Guymer, C.; Wood, J.P.M.; Daskalaki, E.; Chidlow, G.; Zhang, C.; Balasubramanian, R.; Cardozo, B.H.; Foxworth, N.E.; Deering, K.E.; et al. Disturbed glucose and pyruvate metabolism in glaucoma with neuroprotection by pyruvate or rapamycin. Proc. Natl. Acad. Sci. USA 2020, 117, 33619–33627. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, K.A.; Harder, J.M.; Fornarola, L.B.; Freeman, R.S.; Clark, A.F.; Pang, I.H.; John, S.W.; Libby, R.T. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol. Dis. 2012, 46, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, K.A.; Harder, J.M.; John, S.W.; Shrager, P.; Libby, R.T. DLK-dependent signaling is important for somal but not axonal degeneration of retinal ganglion cells following axonal injury. Neurobiol. Dis. 2014, 69, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Syc-Mazurek, S.B.; Fernandes, K.A.; Wilson, M.P.; Shrager, P.; Libby, R.T. Together JUN and DDIT3 (CHOP) control retinal ganglion cell death after axonal injury. Mol. Neurodegener. 2017, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, K.A.; Mitchell, K.L.; Patel, A.; Marola, O.J.; Shrager, P.; Zack, D.J.; Libby, R.T.; Welsbie, D.S. Role of SARM1 and DR6 in retinal ganglion cell axonal and somal degeneration following axonal injury. Exp. Eye Res. 2018, 171, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, A.; Nakamura, M.; Nakanishi, Y.; Yamada, Y.; Negi, A. Long-term glial reactivity in rat retinas ipsilateral and contralateral to experimental glaucoma. Exp. Eye Res. 2005, 81, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.L.; Pasini, S.; Lambert, W.S.; D’Alessandro, K.B.; Yao, V.; Risner, M.L.; Calkins, D.J. Redistribution of metabolic resources through astrocyte networks mitigates neurodegenerative stress. Proc. Natl. Acad. Sci. USA 2020, 117, 18810–18821. [Google Scholar] [CrossRef]
- Gallego, B.I.; Salazar, J.J.; de Hoz, R.; Rojas, B.; Ramirez, A.I.; Salinas-Navarro, M.; Ortin-Martinez, A.; Valiente-Soriano, F.J.; Aviles-Trigueros, M.; Villegas-Perez, M.P.; et al. IOP induces upregulation of GFAP and MHC-II and microglia reactivity in mice retina contralateral to experimental glaucoma. J. Neuroinflamm. 2012, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Armas, R.; Garway-Heath, D.F.; Bunce, C.V.; Poinoosawmy, D.; Hitchings, R.A. Clinical factors influencing the visual prognosis of the fellow eyes of normal tension glaucoma patients with unilateral field loss. Br. J. Ophthalmol. 1999, 83, 1002–1005. [Google Scholar] [CrossRef]
- Calugaru, M.; Marin, C. The partner eye in unilateral malignant glaucoma. Klin. Mon. Augenheilkd. 1991, 198, 223–227. [Google Scholar]
- Susanna, R., Jr.; Galvao-Filho, R.P. Study of the contralateral eye in patients with glaucoma and a unilateral perimetric defect. J. Glaucoma 2000, 9, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.P.; Park, R.J. Visual field progression in patients with initially unilateral visual field loss from chronic open-angle glaucoma. Ophthalmology 2000, 107, 1688–1692. [Google Scholar] [CrossRef]
- Yarangumeli, A.; Davutluoglu, B.; Koz, O.G.; Elhan, A.H.; Yaylaci, M.; Kural, G. Glaucomatous damage in normotensive fellow eyes of patients with unilateral hypertensive pseudoexfoliation glaucoma: Normotensive pseudoexfoliation glaucoma? Clin. Exp. Ophthalmol. 2006, 34, 15–19. [Google Scholar] [CrossRef]
- Rao, A. Clinical and Optical Coherence Tomography Features in Unilateral versus Bilateral Pseudoexfoliation Syndrome. J. Ophthalmic Vis. Res. 2012, 7, 197–202. [Google Scholar]
- Friedman, D.S.; Chew, P.T.; Gazzard, G.; Ang, L.P.; Lai, Y.F.; Quigley, H.A.; Seah, S.K.; Aung, T. Long-term outcomes in fellow eyes after acute primary angle closure in the contralateral eye. Ophthalmology 2006, 113, 1087–1091. [Google Scholar] [CrossRef]
- Tesluk, G.C.; Spaeth, G.L. The occurrence of primary open-angle glaucoma in the fellow eye of patients with unilateral angle-cleavage glaucoma. Ophthalmology 1985, 92, 904–911. [Google Scholar] [CrossRef]
- Bakalash, S.; Kipnis, J.; Yoles, E.; Schwartz, M. Resistance of retinal ganglion cells to an increase in intraocular pressure is immune-dependent. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2648–2653. [Google Scholar]
- Pfister, G.; Stroh, C.M.; Perschinka, H.; Kind, M.; Knoflach, M.; Hinterdorfer, P.; Wick, G. Detection of HSP60 on the membrane surface of stressed human endothelial cells by atomic force and confocal microscopy. J. Cell Sci. 2005, 118, 1587–1594. [Google Scholar] [CrossRef]
- Sakai, M.; Sakai, H.; Nakamura, Y.; Fukuchi, T.; Sawaguchi, S. Immunolocalization of heat shock proteins in the retina of normal monkey eyes and monkey eyes with laser-induced glaucoma. Jpn. J. Ophthalmol. 2003, 47, 42–52. [Google Scholar] [CrossRef]
- Bell, K.; Gramlich, O.W.; Von Thun Und Hohenstein-Blaul, N.; Beck, S.; Funke, S.; Wilding, C.; Pfeiffer, N.; Grus, F.H. Does autoimmunity play a part in the pathogenesis of glaucoma? Prog. Retin. Eye Res. 2013, 36, 199–216. [Google Scholar] [CrossRef]
- Yang, X.; Zeng, Q.; Goktas, E.; Gopal, K.; Al-Aswad, L.; Blumberg, D.M.; Cioffi, G.A.; Liebmann, J.M.; Tezel, G. T-Lymphocyte Subset Distribution and Activity in Patients with Glaucoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 877–888. [Google Scholar] [CrossRef]
- Jiang, B.; Harper, M.M.; Kecova, H.; Adamus, G.; Kardon, R.H.; Grozdanic, S.D.; Kuehn, M.H. Neuroinflammation in advanced canine glaucoma. Mol. Vis. 2010, 16, 2092–2108. [Google Scholar] [PubMed]
- Cebula, A.; Kuczma, M.; Szurek, E.; Pietrzak, M.; Savage, N.; Elhefnawy, W.R.; Rempala, G.; Kraj, P.; Ignatowicz, L. Dormant pathogenic CD4(+) T cells are prevalent in the peripheral repertoire of healthy mice. Nat. Commun. 2019, 10, 4882. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.; Holz, A.; Ludwig, K.; Pfeiffer, N.; Grus, F.H. Elevated Regulatory T Cell Levels in Glaucoma Patients in Comparison to Healthy Controls. Curr. Eye Res. 2017, 42, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Gramlich, O.W.; Beck, S.; von Thun Und Hohenstein-Blaul, N.; Boehm, N.; Ziegler, A.; Vetter, J.M.; Pfeiffer, N.; Grus, F.H. Enhanced insight into the autoimmune component of glaucoma: IgG autoantibody accumulation and pro-inflammatory conditions in human glaucomatous retina. PLoS ONE 2013, 8, e57557. [Google Scholar] [CrossRef]
- Jaczewska, J.; Abdulreda, M.H.; Yau, C.Y.; Schmitt, M.M.; Schubert, I.; Berggren, P.O.; Weber, C.; Koenen, R.R.; Moy, V.T.; Wojcikiewicz, E.P. TNF-alpha and IFN-gamma promote lymphocyte adhesion to endothelial junctional regions facilitating transendothelial migration. J. Leukoc. Biol. 2014, 95, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Wax, M.B.; Tezel, G.; Yang, J.; Peng, G.; Patil, R.V.; Agarwal, N.; Sappington, R.M.; Calkins, D.J. Induced autoimmunity to heat shock proteins elicits glaucomatous loss of retinal ganglion cell neurons via activated T-cell-derived fas-ligand. J. Neurosci. 2008, 28, 12085–12096. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol. 2010, 120, 277–286. [Google Scholar] [CrossRef]
- Astafurov, K.; Elhawy, E.; Ren, L.; Dong, C.Q.; Igboin, C.; Hyman, L.; Griffen, A.; Mittag, T.; Danias, J. Oral microbiome link to neurodegeneration in glaucoma. PLoS ONE 2014, 9, e104416. [Google Scholar] [CrossRef]
- Roh, M.; Zhang, Y.; Murakami, Y.; Thanos, A.; Lee, S.C.; Vavvas, D.G.; Benowitz, L.I.; Miller, J.W. Etanercept, a widely used inhibitor of tumor necrosis factor-alpha (TNF-alpha), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS ONE 2012, 7, e40065. [Google Scholar] [CrossRef]
- Stein, J.D.; Talwar, N.; Kang, J.H.; Okereke, O.I.; Wiggs, J.L.; Pasquale, L.R. Bupropion use and risk of open-angle glaucoma among enrollees in a large U.S. managed care network. PLoS ONE 2015, 10, e0123682. [Google Scholar] [CrossRef]
- Su, W.; Li, Z.; Jia, Y.; Zhuo, Y. Rapamycin is neuroprotective in a rat chronic hypertensive glaucoma model. PLoS ONE 2014, 9, e99719. [Google Scholar] [CrossRef] [PubMed]
- Stallone, G.; Infante, B.; Di Lorenzo, A.; Rascio, F.; Zaza, G.; Grandaliano, G. mTOR inhibitors effects on regulatory T cells and on dendritic cells. J. Transl. Med. 2016, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- McPherson, S.W.; Heuss, N.D.; Gregerson, D.S. Local “on-demand” generation and function of antigen-specific Foxp3+ regulatory T cells. J. Immunol. 2013, 190, 4971–4981. [Google Scholar] [CrossRef]
- Krishnan, A.; Fei, F.; Jones, A.; Busto, P.; Marshak-Rothstein, A.; Ksander, B.R.; Gregory-Ksander, M. Overexpression of Soluble Fas Ligand following Adeno-Associated Virus Gene Therapy Prevents Retinal Ganglion Cell Death in Chronic and Acute Murine Models of Glaucoma. J. Immunol. 2016, 197, 4626–4638. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Kocab, A.J.; Zacks, D.N.; Marshak-Rothstein, A.; Gregory-Ksander, M. A small peptide antagonist of the Fas receptor inhibits neuroinflammation and prevents axon degeneration and retinal ganglion cell death in an inducible mouse model of glaucoma. J. Neuroinflamm. 2019, 16, 184. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shestopalov, V.I.; Spurlock, M.; Gramlich, O.W.; Kuehn, M.H. Immune Responses in the Glaucomatous Retina: Regulation and Dynamics. Cells 2021, 10, 1973. https://doi.org/10.3390/cells10081973
Shestopalov VI, Spurlock M, Gramlich OW, Kuehn MH. Immune Responses in the Glaucomatous Retina: Regulation and Dynamics. Cells. 2021; 10(8):1973. https://doi.org/10.3390/cells10081973
Chicago/Turabian StyleShestopalov, Valery I., Markus Spurlock, Oliver W. Gramlich, and Markus H. Kuehn. 2021. "Immune Responses in the Glaucomatous Retina: Regulation and Dynamics" Cells 10, no. 8: 1973. https://doi.org/10.3390/cells10081973
APA StyleShestopalov, V. I., Spurlock, M., Gramlich, O. W., & Kuehn, M. H. (2021). Immune Responses in the Glaucomatous Retina: Regulation and Dynamics. Cells, 10(8), 1973. https://doi.org/10.3390/cells10081973