
Translating Treg Therapy for Inflammatory Bowel Disease in Humanized Mice

 , ,
, ,
Abstract
:1. Introduction
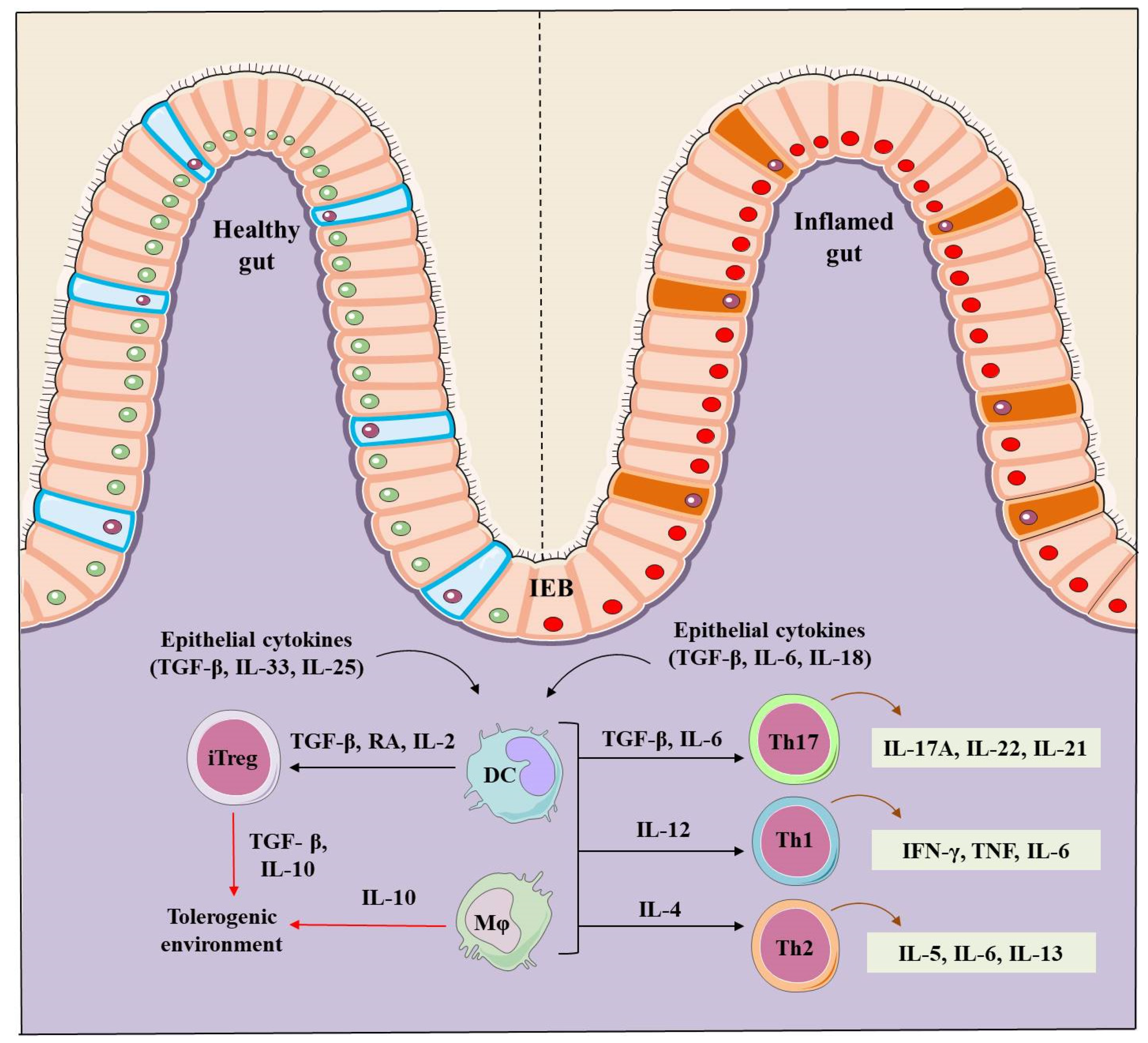
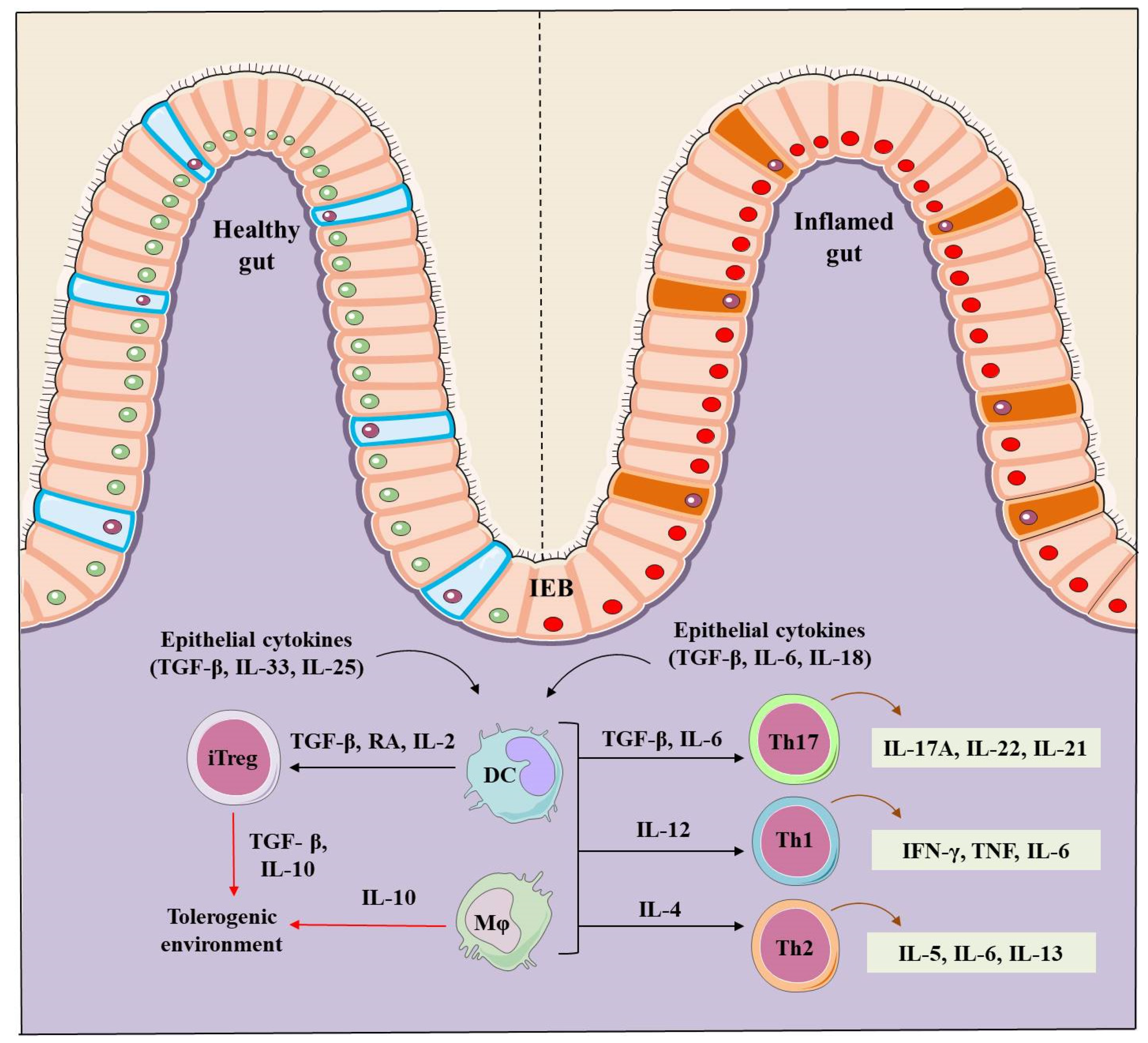
2. IBD Pathogenesis
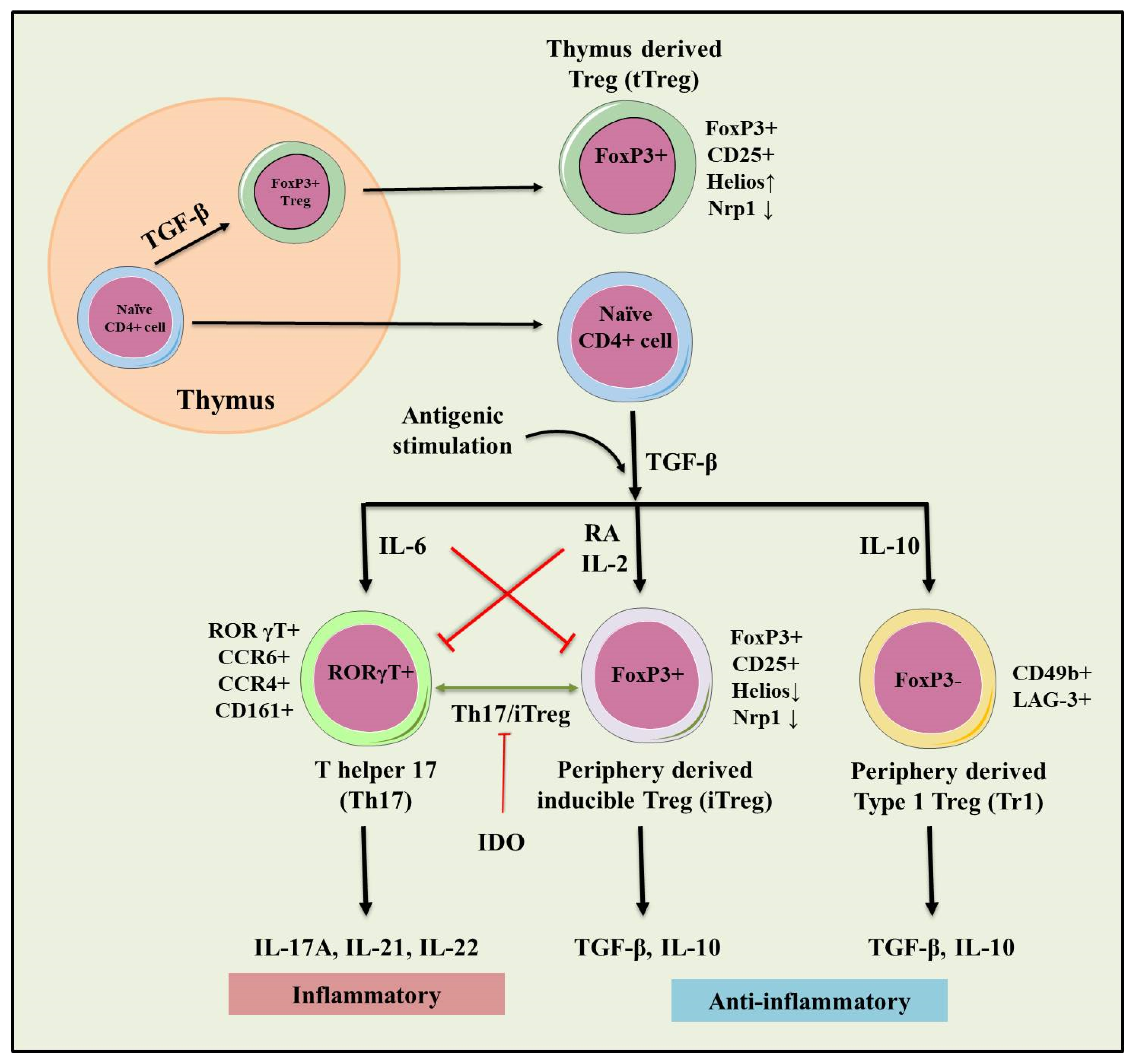
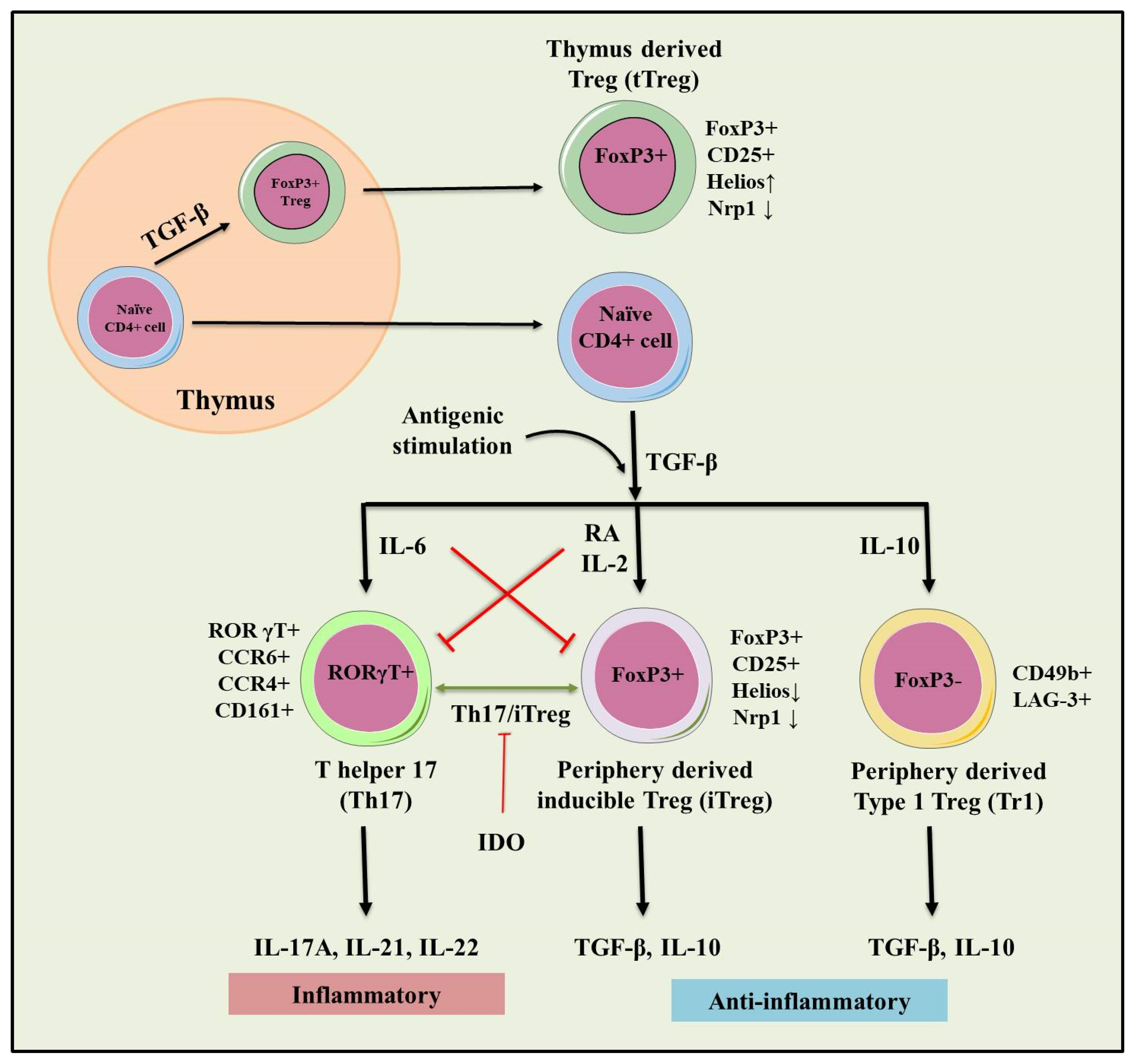
3. Regulatory T Cells (Tregs)
3.1. tTregs and pTre
3.2. Type 1 T regulatory (Tr1) Cells
4. Role of Tregs in IBD
5. Therapeutic Role of Tregs in IBD
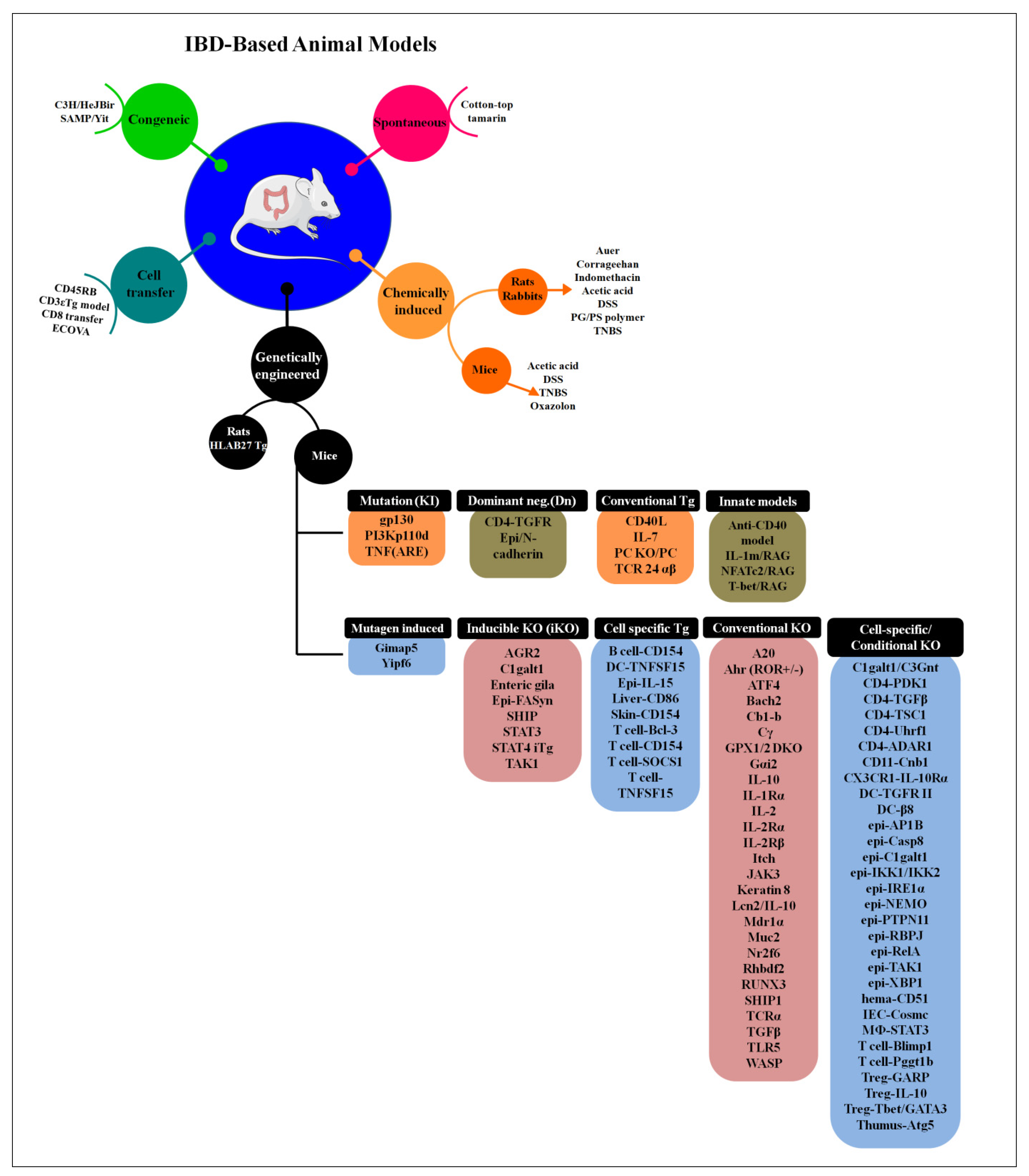
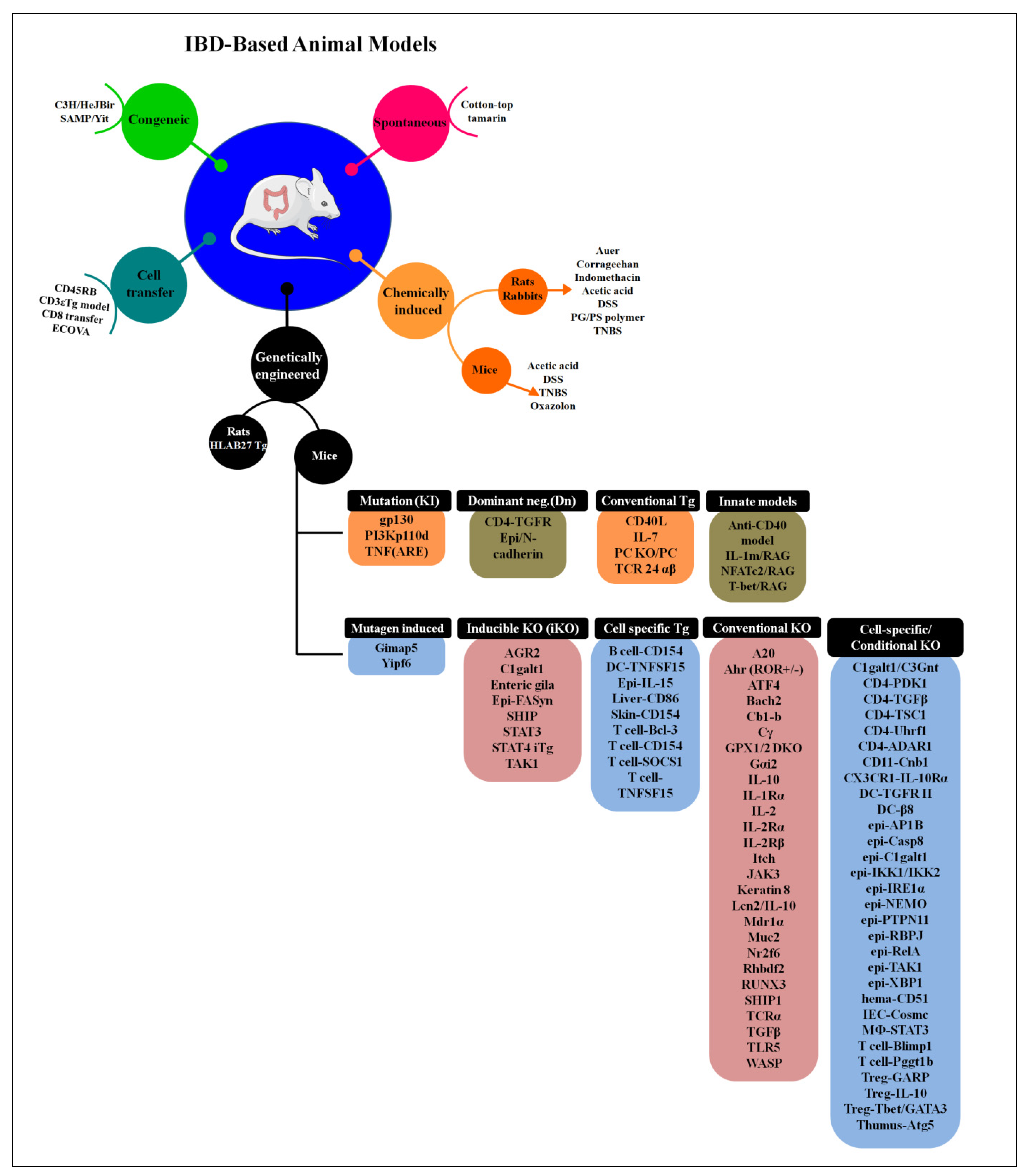
6. Animal Models to Study IBD
6.1. Bacteria-Infected Models
6.2. Genetically Modified Animal Models
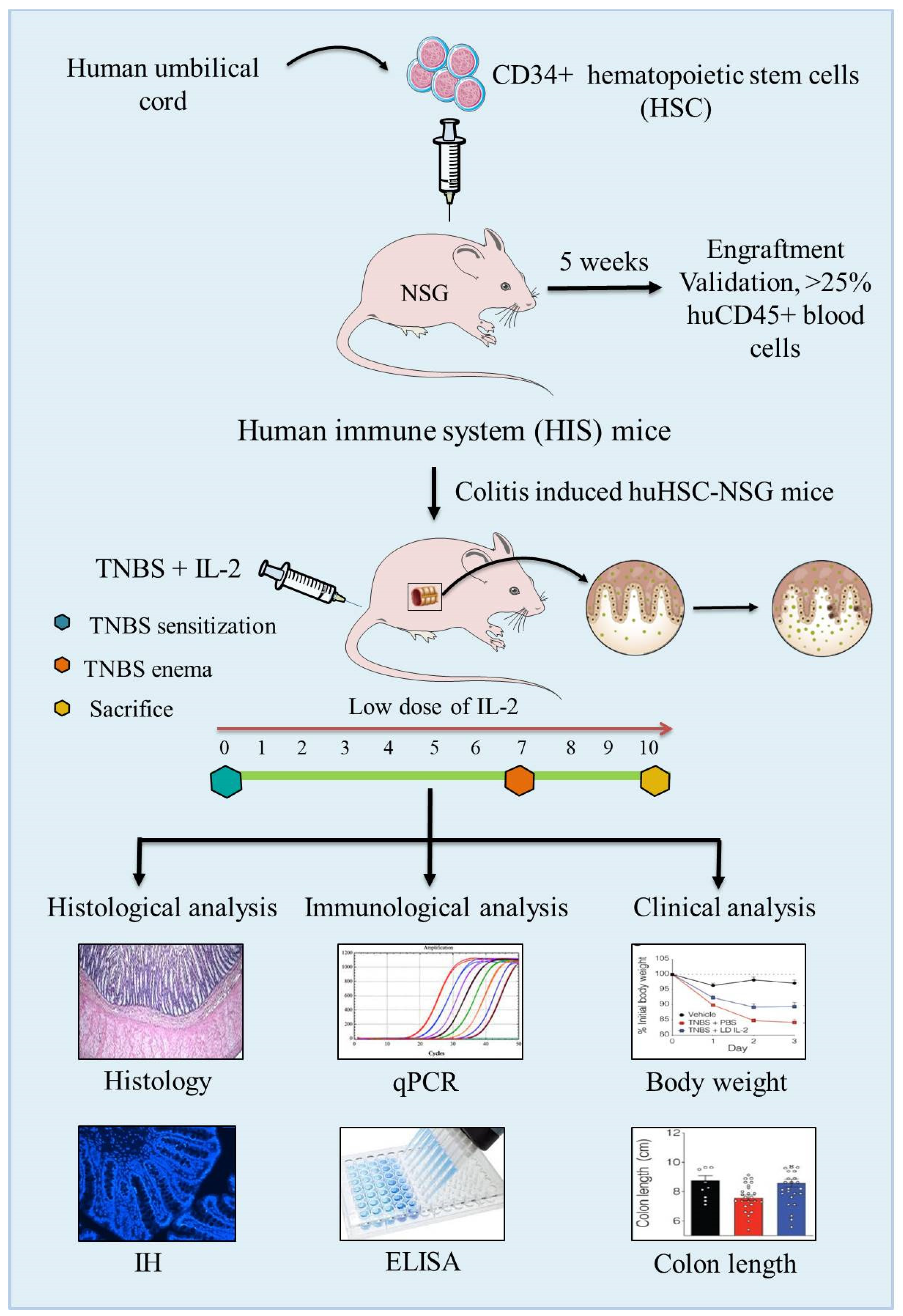
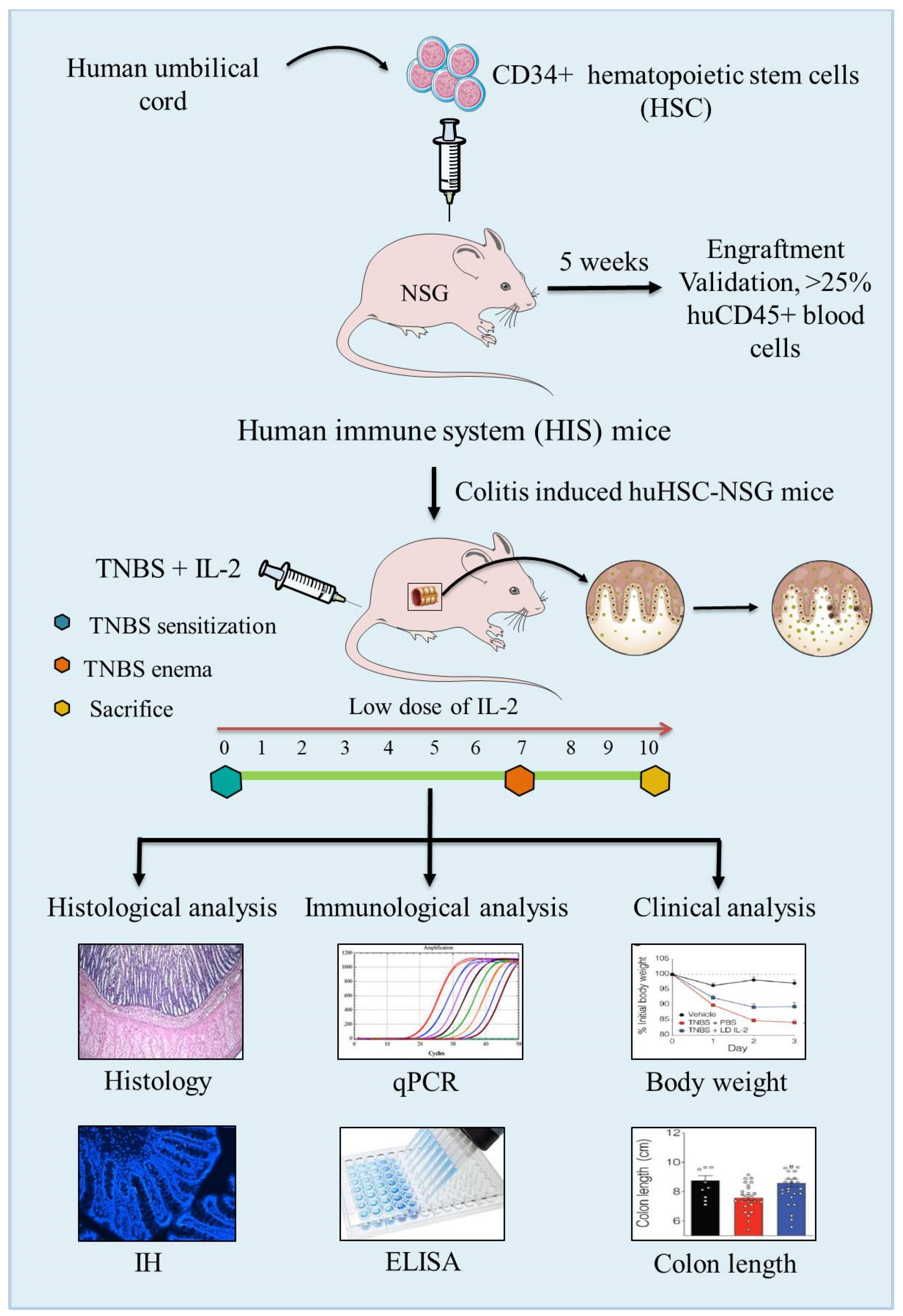
6.3. Humanized Mouse Models
7. Discussion
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IBD | Inflammatory bowel disease |
| Tregs | Regulatory T-cells |
| CD | Crohn’s disease |
| UC | Ulcerative colitis |
| Th cells | T-helper cells |
| HIS | Human immune system |
| DCs | Dendritic cells |
| ILCs | Innate lymphoid cells |
| TNF | Tumor necrosis factor (TNF) |
| IFN-γ | Interferon-gamma |
| TGF-β | Transforming growth factor |
| TLA1 | TNF like cytokine 1A |
| DR3 | Death domain receptor 3 |
| TNBS | 2,4,6-trinitrobenzene sulfonic acid |
| DSS | Dextran sulfate sodium |
| NOD2 | Nucleotide-binding oligomerization domain 2 |
| ATG16L1 | Aumiddlehagy-related 16-like 1 |
| CCR | Chemokine receptor |
| RA | Retinoic acid |
| iTregs | Inducible Treg cells |
| Foxp3 | Forkhead box P3 |
| tTregs | Thymus-derived Treg cells |
| pTregs | Peripheral Treg cells |
| Nrp1 | Neuropilin-1 |
| TSDR | Treg specific demethylated region |
| Tr1 | Type 1 T-regulatory cells |
| GATA3 | GATA binding protein 3 |
| GM | Genetically modified |
| TLR | Toll-like receptor |
| HSCs | Hemamiddleoietic stem cells |
References
- Seyedian, S.S.; Nokhostin, F.; Malamir, M.D. A review of the diagnosis, prevention, and treatment methods of inflammatory bowel disease. J. Med. Life 2019, 12, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Yeshi, K.; Ruscher, R.; Hunter, L.; Daly, N.L.; Loukas, A.; Wangchuk, P. Revisiting Inflammatory Bowel Disease: Pathology, Treatments, Challenges and Emerging Therapeutics Including Drug Leads from Natural Products. J. Clin. Med. 2020, 9, 1273. [Google Scholar] [CrossRef]
- de Souza, H.S.P.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef] [Green Version]
- Loddo, I.; Romano, C. Inflammatory Bowel Disease: Genetics, Epigenetics, and Pathogenesis. Front. Immunol. 2015, 6, 551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imam, T.; Park, S.; Kaplan, M.H.; Olson, M.R. Effector T Helper Cell Subsets in Inflammatory Bowel Diseases. Front. Immunol. 2018, 9, 1212. [Google Scholar] [CrossRef] [PubMed]
- Tindemans, I.; Joosse, M.E.; Samsom, J.N. Dissecting the heterogeneity in T-cell mediated inflammation in IBD. Cells 2020, 9, 110. [Google Scholar] [CrossRef] [Green Version]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S. Naturally arising Foxp3− expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat. Immunol. 2005, 6, 345–352. [Google Scholar] [CrossRef]
- Pereira, L.M.S.; Gomes, S.T.M.; Ishak, R.; Vallinoto, A.C.R. Regulatory T Cell and Forkhead Box Protein 3 as Modulators of Immune Homeostasis. Front. Immunol. 2017, 8, 605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Hu, B.; Xu, D.; Liew, F.Y. CD4+CD25+ regulatory T cells cure murine colitis: The role of IL-10, TGF-beta, and CTLA4. J. Immunol. 2003, 171, 5012–5017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wing, K.; Sakaguchi, S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat. Immunol. 2010, 11, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Fanelli, G.; Albany, C.J.; Giganti, G.; Lombardi, G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Mohammadnia-Afrouzi, M.; Hosseini, A.; Khalili, A.; Abediankenari, S.; Hosseini, V.; Maleki, I. Decrease of CD4(+) CD25(+) CD127(low) Foxp3(+) regulatory T cells with impaired suppressive function in untreated ulcerative colitis patients. Autoimmunity 2015, 48, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Boschetti, G.; Kanjarawi, R.; Bardel, E.; Collardeau-Frachon, S.; Duclaux-Loras, R.; Moro-Sibilot, L.; Almeras, T.; Flourié, B.; Nancey, S.; Kaiserlian, D. Gut Inflammation in Mice Triggers Proliferation and Function of Mucosal Foxp3+ Regulatory T Cells but Impairs Their Conversion from CD4+ T Cells. J. Crohns. Colitis. 2016, 11, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sznurkowska, K.; Luty, J.; Bryl, E.; Witkowski, J.M.; Hermann-Okoniewska, B.; Landowski, P.; Kosek, M.; Szlagatys-Sidorkiewicz, A. Enhancement of Circulating and Intestinal T Regulatory Cells and Their Expression of Helios and Neuropilin-1 in Children with Inflammatory Bowel Disease. J. Inflamm. Res. 2020, 13, 995–1005. [Google Scholar] [CrossRef]
- Bacchetta, R.; Barzaghi, F.; Roncarolo, M.-G. From IPEX syndrome to FOXP3 mutation: A lesson on immune dysregulation. Ann. N. Y. Acad. Sci. 2018, 1417, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Pedros, C.; Duguet, F.; Saoudi, A.; Chabod, M. Disrupted regulatory T cell homeostasis in inflammatory bowel diseases. World J. Gastroenterol. 2016, 22, 974–995. [Google Scholar] [CrossRef]
- Mayne, C.G.; Williams, C.B. Induced and natural regulatory T cells in the development of inflammatory bowel disease. Inflamm. Bowel Dis. 2013, 19, 1772–1788. [Google Scholar] [CrossRef] [Green Version]
- Rocamora-Reverte, L.; Tuzlak, S.; von Raffay, L.; Tisch, M.; Fiegl, H.; Drach, M.; Reichardt, H.M.; Villunger, A.; Tischner, D.; Wiegers, G.J. Glucocorticoid receptor-deficient Foxp3+ regulatory T cells fail to control experimental inflammatory bowel disease. Front. Immunol. 2019, 10, 472. [Google Scholar] [CrossRef]
- Giuffrida, P.; Cococcia, S.; Delliponti, M.; Lenti, M.V.; Di Sabatino, A. Controlling gut inflammation by restoring anti-inflammatory pathways in inflammatory bowel disease. Cells 2019, 8, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clough, J.N.; Omer, O.S.; Tasker, S.; Lord, G.M.; Irving, P.M. Regulatory T-cell therapy in Crohn’s disease: Challenges and advances. Gut 2020, 69, 942–952. [Google Scholar] [CrossRef] [Green Version]
- Mottet, C.; Uhlig, H.H.; Powrie, F. Cutting edge: Cure of colitis by CD4+CD25+ regulatory T cells. J. Immunol. 2003, 170, 3939–3943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canavan, J.B.; Scottà, C.; Vossenkämper, A.; Goldberg, R.; Elder, M.J.; Shoval, I.; Marks, E.; Stolarczyk, E.; Lo, J.W.; Powell, N.; et al. Developing in vitro expanded CD45RA+ regulatory T cells as an adoptive cell therapy for Crohn’s disease. Gut 2016, 65, 584–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iftekhar, A.; Sigal, M. Defence and adaptation mechanisms of the intestinal epithelium upon infection. Int. J. Med. Microbiol. 2021, 311, 151486. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.O. Fight them or feed them: How the intestinal mucus layer manages the gut microbiota. Gastroenterol. Rep. 2019, 7, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakshani, C.R.; Morales-Garcia, A.L.; Althaus, M.; Wilcox, M.D.; Pearson, J.P.; Bythell, J.C.; Burgess, J.G. Evolutionary conservation of the antimicrobial function of mucus: A first defence against infection. npj Biofilms Microbiomes 2018, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Zhang, J. Role of intestinal microbiota and metabolites on gut homeostasis and human diseases. Bmc Immunol. 2017, 18, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagg, A.J. Intestinal dendritic cells in health and gut inflammation. Front. Immunol. 2018, 9, 2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denning, T.L.; Wang, Y.C.; Patel, S.R.; Williams, I.R.; Pulendran, B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 2007, 8, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ye, Q.; Zeng, X.; Qiao, S. Functions of macrophages in the maintenance of intestinal homeostasis. J. Immunol. Res. 2019, 2019, 1512969. [Google Scholar] [CrossRef] [Green Version]
- Pandiyan, P.; Bhaskaran, N.; Zou, M.; Schneider, E.; Jayaraman, S.; Huehn, J. Microbiome dependent regulation of Tregs and Th17 cells in mucosa. Front. Immunol. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef] [PubMed]
- Okumura, R.; Takeda, K. Maintenance of gut homeostasis by the mucosal immune system. Proc. Jpn. Acad. Ser. B 2016, 92, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Yue, B.; Luo, X.; Yu, Z.; Mani, S.; Wang, Z.; Dou, W. Inflammatory bowel disease: A potential result from the collusion between gut microbiota and mucosal immune system. Microorganisms 2019, 7, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Chen, Z. Inflammatory bowel disease related innate immunity and adaptive immunity. Am. J. Transl. Res. 2016, 8, 2490–2497. [Google Scholar] [PubMed]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine networks in the pathophysiology of inflammatory bowel disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Guan, Q.; Zhang, J. Recent Advances: The Imbalance of Cytokines in the Pathogenesis of Inflammatory Bowel Disease. Mediat. Inflamm. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, N.; Hisamatsu, T.; Honda, H.; Kobayashi, T.; Chinen, H.; Takayama, T.; Kitazume, M.; Okamoto, S.; Koganei, K.; Sugita, A.; et al. TL1A Produced by Lamina Propria Macrophages Induces Th1 and Th17 Immune Responses in Cooperation with IL-23 in Patients with Crohn’s Disease. Inflamm. Bowel Dis. 2010, 16, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Sidhu-Varma, M.; Shih, D.Q.; Targan, S.R. Differential Levels of Tl1a Affect the Expansion and Function of Regulatory T Cells in Modulating Murine Colitis. Inflamm. Bowel Dis. 2016, 22, 548–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takedatsu, H.; Michelsen, K.S.; Wei, B.; Landers, C.J.; Thomas, L.S.; Dhall, D.; Braun, J.; Targan, S.R. TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T-helper 1 and T-helper 17 activation. Gastroenterology 2008, 135, 552–567. [Google Scholar] [CrossRef] [Green Version]
- Meylan, F.; Song, Y.J.; Fuss, I.; Villarreal, S.; Kahle, E.; Malm, I.J.; Acharya, K.; Ramos, H.L.; Lo, L.; Mentink-Kane, M.M.; et al. The TNF-family cytokine TL1A drives IL-13-dependent small intestinal inflammation. Mucosal. Immunol. 2011, 4, 172–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Kim, N.-M.; Jiang, Y.-W.; Zhang, H.; Zheng, D.; Zhu, F.-X.; Liang, R.; Li, B.; Xu, H.-X. Cambogin suppresses dextran sulphate sodium-induced colitis by enhancing Treg cell stability and function. Br. J. Pharm. 2018, 175, 1085–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, M.H.; Kamada, N.; Warner, N.; Kim, Y.-G.; Nuñez, G. The ever-expanding function of NOD2: Autophagy, viral recognition, and T cell activation. Trends Immunol. 2011, 32, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuballa, P.; Huett, A.; Rioux, J.D.; Daly, M.J.; Xavier, R.J. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS ONE 2008, 3, e3391. [Google Scholar] [CrossRef]
- Cooney, R.; Baker, J.; Brain, O.; Danis, B.; Pichulik, T.; Allan, P.; Ferguson, D.J.; Campbell, B.J.; Jewell, D.; Simmons, A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 2010, 16, 90–97. [Google Scholar] [CrossRef]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.-G.; Magalhães, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Barreau, F.; Meinzer, U.; Chareyre, F.; Berrebi, D.; Niwa-Kawakita, M.; Dussaillant, M.; Foligne, B.; Ollendorff, V.; Heyman, M.; Bonacorsi, S.; et al. CARD15/NOD2 is required for Peyer’s patches homeostasis in mice. PLoS ONE 2007, 2, e523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabat, A.; Harrison, O.; Riffelmacher, T.; Moghaddam, A.E.; Pearson, C.; Laing, A.; Abeler-Dörner, L.; Forman, S.; Grencis, R.; Sattentau, Q.; et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. eLife 2016, 5, e12444. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Arakaki, R.; Saito, M.; Tsunematsu, T.; Kudo, Y.; Ishimaru, N. Role of regulatory T cell in the pathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.A.; Meingassner, J.G.; Lipp, M.; Moore, H.D.; Rot, A. CCR7 is required for the in vivo function of CD4+ CD25+ regulatory T cells. J. Exp. Med. 2007, 204, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Bromley, S.K.; Means, T.K.; Jones, K.J.; Hayashi, F.; Bhan, A.K.; Luster, A.D. CCR4-dependent regulatory T cell function in inflammatory bowel disease. J. Exp. Med. 2007, 204, 1327–1334. [Google Scholar] [CrossRef] [Green Version]
- McNamee, E.N.; Masterson, J.C.; Veny, M.; Collins, C.B.; Jedlicka, P.; Byrne, F.R.; Ng, G.Y.; Rivera-Nieves, J. Chemokine receptor CCR7 regulates the intestinal TH1/TH17/Treg balance during Crohn’s-like murine ileitis. J. Leukoc. Biol. 2015, 97, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Kuk, W.; Rivera-Nieves, J.; Lopez-Ramirez, M.A.; Eckmann, L.; Ginsberg, M.H. β7 Integrin Inhibition Can Increase Intestinal Inflammation by Impairing Homing of CD25(hi)Foxp3(+) Regulatory T Cells. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 369–385. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, D.; Tsun, A.; Li, B. Foxp3+ regulatory T cells and their functional regulation. Cell Mol. Immunol. 2015, 12, 558–565. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription FactorFoxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Putnam, A.L.; Xu-Yu, Z.; Szot, G.L.; Lee, M.R.; Zhu, S.; Gottlieb, P.A.; Kapranov, P.; Gingeras, T.R.; Fazekas de St Groth, B.; et al. CD127 expression inversely correlates with Foxp3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 2006, 203, 1701–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trzonkowski, P.; Bieniaszewska, M.; Juścińska, J.; Dobyszuk, A.; Krzystyniak, A.; Marek, N.; Myśliwska, J.; Hellmann, A. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+ CD25+ CD127−T regulatory cells. Clin. Immunol. 2009, 133, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Haribhai, D.; Williams, J.B.; Jia, S.; Nickerson, D.; Schmitt, E.G.; Edwards, B.; Ziegelbauer, J.; Yassai, M.; Li, S.-H.; Relland, L.M. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity 2011, 35, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Josefowicz, S.Z.; Niec, R.E.; Kim, H.Y.; Treuting, P.; Chinen, T.; Zheng, Y.; Umetsu, D.T.; Rudensky, A.Y. Extrathymically generated regulatory T cells control mucosal TH 2 inflammation. Nature 2012, 482, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Darrigues, J.; van Meerwijk, J.P.; Romagnoli, P. Age-dependent changes in regulatory T lymphocyte development and function: A mini-review. Gerontology 2018, 64, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Fujikado, N.; Kolodin, D.; Benoist, C.; Mathis, D. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 2015, 348, 589–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, W.; Beckett, O.; Ma, Q.; Li, M.O. Transforming growth factor-β signaling curbs thymic negative selection promoting regulatory T cell development. Immunity 2010, 32, 642–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretschmer, K.; Apostolou, I.; Hawiger, D.; Khazaie, K.; Nussenzweig, M.C.; von Boehmer, H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 2005, 6, 1219–1227. [Google Scholar] [CrossRef]
- Knoechel, B.; Lohr, J.; Kahn, E.; Bluestone, J.A.; Abbas, A.K. Sequential development of interleukin 2–dependent effector and regulatory T cells in response to endogenous systemic antigen. J. Exp. Med. 2005, 202, 1375–1386. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.; Bluestone, J.A.; Stephan, S. Peripherally induced tregs–role in immune homeostasis and autoimmunity. Front. Immunol. 2013, 4, 232. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Josefowicz, S.; Chaudhry, A.; Peng, X.P.; Forbush, K.; Rudensky, A.Y. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 2010, 463, 808–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.-j.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+ CD25− naive T cells to CD4+ CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Schlenner, S.M.; Weigmann, B.; Ruan, Q.; Chen, Y.; von Boehmer, H. Smad3 binding to the Foxp3 enhancer is dispensable for the development of regulatory T cells with the exception of the gut. J. Exp. Med. 2012, 209, 1529–1535. [Google Scholar] [CrossRef] [Green Version]
- Tai, X.; Cowan, M.; Feigenbaum, L.; Singer, A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat. Immunol. 2005, 6, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-M.; Hall, J.A.; Blank, R.B.; Bouladoux, N.; Oukka, M.; Mora, J.R.; Belkaid, Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 2007, 204, 1775–1785. [Google Scholar] [CrossRef] [Green Version]
- Benson, M.J.; Pino-Lagos, K.; Rosemblatt, M.; Noelle, R.J. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J. Exp. Med. 2007, 204, 1765–1774. [Google Scholar] [CrossRef] [Green Version]
- Shevach, E.M.; Thornton, A.M. tTregs, pTregs, and iTregs: Similarities and differences. Immunol. Rev. 2014, 259, 88–102. [Google Scholar] [CrossRef] [Green Version]
- Shevyrev, D.; Tereshchenko, V. Treg heterogeneity, function, and homeostasis. Front. Immunol. 2020, 10, 3100. [Google Scholar] [CrossRef] [Green Version]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef]
- Collison, L.W.; Pillai, M.R.; Chaturvedi, V.; Vignali, D.A.A. Regulatory T cell suppression is potentiated by target T cells in a cell contact, IL-35- and IL-10-dependent manner. J. Immunol. 2009, 182, 6121–6128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, W.J.; Verbsky, J.W.; Barchet, W.; Colonna, M.; Atkinson, J.P.; Ley, T.J. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 2004, 21, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Gondek, D.C.; Lu, L.-F.; Quezada, S.A.; Sakaguchi, S.; Noelle, R.J. Cutting Edge: Contact-Mediated Suppression by CD4+CD25+ Regulatory Cells Involves a Granzyme B-Dependent, Perforin-Independent Mechanism. J. Immunol. 2005, 174, 1783–1786. [Google Scholar] [CrossRef] [Green Version]
- Karreci, E.S.; Eskandari, S.K.; Dotiwala, F.; Routray, S.K.; Kurdi, A.T.; Assaker, J.P.; Luckyanchykov, P.; Mihali, A.B.; Maarouf, O.; Borges, T.J. Human regulatory T cells undergo self-inflicted damage via granzyme pathways upon activation. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Lin, X.; Chen, M.; Liu, Y.; Guo, Z.; He, X.; Brand, D.; Zheng, S.G. Advances in distinguishing natural from induced Foxp3(+) regulatory T cells. Int. J. Clin. Exp. Pathol. 2013, 6, 116–123. [Google Scholar]
- Singh, K.; Hjort, M.; Thorvaldson, L.; Sandler, S. Concomitant analysis of Helios and Neuropilin-1 as a marker to detect thymic derived regulatory T cells in naive mice. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgoffe, G.M.; Woo, S.-R.; Turnis, M.E.; Gravano, D.M.; Guy, C.; Overacre, A.E.; Bettini, M.L.; Vogel, P.; Finkelstein, D.; Bonnevier, J.; et al. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature 2013, 501, 252–256. [Google Scholar] [CrossRef]
- Thornton, A.M.; Korty, P.E.; Tran, D.Q.; Wohlfert, E.A.; Murray, P.E.; Belkaid, Y.; Shevach, E.M. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol. 2010, 184, 3433–3441. [Google Scholar] [CrossRef] [Green Version]
- Gottschalk, R.A.; Corse, E.; Allison, J.P. Expression of Helios in peripherally induced Foxp3+ regulatory T cells. J. Immunol. 2012, 188, 976–980. [Google Scholar] [CrossRef] [Green Version]
- Elkord, E. Helios should not be cited as a marker of human thymus-derived Tregs. Commentary: Helios+ and Helios− cells coexist within the natural Foxp3+ T regulatory cell subset in humans. Front. Immunol. 2016, 7, 276. [Google Scholar] [CrossRef] [Green Version]
- Szurek, E.; Cebula, A.; Wojciech, L.; Pietrzak, M.; Rempala, G.; Kisielow, P.; Ignatowicz, L. Differences in expression level of helios and neuropilin-1 do not distinguish thymus-derived from extrathymically-induced CD4+ Foxp3+ regulatory T cells. PLoS ONE 2015, 10, e0141161. [Google Scholar]
- Lee, S.; Park, K.; Kim, J.; Min, H.; Seong, R.H. Foxp3 expression in induced regulatory T cells is stabilized by C/EBP in inflammatory environments. Embo Rep. 2018, 19, e45995. [Google Scholar] [CrossRef] [PubMed]
- Polansky, J.K.; Kretschmer, K.; Freyer, J.; Floess, S.; Garbe, A.; Baron, U.; Olek, S.; Hamann, A.; von Boehmer, H.; Huehn, J. DNA methylation controls Foxp3 gene expression. Eur. J. Immunol. 2008, 38, 1654–1663. [Google Scholar] [CrossRef] [PubMed]
- Opstelten, R.; de Kivit, S.; Slot, M.C.; van den Biggelaar, M.; Iwaszkiewicz-Grześ, D.; Gliwiński, M.; Scott, A.M.; Blom, B.; Trzonkowski, P.; Borst, J. GPA33: A marker to identify stable human regulatory T cells. J. Immunol. 2020, 204, 3139–3148. [Google Scholar] [CrossRef]
- Levings, M.K.; Gregori, S.; Tresoldi, E.; Cazzaniga, S.; Bonini, C.; Roncarolo, M.G. Differentiation of Tr1 cells by immature dendritic cells requires IL-10 but not CD25+CD4+ Tr cells. Blood 2005, 105, 1162–1169. [Google Scholar] [CrossRef] [Green Version]
- Groux, H.; O’Garra, A.; Bigler, M.; Rouleau, M.; Antonenko, S.; De Vries, J.E.; Roncarolo, M.G. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997, 389, 737–742. [Google Scholar] [CrossRef]
- Zeng, H.; Zhang, R.; Jin, B.; Chen, L. Type 1 regulatory T cells: A new mechanism of peripheral immune tolerance. Cell Mol. Immunol. 2015, 12, 566–571. [Google Scholar] [CrossRef] [Green Version]
- Gagliani, N.; Magnani, C.F.; Huber, S.; Gianolini, M.E.; Pala, M.; Licona-Limon, P.; Guo, B.; De’Broski, R.H.; Bulfone, A.; Trentini, F. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med. 2013, 19, 739. [Google Scholar] [CrossRef]
- Barrat, F.J.; Cua, D.J.; Boonstra, A.; Richards, D.F.; Crain, C.; Savelkoul, H.F.; de Waal-Malefyt, R.; Coffman, R.L.; Hawrylowicz, C.M.; O’Garra, A. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J. Exp. Med. 2002, 195, 603–616. [Google Scholar] [CrossRef]
- Levings, M.K.; Sangregorio, R.; Galbiati, F.; Squadrone, S.; de Waal Malefyt, R.; Roncarolo, M.-G. IFN-α and IL-10 induce the differentiation of human type 1 T regulatory cells. J. Immunol. 2001, 166, 5530–5539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wildin, R.S.; Ramsdell, F.; Peake, J.; Faravelli, F.; Casanova, J.L.; Buist, N.; Levy-Lahad, E.; Mazzella, M.; Goulet, O.; Perroni, L.; et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 2001, 27, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef]
- Boehm, F.; Martin, M.; Kesselring, R.; Schiechl, G.; Geissler, E.K.; Schlitt, H.-J.; Fichtner-Feigl, S. Deletion of Foxp3+ regulatory T cells in genetically targeted mice supports development of intestinal inflammation. BMC Gastroenterol. 2012, 12, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maul, J.; Loddenkemper, C.; Mundt, P.; Berg, E.; Giese, T.; Stallmach, A.; Zeitz, M.; Duchmann, R. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology 2005, 128, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Holmén, N.; Lundgren, A.; Lundin, S.; Bergin, A.-M.; Rudin, A.; Sjövall, H.; Ohman, L. Functional CD4+CD25high regulatory T cells are enriched in the colonic mucosa of patients with active ulcerative colitis and increase with disease activity. Inflamm. Bowel Dis. 2006, 12, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Hovhannisyan, Z.; Treatman, J.; Littman, D.R.; Mayer, L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology 2011, 140, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, X.P.; Zhao, Z.B.; Chen, J.H.; Yu, C.G. Expression of CD4+ forkhead box P3 (FOXP3)+ regulatory T cells in inflammatory bowel disease. J. Dig. Dis 2011, 12, 286–294. [Google Scholar] [CrossRef]
- Eastaff-Leung, N.; Mabarrack, N.; Barbour, A.; Cummins, A.; Barry, S. Foxp3+ Regulatory T Cells, Th17 Effector Cells, and Cytokine Environment in Inflammatory Bowel Disease. J. Clin. Immunol. 2009, 30, 80–89. [Google Scholar] [CrossRef]
- Li, J.; Ueno, A.; Iacucci, M.; Fort Gasia, M.; Jijon, H.B.; Panaccione, R.; Kaplan, G.G.; Beck, P.L.; Luider, J.; Barkema, H.W.; et al. Crossover Subsets of CD4+ T Lymphocytes in the Intestinal Lamina Propria of Patients with Crohn’s Disease and Ulcerative Colitis. Dig. Dis. Sci. 2017, 62, 2357–2368. [Google Scholar] [CrossRef]
- Ueno, A.; Jijon, H.; Chan, R.; Ford, K.; Hirota, C.; Kaplan, G.G.; Beck, P.L.; Iacucci, M.; Fort Gasia, M.; Barkema, H.W.; et al. Increased prevalence of circulating novel IL-17 secreting Foxp3 expressing CD4+ T cells and defective suppressive function of circulating Foxp3+ regulatory cells support plasticity between Th17 and regulatory T cells in inflammatory bowel disease patients. Inflamm. Bowel Dis. 2013, 19, 2522–2534. [Google Scholar] [CrossRef]
- Yang, B.H.; Hagemann, S.; Mamareli, P.; Lauer, U.; Hoffmann, U.; Beckstette, M.; Föhse, L.; Prinz, I.; Pezoldt, J.; Suerbaum, S.; et al. Foxp3+ T cells expressing RORγt represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol. 2016, 9, 444–457. [Google Scholar] [CrossRef]
- Di Giovangiulio, M.; Rizzo, A.; Franzè, E.; Caprioli, F.; Facciotti, F.; Onali, S.; Favale, A.; Stolfi, C.; Fehling, H.-J.; Monteleone, G.; et al. Tbet Expression in Regulatory T Cells Is Required to Initiate Th1-Mediated Colitis. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Ogino, H.; Nakamura, K.; Ihara, E.; Akiho, H.; Takayanagi, R. CD4+CD25+ Regulatory T Cells Suppress Th17-Responses in an Experimental Colitis Model. Dig. Dis. Sci. 2011, 56, 376–386. [Google Scholar] [CrossRef]
- Uhlig, H.H.; Coombes, J.; Mottet, C.; Izcue, A.; Thompson, C.; Fanger, A.; Tannapfel, A.; Fontenot, J.D.; Ramsdell, F.; Powrie, F. Characterization of Foxp3+ CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J. Immunol. 2006, 177, 5852–5860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, O.J.; Srinivasan, N.; Pott, J.; Schiering, C.; Krausgruber, T.; Ilott, N.E.; Maloy, K.J. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3+ Treg cell function in the intestine. Mucosal Immunol. 2015, 8, 1226–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, B.; Banz, A.; Bienvenu, B.; Cordier, C.; Dautigny, N.; Bécourt, C.; Lucas, B. Suppression of CD4+ T lymphocyte effector functions by CD4+ CD25+ cells in vivo. J. Immunol. 2004, 172, 3391–3398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin–EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, L.; Stahl, M.; Han, X.; Nazli, A.; MacDonald, K.N.; Wong, M.Q.; Tsai, K.; Dizzell, S.; Jacobson, K.; Bressler, B. Suppressive and gut-reparative functions of human type 1 T regulatory cells. Gastroenterology 2019, 157, 1584–1598. [Google Scholar] [CrossRef] [Green Version]
- Ananthakrishnan, A.N.; Cagan, A.; Cai, T.; Gainer, V.S.; Shaw, S.Y.; Savova, G.; Churchill, S.; Karlson, E.W.; Kohane, I.; Liao, K.P. Comparative effectiveness of infliximab and adalimumab in Crohn’s disease and ulcerative colitis. Inflamm. Bowel Dis. 2016, 22, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Vulliemoz, M.; Brand, S.; Juillerat, P.; Mottet, C.; Ben-Horin, S.; Michetti, P. TNF-Alpha Blockers in Inflammatory Bowel Diseases: Practical Recommendations and a User’s Guide: An Update. Digestion 2020, 101, 20–30. [Google Scholar] [CrossRef]
- Ford, A.C.; Peyrin-Biroulet, L. Opportunistic Infections with Anti-Tumor Necrosis Factor-α Therapy in Inflammatory Bowel Disease: Meta-Analysis of Randomized Controlled Trials. Am. J. Gastroenterol. 2013, 108, 1268–1276. [Google Scholar] [CrossRef]
- Fellermann, K. Adverse events of tumor necrosis factor inhibitors. Dig. Dis. 2013, 31, 374–378. [Google Scholar] [CrossRef]
- Kirchgesner, J.; Lemaitre, M.; Carrat, F.; Zureik, M.; Carbonnel, F.; Dray-Spira, R. Risk of serious and opportunistic infections associated with treatment of inflammatory bowel diseases. Gastroenterology 2018, 155, 337–346.e310. [Google Scholar] [CrossRef] [Green Version]
- Deepak, P.; Sifuentes, H.; Sherid, M.; Stobaugh, D.; Sadozai, Y.; Ehrenpreis, E.D. T-cell non-Hodgkin’s lymphomas reported to the FDA AERS with tumor necrosis factor-alpha (TNF-α) inhibitors: Results of the REFURBISH study. Off. J. Am. Coll. Gastroenterol. ACG 2013, 108, 99–105. [Google Scholar] [CrossRef]
- Lemaitre, M.; Kirchgesner, J.; Rudnichi, A.; Carrat, F.; Zureik, M.; Carbonnel, F.; Dray-Spira, R. Association between use of thiopurines or tumor necrosis factor antagonists alone or in combination and risk of lymphoma in patients with inflammatory bowel disease. JAMA 2017, 318, 1679–1686. [Google Scholar] [CrossRef]
- Eickstaedt, J.B.; Killpack, L.; Tung, J.; Davis, D.; Hand, J.L.; Tollefson, M.M. Psoriasis and psoriasiform eruptions in pediatric patients with inflammatory bowel disease treated with anti–tumor necrosis factor alpha agents. Pediatric Dermatol. 2017, 34, 253–260. [Google Scholar] [CrossRef]
- Decock, A.; Van Assche, G.; Vermeire, S.; Wuyts, W.; Ferrante, M. Sarcoidosis-like lesions: Another paradoxical reaction to anti-TNF therapy? J. Crohns. Colitis 2017, 11, 378–383. [Google Scholar] [CrossRef]
- Alivernini, S.; Pugliese, D.; Tolusso, B.; Bui, L.; Petricca, L.; Guidi, L.; Mirone, L.; Rapaccini, G.L.; Federico, F.; Ferraccioli, G. Paradoxical arthritis occurring during anti-TNF in patients with inflammatory bowel disease: Histological and immunological features of a complex synovitis. Rmd Open 2018, 4, e000667. [Google Scholar] [CrossRef]
- Perše, M.; Unkovič, A. The role of TNF in the pathogenesis of inflammatory bowel disease. In Biological Therapy for Inflammatory Bowel Disease; IntechOpen: London, UK, 2019. [Google Scholar]
- Shovman, O.; Tamar, S.; Amital, H.; Watad, A.; Shoenfeld, Y. Diverse patterns of anti-TNF-α-induced lupus: Case series and review of the literature. Clin. Rheumatol. 2018, 37, 563–568. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Wolf, D.C.; D’Haens, G.; Vermeire, S.; Hanauer, S.B.; Ghosh, S.; Smith, H.; Cravets, M.; Frohna, P.A. Ozanimod induction and maintenance treatment for ulcerative colitis. N. Engl. J. Med. 2016, 374, 1754–1762. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Sands, B.E.; Danese, S.; D’Haens, G.R.; Vermeire, S.; Schreiber, S.; Feagan, B.G.; Reinisch, W.; Friedman, G.; Woodworth, D.A. Efficacy and safety of oral tofacitinib as maintenance therapy in patients with moderate to severe ulcerative colitis: Results from a phase 3 randomised controlled trial. Gastroenterology 2017, 152, S199. [Google Scholar] [CrossRef]
- D’Haens, G.R.; Panaccione, R.; Higgins, P.; Colombel, J.-F.; Feagan, B.G.; Moscariello, M.; Chan, G.; Healey, P.J.; Niezychowski, W.; Wang, W. 856 Efficacy and safety of oral tofacitinib for maintenance therapy in patients with moderate to severe Crohn’s disease: Results of a phase 2B randomized placebo-controlled trial. Gastroenterology 2016, 150, S183. [Google Scholar] [CrossRef]
- Villablanca, E.J.; Cassani, B.; Von Andrian, U.H.; Mora, J.R. Blocking lymphocyte localization to the gastrointestinal mucosa as a therapeutic strategy for inflammatory bowel diseases. Gastroenterology 2011, 140, 1776–1784.e1775. [Google Scholar] [CrossRef] [Green Version]
- Feagan, B.G.; Rutgeerts, P.; Sands, B.E.; Hanauer, S.; Colombel, J.-F.; Sandborn, W.J.; Van Assche, G.; Axler, J.; Kim, H.-J.; Danese, S. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2013, 369, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Sands, B.E.; Feagan, B.G.; Rutgeerts, P.; Colombel, J.-F.; Sandborn, W.J.; Sy, R.; D’Haens, G.; Ben-Horin, S.; Xu, J.; Rosario, M. Effects of vedolizumab induction therapy for patients with Crohn’s disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology 2014, 147, 618–627.e613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombel, J.-F.; Sands, B.E.; Rutgeerts, P.; Sandborn, W.; Danese, S.; D’Haens, G.; Panaccione, R.; Loftus, E.V.; Sankoh, S.; Fox, I. The safety of vedolizumab for ulcerative colitis and Crohn’s disease. Gut 2017, 66, 839–851. [Google Scholar] [CrossRef]
- Loftus Jr, E.V.; Colombel, J.-F.; Feagan, B.G.; Vermeire, S.; Sandborn, W.J.; Sands, B.E.; Danese, S.; D’Haens, G.R.; Kaser, A.; Panaccione, R. Long-term efficacy of vedolizumab for ulcerative colitis. J. Crohns. Colitis 2017, 11, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Monteleone, G.; Neurath, M.F.; Ardizzone, S.; Di Sabatino, A.; Fantini, M.C.; Castiglione, F.; Scribano, M.L.; Armuzzi, A.; Caprioli, F.; Sturniolo, G.C.; et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N. Engl. J. Med. 2015, 372, 1104–1113. [Google Scholar] [CrossRef] [Green Version]
- Marafini, I.; Monteleone, I.; Dinallo, V.; Di Fusco, D.; De Simone, V.; Laudisi, F.; Fantini, M.C.; Di Sabatino, A.; Pallone, F.; Monteleone, G. CCL20 is negatively regulated by TGF-β1 in intestinal epithelial cells and reduced in Crohn’s disease patients with a successful response to Mongersen, a Smad7 antisense oligonucleotide. J. Crohns. Colitis 2017, 11, 603–609. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sands, B.E.; Rossiter, G.; Li, X.; Usiskin, K.; Zhan, X.; Colombel, J.-F. Effects of mongersen (GED-0301) on endoscopic and clinical outcomes in patients with active Crohn’s disease. Gastroenterology 2018, 154, 61–64.e66. [Google Scholar] [CrossRef]
- Danese, S.; Furfaro, F.; Vetrano, S. Targeting S1P in inflammatory bowel disease: New avenues for modulating intestinal leukocyte migration. J. Crohns. Colitis 2018, 12, S678–S686. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, C.L.; Kimball, A.B.; Papp, K.A.; Yeilding, N.; Guzzo, C.; Wang, Y.; Li, S.; Dooley, L.T.; Gordon, K.B.; Investigators, P.S. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008, 371, 1665–1674. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Fedorak, R.N.; Scherl, E.; Fleisher, M.R.; Katz, S.; Johanns, J.; Blank, M.; Rutgeerts, P.; Ustekinumab Crohn’s Disease Study Group. A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn’s disease. Gastroenterology 2008, 135, 1130–1141. [Google Scholar] [CrossRef] [Green Version]
- Sandborn, W.J.; Gasink, C.; Gao, L.-L.; Blank, M.A.; Johanns, J.; Guzzo, C.; Sands, B.E.; Hanauer, S.B.; Targan, S.; Rutgeerts, P. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N. Engl. J. Med. 2012, 367, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Hibi, T.; Imai, Y.; Murata, Y.; Matsushima, N.; Zheng, R.; Gasink, C. Efficacy and safety of ustekinumab in Japanese patients with moderately to severely active Crohn’s disease: A subpopulation analysis of phase 3 induction and maintenance studies. Intest Res. 2017, 15, 475. [Google Scholar] [CrossRef] [Green Version]
- Nigam, G.B.; Limdi, J.K. An update on the role of anti-IL-12/IL23 agents in the management of inflammatory bowel disease. Br. Med. Bull. 2021, 138, 29–40. [Google Scholar] [CrossRef]
- Jefremow, A.; Neurath, M.F. All are Equal, Some are More Equal: Targeting IL 12 and 23 in IBD—A Clinical Perspective. Immunotargets Ther. 2020, 9, 289. [Google Scholar] [CrossRef]
- Kashani, A.; Schwartz, D.A. The Expanding Role of Anti–IL-12 and/or Anti–IL-23 Antibodies in the Treatment of Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2019, 15, 255. [Google Scholar]
- Misselwitz, B.; Juillerat, P.; Sulz, M.C.; Siegmund, B.; Brand, S. Emerging treatment options in inflammatory Bowel disease: Janus Kinases, stem cells, and more. Digestion 2020, 101, 69–82. [Google Scholar] [CrossRef]
- Sun, X.; He, S.; Lv, C.; Sun, X.; Wang, J.; Zheng, W.; Wang, D. Analysis of murine and human Treg subsets in inflammatory bowel disease. Mol. Med. Rep. 2017, 16, 2893–2898. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.-H.; Zhang, J.; Chen, X.; Xie, Y.-F.; Pang, Y.-H.; Liu, X.-J. Increased CD4 + CD45RA - Foxp3 low cells alter the balance between Treg and Th17 cells in colitis mice. World J. Gastroenterol. 2016, 22, 9356. [Google Scholar] [CrossRef]
- Gong, Y.; Lin, Y.; Zhao, N.; He, X.; Lu, A.; Wei, W.; Jiang, M. The Th17/Treg Immune Imbalance in Ulcerative Colitis Disease in a Chinese Han Population. Mediat. Inflamm. 2016, 2016, 7089137. [Google Scholar] [CrossRef] [Green Version]
- Geng, X.; Xue, J. Expression of Treg/Th17 cells as well as related cytokines in patients with inflammatory bowel disease. Pak. J. Med. Sci. 2016, 32, 1164–1168. [Google Scholar] [CrossRef]
- Chaudhry, A.; Samstein, R.M.; Treuting, P.; Liang, Y.; Pils, M.C.; Heinrich, J.-M.; Jack, R.S.; Wunderlich, F.T.; Brüning, J.C.; Müller, W.; et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 2011, 34, 566–578. [Google Scholar] [CrossRef] [Green Version]
- Song-Zhao, G.X.; Maloy, K.J. Experimental mouse models of T cell-dependent inflammatory bowel disease. Methods Mol. Biol. 2014, 1193, 199–211. [Google Scholar] [CrossRef]
- Ogino, H.; Nakamura, K.; Iwasa, T.; Ihara, E.; Akiho, H.; Motomura, Y.; Akahoshi, K.; Igarashi, H.; Kato, M.; Kotoh, K.; et al. Regulatory T cells expanded by rapamycin in vitro suppress colitis in an experimental mouse model. J. Gastroenterol. 2012, 47, 366–376. [Google Scholar] [CrossRef]
- Zhou, P.; Borojevic, R.; Streutker, C.; Snider, D.; Liang, H.; Croitoru, K. Expression of dual TCR on DO11.10 T cells allows for ovalbumin-induced oral tolerance to prevent T cell-mediated colitis directed against unrelated enteric bacterial antigens. J. Immunol. 2004, 172, 1515–1523. [Google Scholar] [CrossRef] [Green Version]
- Desreumaux, P.; Foussat, A.; Allez, M.; Beaugerie, L.; Hébuterne, X.; Bouhnik, Y.; Nachury, M.; Brun, V.; Bastian, H.; Belmonte, N.; et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology 2012, 143, 1207–1217.e1202. [Google Scholar] [CrossRef]
- Mizoguchi, A. Animal models of inflammatory bowel disease. Prog. Mol. Biol. Transl. Sci. 2012, 105, 263–320. [Google Scholar] [CrossRef]
- Cominelli, F.; Arseneau, K.O.; Rodriguez-Palacios, A.; Pizarro, T.T. Uncovering Pathogenic Mechanisms of Inflammatory Bowel Disease Using Mouse Models of Crohn’s Disease-Like Ileitis: What is the Right Model? Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Corridoni, D.; Arseneau, K.O.; Cominelli, F. Inflammatory bowel disease. Immunol. Lett 2014, 161, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 2017, 12, 1295–1309. [Google Scholar] [CrossRef]
- Hernández-Chirlaque, C.; Aranda, C.J.; Ocón, B.; Capitán-Cañadas, F.; Ortega-González, M.; Carrero, J.J.; Suárez, M.D.; Zarzuelo, A.; Sánchez de Medina, F.; Martínez-Augustin, O. Germ-free and Antibiotic-treated Mice are Highly Susceptible to Epithelial Injury in DSS Colitis. J. Crohns. Colitis 2016, 10, 1324–1335. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, E.; Low, D.; Ezaki, Y.; Okada, T. Recent updates on the basic mechanisms and pathogenesis of inflammatory bowel diseases in experimental animal models. Intest. Res. 2020, 18, 16. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, A.; Takeuchi, T.; Himuro, H.; Okada, T.; Mizoguchi, E. Genetically engineered mouse models for studying inflammatory bowel disease. J. Pathol. 2015, 238, 14. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, E.A.; King, K.Y.; Baldridge, M.T. Mouse Microbiota Models: Comparing Germ-Free Mice and Antibiotics Treatment as Tools for Modifying Gut Bacteria. Front. Physiol 2018, 9, 1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.A.; Bouladoux, N.; Sun, C.M.; Wohlfert, E.A.; Blank, R.B.; Zhu, Q.; Grigg, M.E.; Berzofsky, J.A.; Belkaid, Y. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity 2008, 29, 637–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. [Google Scholar] [CrossRef]
- Håkansson, Å.; Tormo-Badia, N.; Baridi, A.; Xu, J.; Molin, G.; Hagslätt, M.L.; Karlsson, C.; Jeppsson, B.; Cilio, C.M.; Ahrné, S. Immunological alteration and changes of gut microbiota after dextran sulfate sodium (DSS) administration in mice. Clin. Exp. Med. 2015, 15, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.C.; Jobin, C. The complex interplay between inflammation, the microbiota and colorectal cancer. Gut Microbes 2013, 4, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Yang, I.; Eibach, D.; Kops, F.; Brenneke, B.; Woltemate, S.; Schulze, J.; Bleich, A.; Gruber, A.D.; Muthupalani, S.; Fox, J.G.; et al. Intestinal microbiota composition of interleukin-10 deficient C57BL/6J mice and susceptibility to Helicobacter hepaticus-induced colitis. PLoS ONE 2013, 8, e70783. [Google Scholar] [CrossRef] [Green Version]
- Dennis, K.L.; Wang, Y.; Blatner, N.R.; Wang, S.; Saadalla, A.; Trudeau, E.; Roers, A.; Weaver, C.T.; Lee, J.J.; Gilbert, J.A.; et al. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10-producing T cells. Cancer Res. 2013, 73, 8. [Google Scholar] [CrossRef] [Green Version]
- Ettreiki, C.; Gadonna-Widehem, P.; Mangin, I.; Coëffier, M.; Delayre-Orthez, C.; Anton, P.M. Juvenile ferric iron prevents microbiota dysbiosis and colitis in adult rodents. World J. Gastroenterol. 2012, 18, 2619–2629. [Google Scholar] [CrossRef]
- Poussier, P.; Ning, T.; Chen, J.; Banerjee, D.; Julius, M. Intestinal inflammation observed in IL-2R/IL-2 mutant mice is associated with impaired intestinal T lymphopoiesis. Gastroenterology 2000, 118, 880–891. [Google Scholar] [CrossRef]
- Long, S.A.; Cerosaletti, K.; Bollyky, P.L.; Tatum, M.; Shilling, H.; Zhang, S.; Zhang, Z.-Y.; Pihoker, C.; Sanda, S.; Greenbaum, C. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4+ CD25+ regulatory T-cells of type 1 diabetic subjects. Diabetes 2010, 59, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Rubino, S.J.; Selvanantham, T.; Girardin, S.E.; Philpott, D.J. Nod-like receptors in the control of intestinal inflammation. Curr. Opin. Immunol. 2012, 24, 398–404. [Google Scholar] [CrossRef]
- Maggio-Price, L.; Shows, D.; Waggie, K.; Burich, A.; Zeng, W.; Escobar, S.; Morrissey, P.; Viney, J.L. Helicobacter bilis infection accelerates and H. hepaticus infection delays the development of colitis in multiple drug resistance-deficient (mdr1a-/-) mice. Am. J. Pathol. 2002, 160, 739–751. [Google Scholar] [CrossRef]
- Hapfelmeier, S.; Stecher, B.; Barthel, M.; Kremer, M.; Müller, A.J.; Heikenwalder, M.; Stallmach, T.; Hensel, M.; Pfeffer, K.; Akira, S. The Salmonella pathogenicity island (SPI)-2 and SPI-1 type III secretion systems allow Salmonella serovar typhimurium to trigger colitis via MyD88-dependent and MyD88-independent mechanisms. J. Immunol. 2005, 174, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Uematsu, S.; Jang, M.H.; Chevrier, N.; Guo, Z.; Kumagai, Y.; Yamamoto, M.; Kato, H.; Sougawa, N.; Matsui, H.; Kuwata, H. Detection of pathogenic intestinal bacteria by Toll-like receptor 5 on intestinal CD11c+ lamina propria cells. Nat. Immunol. 2006, 7, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.; Pasparakis, M.; Pizarro, T.T.; Cominelli, F.; Kollias, G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: Implications for joint and gut-associated immunopathologies. Immunity 1999, 10, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Burich, A.; Hershberg, R.; Waggie, K.; Zeng, W.; Brabb, T.; Westrich, G.; Viney, J.L.; Maggio-Price, L. Helicobacter-induced inflammatory bowel disease in IL-10-and T cell-deficient mice. Am. J. Physiol.-Gastrointest. Liver Physiol. 2001, 281, G764–G778. [Google Scholar] [CrossRef]
- Goettel, J.A.; Kotlarz, D.; Emani, R.; Canavan, J.B.; Konnikova, L.; Illig, D.; Frei, S.M.; Field, M.; Kowalik, M.; Peng, K.; et al. Low-Dose Interleukin-2 Ameliorates Colitis in a Preclinical Humanized Mouse Model. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 193–195. [Google Scholar] [CrossRef] [Green Version]
- Glocker, E.O.; Kotlarz, D.; Boztug, K.; Gertz, E.M.; Schaffer, A.A.; Noyan, F.; Perro, M.; Diestelhorst, J.; Allroth, A.; Murugan, D.; et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 2009, 361, 2033–2045. [Google Scholar] [CrossRef] [Green Version]
- Neven, B.; Mamessier, E.; Bruneau, J.; Kaltenbach, S.; Kotlarz, D.; Suarez, F.; Masliah-Planchon, J.; Billot, K.; Canioni, D.; Frange, P. A Mendelian predisposition to B-cell lymphoma caused by IL-10R deficiency. Blood J. Am. Soc. Hematol. 2013, 122, 3713–3722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shouval, D.S.; Ebens, C.L.; Murchie, R.; McCann, K.; Rabah, R.; Klein, C.; Muise, A.; Snapper, S.B. Large B-cell lymphoma in an adolescent patient with IL-10 receptor deficiency and history of infantile inflammatory bowel disease. J. Pediatric Gastroenterol. Nutr. 2016, 63, e15. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, R.K.; Li, J.; Jacobse, J.; Snapper, S.B.; Shouval, D.S.; Goettel, J.A. Humanized mouse models of genetic immune disorders and hematological malignancies. Biochem. Pharm. 2020, 174, 113671. [Google Scholar] [CrossRef]
- Tyagi, R.K.; Jacobse, J.; Li, J.; Allaman, M.M.; Otipoby, K.L.; Sampson, E.R.; Wilson, K.T.; Goettel, J.A. HLA-Restriction of Human Treg Cells Is Not Required for Therapeutic Efficacy of Low-Dose IL-2 in Humanized Mice. Front. Immunol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.H.; Cantrell, D.A. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu. Rev. Immunol. 2018, 36, 22. [Google Scholar] [CrossRef]
- Śledzińska, A.; Mucha, M.V.d.; Bergerhoff, K.; Hotblack, A.; Demane, D.F.; Ghorani, E.; Akarca, A.U.; Marzolini, M.A.V.; Solomon, I.; Vargas, F.A.; et al. Regulatory T Cells Restrain Interleukin-2- and Blimp-1-Dependent Acquisition of Cytotoxic Function by CD4 + T Cells. Immunity 2020, 52, 15. [Google Scholar] [CrossRef]
- Landman, S.; de Oliveira, V.L.; van Erp, P.E.J.; Fasse, E.; Bauland, S.C.G.; Joosten, I.; Koenen, H.J.P.M. Intradermal injection of low dose human regulatory T cells inhibits skin inflammation in a humanized mouse model. Sci. Rep. 2018, 8, 10044. [Google Scholar] [CrossRef]
- Zhang, B.; Duan, Z.; Zhao, Y. Mouse models with human immunity and their application in biomedical research. J. Cell. Mol. Med. 2009, 13, 1043–1058. [Google Scholar] [CrossRef] [Green Version]
- McCune, J.M.; Namikawa, R.; Kaneshima, H.; Shultz, L.D.; Lieberman, M.; Weissman, I.L. The SCID-hu mouse: Murine model for the analysis of human hematolymphoid differentiation and function. Science 1988, 241, 1632–1639. [Google Scholar] [CrossRef]
- Mosier, D.E.; Gulizia, R.J.; Baird, S.M.; Wilson, D.B. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature 1988, 335, 256–259. [Google Scholar] [CrossRef]
- Tournoy, K.G.; Depraetere, S.; Pauwels, R.A.; Leroux-Roels, G.G. Mouse strain and conditioning regimen determine survival and function of human leucocytes in immunodeficient mice. Clin. Exp. Immunol. 2000, 119, 9. [Google Scholar] [CrossRef]
- Ito, R.; Takahashi, T.; Katano, I.; Ito, M. Current advances in humanized mouse models. Cell Mol. Immunol. 2012, 9, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Shultz, L.D.; Brehm, M.A.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Pearson, T.; King, M.; Giassi, L.; Carney, L.; Gott, B.; Lyons, B.; Rossini, A.A.; Greiner, D.L. Humanized NOD/LtSz-scid IL2 receptor common gamma chain knockout mice in diabetes research. Ann. N. Y. Acad. Sci. 2007, 1103, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K. NOD/SCID/γ c null mouse: An excellent recipient mouse model for engraftment of human cells. Blood J. Am. Soc. Hematol. 2002, 100, 3175–3182. [Google Scholar]
- King, M.; Pearson, T.; Shultz, L.D.; Leif, J.; Bottino, R.; Trucco, M.; Atkinson, M.A.; Wasserfall, C.; Herold, K.C.; Woodland, R.T. A new Hu-PBL model for the study of human islet alloreactivity based on NOD-scid mice bearing a targeted mutation in the IL-2 receptor gamma chain gene. Clin. Immunol. 2008, 126, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Harrison, O.J.; Powrie, F.M. Regulatory T cells and immune tolerance in the intestine. Cold Spring Harb. Perspect. Biol. 2013, 5, a018341. [Google Scholar] [CrossRef]
- Sharma, A.; Rudra, D. Emerging functions of regulatory T cells in tissue homeostasis. Front. Immunol. 2018, 9, 883. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, L.-Y.; Wu, J.; Xu, L.-F.; Sun, M. The JAK2 inhibitor AG490 regulates the Treg/Th17 balance and alleviates DSS-induced intestinal damage in IBD rats. Clin. Exp. Pharm. Physiol 2020. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Liang, W.; Wang, T.; Sui, J.; Wang, J.; Deng, Z.; Chen, D. Saponins regulate intestinal inflammation in colon cancer and IBD. Pharm. Res. 2019, 144, 66–72. [Google Scholar] [CrossRef]
- Lee, S.H.; Kwon, J.E.; Cho, M.-L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef] [Green Version]
- Hazel, K.; O’Connor, A. Emerging treatments for inflammatory bowel disease. Ther. Adv. Chronic Dis. 2020, 11, 2040622319899297. [Google Scholar] [CrossRef] [PubMed]
- Hitotsumatsu, O.; Ahmad, R.-C.; Tavares, R.; Wang, M.; Philpott, D.; Turer, E.E.; Lee, B.L.; Shiffin, N.; Advincula, R.; Malynn, B.A.; et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity 2008, 28, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantini, M.C.; Monteleone, G. Update on the Therapeutic Efficacy of Tregs in IBD: Thumbs up or Thumbs down? Inflamm. Bowel Dis. 2017, 23, 1682–1688. [Google Scholar] [CrossRef]
- Weigmann, B.; Schughart, N.; Wiebe, C.; Sudowe, S.; Lehr, H.A.; Jonuleit, H.; Vogel, L.; Becker, C.; Neurath, M.F.; Grabbe, S.; et al. Allergen-induced IgE-dependent gut inflammation in a human PBMC-engrafted murine model of allergy. J. Allergy Clin. Immunol. 2012, 129, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Nolte, T.; Zadeh-Khorasani, M.; Safarov, O.; Rueff, F.; Gülberg, V.; Herbach, N.; Wollenberg, A.; Mueller, T.; Siebeck, M.; Wolf, E.; et al. Oxazolone and ethanol induce colitis in non-obese diabetic-severe combined immunodeficiency interleukin-2Rγ(null) mice engrafted with human peripheral blood mononuclear cells. Clin. Exp. Immunol. 2013, 172, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Goettel, J.; Shouval, D.; Lexmond, W.; Muise, A.; Fiebiger, E.; Snapper, S. Development of Novel Humanized Murine Models to Assess Mucosal Homeostasis: Human anti-CD3 Antibody or TNBS Administration Leads to Small and Large Bowel Inflammation Respectively in Immunodeficient Mice Transferred With Human T Cells. Gastroenterology 2013, 144, S-32. [Google Scholar] [CrossRef]
- Goettel, J.; Biswas, B.; Lexmond, W.; Sun, J.; Ouahed, J.; McCann, K.; Shouval, D.; Milford, E.; Fiebiger, E.; Muise, A.; et al. Human Hematopoietic Stem Cells With a Defined Immunodeficiency and Enteropathy Transfer Clinical Phenotype to a Novel Humanized Mouse Strain. Gastroenterology 2014, 146, S-81. [Google Scholar] [CrossRef]
- Goettel, J.A.; Gandhi, R.; Kenison, J.E.; Yeste, A.; Murugaiyan, G.; Sambanthamoorthy, S.; Griffith, A.E.; Patel, B.; Shouval, D.S.; Weiner, H.L.; et al. AHR Activation Is Protective against Colitis Driven by T Cells in Humanized Mice. Cell Rep. 2016, 17, 1318–1329. [Google Scholar] [CrossRef] [Green Version]
- Jodeleit, H.; Winkelmann, P.; Caesar, J.; Sterz, S.; Holdt, L.M.; Beigel, F.; Stallhofer, J.; Breiteneicher, S.; Bartnik, E.; Leeuw, T.; et al. Head-to-head study of oxelumab and adalimumab in a mouse model of ulcerative colitis based on NOD/Scid IL2Rγnull mice reconstituted with human peripheral blood mononuclear cells. Dis. Model. Mech. 2021, 14. [Google Scholar] [CrossRef]
- Harshe, R.P.; Xie, A.; Vuerich, M.; Frank, L.A.; Gromova, B.; Zhang, H.; Robles, R.J.; Mukherjee, S.; Csizmadia, E.; Kokkotou, E.; et al. Endogenous antisense RNA curbs CD39 expression in Crohn’s disease. Nat. Commun. 2020, 11, 5894. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Group | Animal Model | Information |
|---|---|---|---|
| 1. | Chemically Induced | Acetic acid (rat) |
|
| DSS |
| ||
| TNBS |
| ||
| Oxazolon |
| ||
| PG/PS polymer (Peptidoglycan polysaccharide) |
| ||
| Auer |
| ||
| Carrageenan (CGN) |
| ||
| Indomethacin |
| ||
| 2. | Adaptive Cell Transfer | CD45RB |
|
| ECOVA |
| ||
| CD8 Transfer |
| ||
| 3. | Bacteria-Infected Model | Citrobacter rodentium |
|
| Helicobactor hepaticus |
| ||
| 4. | Conogenic Model | C3H/HeJBir |
|
| SAMP1/Yit |
| ||
| SAMP1/YitFc |
| ||
| 5. | Spontaneous | Cotton-top tamarin |
|
| Genetic Background of Mice | Induction of Colitis | Human Cells Transplanted | Remarks | References |
|---|---|---|---|---|
| NOD-SCID IL2Rγ−/− (NSG) | Allergen (Birch, grass, Hazelnut) | PBMCs from allergic and non-allergic subjects | The amplified extent of colitis was seen in allergic donors isolated PBMCs engrafted micecompared to healthy donors. | [206] |
| NSG | Oxazolone | PBMCs from healthy, UC and AD subjects | The described model showed the potential to study the efficacy of therapeutics targeting human lymphocytes in a model closely mimicking human ulcerative colitis. | [207] |
| NSG | TNBS | HLA-matched human CD4+ T cells | Adoptive transfer of human CD4+ T cells in humanized animals with TNBS induced small bowel enteropathy and promoted colonic inflammation. | [208] |
| HLA-matched CD34+ human HSCs from healthy and IPEX subjects | The study established the use of human HSCs to transfer disease phenotype in humanized mice to study human immune effectors and pathogenesis of IBD. | [209] | ||
| NSG | TNBS | HLA-matched human CD4+ T cells isolated from a healthy donor | The study developed an experimental humanized murine model to investigate human CD4+ T responses in vivo and identify the ITE (a non-toxic AHR agonist) as a potential therapy to achieving the immune tolerance in the intestine. | [210] |
| NSG | TNBS | PBMCs isolated from healthy donors | Low-dose IL-2 helped in expanding Trges for using as a therapeutic strategy against colitis in humanized mice. | [180] |
| NSG | PBMCs from UC donors were reconstituted in NSG mice and treated with oxelumab | NSG-UC mice treated with oxelumab significantly reduced clinical, colon and histological scores and reduced serum levels of IL-6. | [211] | |
| NSG | TNBS | Antisense+ CD4 cells isolated from a healthy donor | Silencing the endogenous antisense long non-coding RNA restores CD39 levels with enhancing Treg-suppressive function. | [212] |
| NSG | TNBS | CD34+ human HSCs | Low-dose (LD) IL-2 reduced the severity of TNBS induced in HSC CD34+ reconstituted NSG mice and paved the way for developing future therapeutic strategy based on LD IL-2 | [185] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negi, S.; Saini, S.; Tandel, N.; Sahu, K.; Mishra, R.P.N.; Tyagi, R.K. Translating Treg Therapy for Inflammatory Bowel Disease in Humanized Mice. Cells 2021, 10, 1847. https://doi.org/10.3390/cells10081847
Negi S, Saini S, Tandel N, Sahu K, Mishra RPN, Tyagi RK. Translating Treg Therapy for Inflammatory Bowel Disease in Humanized Mice. Cells. 2021; 10(8):1847. https://doi.org/10.3390/cells10081847
Chicago/Turabian StyleNegi, Sushmita, Sheetal Saini, Nikunj Tandel, Kiran Sahu, Ravi P.N. Mishra, and Rajeev K. Tyagi. 2021. "Translating Treg Therapy for Inflammatory Bowel Disease in Humanized Mice" Cells 10, no. 8: 1847. https://doi.org/10.3390/cells10081847
APA StyleNegi, S., Saini, S., Tandel, N., Sahu, K., Mishra, R. P. N., & Tyagi, R. K. (2021). Translating Treg Therapy for Inflammatory Bowel Disease in Humanized Mice. Cells, 10(8), 1847. https://doi.org/10.3390/cells10081847