Abstract
Crohn’s disease and ulcerative colitis, two major forms of inflammatory bowel disease (IBD) in humans, afflicted in genetically predisposed individuals due to dysregulated immune response directed against constituents of gut flora. The defective immune responses mounted against the regulatory mechanisms amplify and maintain the IBD-induced mucosal inflammation. Therefore, restoring the balance between inflammatory and anti-inflammatory immunepathways in the gut may contribute to halting the IBD-associated tissue-damaging immune response. Phenotypic and functional characterization of various immune-suppressive T cells (regulatory T cells; Tregs) over the last decade has been used to optimize the procedures for in vitro expansion of these cells for developing therapeutic interventional strategies. In this paper, we review the mechanisms of action and functional importance of Tregs during the pathogenesis of IBD and modulating the disease induced inflammation as well as role of mouse models including humanized mice repopulated with the human immune system (HIS) to study the IBD. “Humanized” mouse models provide new tools to analyze human Treg ontogeny, immunobiology, and therapy and the role of Tregs in developing interventional strategies against IBD. Overall, humanized mouse models replicate the human conditions and prove a viable tool to study molecular functions of human Tregs to harness their therapeutic potential.
1. Introduction
Inflammatory bowel disease (IBD) is a complex inflammatory chronic and pathological condition that includes Crohn’s disease (CD) and Ulcerative colitis (UC). CD primarily affects the small and large intestine whereas the prime targets for UC are colon and rectum [1]. Clinical manifestations of IBD are characterized by abdominal pain, rectal bleeding, bloody stools, tenesmus, diarrhea, weight loss, and the urgency to evacuate [1,2]. Pathogenesis of IBD involves various environmental, genetic and bacterial factors with dysregulated mucosal immune-mechanisms resulting in the disrupted intestinal homeostasis [3,4], and dysregulated mucosal immune response provokes robust inflammatory response against intestinal flora [2,5].
Immune sentinel subsets of CD4+ T cells such as Th (T helper cells)-1, Th2, Th17, and regulatory T cells (Tregs) play a crucial role in the pathogenesis of IBD. Immunological balance between effector Th cells and Trges is essential for maintaining immune-homeostasis [6,7]. Immunoregulatory Trges are characterized by the expression of transcription factor Forkhead box P3 (Foxp3), and surface marker CD25 [8,9], and are functionally immunosuppressive & important for immune tolerance [10]. Therapeutic arrangement based on Tregs is important to address the systemic inflammatory and autoimmune diseases such as IBD and rheumatoid arthritis [11,12,13]. Further, decreased number of Trges was seen in the patients with IBD than healthy control [14,15,16]. Inhibited generation of functionally impaired Trges contribute to the intestinal inflammation leading to colitis and other biological complications [17,18]. Functionally immune-suppressive Trges have reportedly shown to ameliorate the IBD-induced immune responses [19,20]. Reduced intestinal inflammation drives the control of IBD pathogenesis [21], and increased number of functional Tregs experimentally confirmed their “therapeutic potential” during IBD pathogenesis [22,23,24].
Present article is an orchestrated attempt to validate the functional aspects of Trges during the IBD pathogenesis for developing interventional therapeutic strategies. Further, role of Trges in the maintenance of peripheral immune tolerance with an emphasis on mucosal immunity has been discussed at length. Additionally, various transgenic and immunodeficient mice repopulated with the human immune system; mouse-human chimeric models and their importance to study of IBD pathogenesis and associated biological phenomena have been discussed.
2. IBD Pathogenesis
The epithelial layer of the human gut consists of goblet cells, columnar cells, paneth cells, endocrine cells, M cells, tuft cells, and epithelial resident intestinal stem cells. These cells are responsible for the differentiation of gut microbiota and secretion of various mucus-containing antimicrobial peptides [25,26,27]. The intestinal barrier contains innate immune cells such as dendritic cells (DCs), neutrophils, macrophages and innate lymphoid cells (ILCs) which reside in the state of hypo-responsiveness in a healthy human gut [28,29]. The mucosal macrophages prevent the inter-conversion of Th1 and Th17 cells by producing anti-inflammatory cytokines and thus promoting the differentiation of Trges [30,31,32]. The immune cells’ balance in intestinal mucosa and luminal content is crucial for the normal functioning of the mucosal immune system since dysregulated immune effecters result in the IBD pathogenesis [33,34,35]. The impaired innate immune system is responsible for the functional abnormalities of the adaptive immune system, and interconversion of effector Th cells and Trges causing IBD pathogenesis [35,36]. Therefore, balanced gut mucosal immunity to maintain immune homeostasis and protection is inevitably critical to fighting IBD.
Activated lamina propria (intestinal mucous membrane) secrete a large number of soluble immune mediators, including pro-inflammatory cytokines viz. tumor necrosis factor (TNF), interferon-gamma (IFN-γ), Interleukin (IL)-6, IL-12, IL-21, IL-23, IL-17 and anti-inflammatory cytokines such asIL-10, transforming growth factor (TGF-β) and IL-35in local tissues [37]. The imbalance between these secreted soluble mediators especially inflammatory and anti-inflammatory cytokines secreted by immune system results in IBD pathogenesis [3,37,38,39] (Figure 1). The exogenous administration of TNF and TNF-like cytokine 1Acytokines (TLA1) regulates the balance between Th1 and Th17 cell population in the inflamed colonic tissues [40]. Further, TLA1 modulates Foxp3 expression in Tregs and its function, and murine model of colitis has seen the alleviation of colitis when treated with Tregs expressing low levels of TLA1.TLA1 may promote the maintenance of Treg suppressor function in a death domain receptor 3 (DR3) dependent manner [41]. Passive administration of anti-TLA1 antibodies prevents the development of 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis in mice, partially improves dextran Sulfate sodium (DSS)-induced colitis and decreases the intestinal fibrosis in a chronic colitis model [42,43,44].
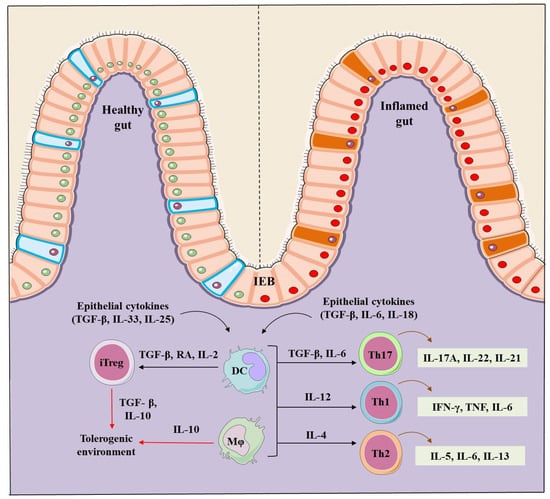
Figure 1.
Regulation of the intestinal homeostasis in healthy and IBD inflamed gut. A healthy intestinal epithelial barrier (IEB) in presence of TGF-β, retinoic acid (RA) and IL-2 promote dendritic cells (DCs) and macrophages (mφ) to stimulate the generation of inducible Trges (iTregs). TGF-β and IL-10 are markers that contribute to the generation of iTregs, and establish and maintain the tolerogenic environment in a healthy gut. On the contrary, IBD induced inflammation induces intestinal epithelial barriers and secrete TGF-β, IL-6 and IL-8, stimulating DCs and mφ to produce the inflammatory Th-17 (IL-17A, IL-22, IL-21), Th-1 (IFN-γ, TNF-α, IL-6) and Th-2 type cells (IL-5, IL-6, IL-13) creating an inflammation focus and diseased intestine.
A meta-analysis has identified more than 200 genomic loci associated with IBD pathogenesis, and 68% of loci have been shared by the UC and CD disease [45]. Genome-wide association studies (GWAS) have helped the scientific community to find out genes responsible for IBD pathology and nucleotide-binding oligomerization domain 2 (NOD2) is the first gene found to be associated with IBD [46]. The associated mutations in NOD2 and polymorphisms within autophagy-related 16-like 1 (ATG16L1) genes have disseminated the pivotal role of autophagy in the pathogenicity of IBD [47,48,49]. NOD2 knockouts exhibited excessive intestinal inflammation as compared to NOD2 sufficient mice [50], and selective deletion of ATG16L1 in T cells ends with spontaneous intestinal inflammation, characterized by a decrease in Foxp3+ Tregs and aberrant expression of Th2 cells [51].
The contribution of gut homing or migration associated molecules such as α4b7 integrin, αEb7 integrin, CD62L, chemokine receptor (CCR)-4 (CCR4), CCR5, CCR7 and CCR9 in the pathogenesis of IBD is well known [52]. Defective or loss of expression of these molecules leads to the impaired Tregs trafficking to the target organs and thus inducing IBD. Loss of CCR7 and CCR4impairs the functions of Tregs in experimental colitis [53,54], and further CCR7 regulates the balance between Th1, Th17, and Trges in Crohn’s-like Murine ileitis [55]. b7 integrin deficiency impaired Tregs homing in IL-10 deficient mice and spontaneously increased IBD-induced inflammation [56]. In essence, all findings indicate the crucial role played by the number of functional Tregs since a compromised number of Tregs contribute to the pathogenesis of IBD and other associated biological impairments.
3. Regulatory T Cells (Tregs)
Tregs are heterogeneous cell populations of CD4+ T cells and possess immunosuppressive attributes. CD4+ Trges expressing high levels of IL-2 receptor α chain (CD25) and master transcription factor Foxp3 are the best-characterized populations with the immunosuppressive phenotype [57]. Foxp3 is essentially required for maintaining their immunosuppressive activity against infections, tumors, intestinal inflammation, allergy, and autoimmunity [8,9,10,58]. The absence of CD127 (IL-7 receptor α-chain) is considered as another feature of Tregs and up-regulates CD25 and Foxp3 expression upon activation. Thus a lower expression of CD127 is important alongwith elevated CD4, CD25, and Foxp3 expression to confirm the functional phenotype of immunoregulatory T cells [59,60]. Two main subsets of Trges are characterized as Foxp3 positive Trges and Foxp3 negative type 1 Treg (Tr1) cells.
3.1. tTregs and pTre
Depending on generation, Foxp3+ Trges are further categorized as naturally occurring thymus-derived Treg (tTreg) cells and Trges developed from conventional CD4+ T cells in the periphery (pTreg). These cells possess immunosuppressive functions and maintain peripheral tolerance [61,62]. tTregs are produced by the thymus at an early stage after birth and maintain tolerance toward self-antigens [63,64]. TGF-β1 directly enhances the Foxp3 promoter and encourages the generation of tTregs [65]. Besides, exposure of naive T-cells to its cognate-antigen leads to the differentiation of pTregs under tolerogenic conditions [66,67,68], and differentiation of pTregs is facilitated by the higher concentrations of TGF-β and higher levels of Foxp3 [69,70,71]. Therefore, TGF-β1 is a key cytokine and plays a crucial role in the differentiation of both subsets of Trges. The induction of Foxp3 in peripheral naive T cells is achieved by a higher concentration of TGF-β, retinoic acid, and CD28 co-stimulation [70,72,73,74]. Both, tTreg and pTregs show similar expression levels of FoxP3, CD25, CTLA-4, GITR, ICOS, CD103, CD127 and a broad T-cell receptor (TCR) repertoire to deploy various suppressive mechanisms to control effector cells [8,59,75,76]. Foxp3+ Trges are also known to secrete IL-10, TGF-β, and IL-35 [77,78] along with granzyme A and B [79,80,81]. Furthermore, tTregs express higher levels of neuropilin-1(Nrp1), TF Ikzf2 (Helios), PD-1, and ecto nucleotidase CD73 than pTregs [82,83]. Helios and Nrp1 are considered as markers for tTregs since their greater expression is seen in tTregs as compared to pTregs [83,84,85]. Interestingly, under in-vivo conditions, pTregs could express helios [86], and a fraction of the human tTreg population did not express helios [87]. Moreover, tTregs are not differentiated based on helios and Nrp1 expression in mice [88]. Treg specific demethylated region (TSDR) is highly demthylated in tTregs, and partially demethylated along with an unstable expression of Foxp3 and CD25 in pTregs [69,89,90]. Apart from TSDR, Ig superfamily surface protein GPA33along with other Treg cell markers was recently used to identify Trges of thymus origin since this molecule is stably expressed on tTregs [91].
3.2. Type 1 T regulatory (Tr1) Cells
Tr1 cells are unique Foxp3− regulatory T cells that develop in the periphery and secrete elevated levels of immunosuppressive cytokines such as IL-10 and TGF-β [92,93]. tTreg and pTregs constitutively express Foxp3 and CD25 but these markers are expressed by Tr1 cells only in the activated state [94]. Tr1 cells are characterized by the co-expression of surface markers, CD49b and LAG-3 [95], and can be distinct due to the cytokine expression of IL-2, IL-10, IFN-γ, IL-5 and Il-17 [94] as well as granzyme B and perforin via cell death mechanism [93]. And, experimental evidences have confirmed the immunosuppressive function of Tr1 cells mediated by IL-10 [96,97].
4. Role of Tregs in IBD
Trges play a vital role in maintaining gut immune homeostasis and regulate pro-inflammatory responses elicited by the adaptive and innate immune effectors [52]. Scurfy mouse strain showing the severe autoimmune phenotype with a genetic defect in the Foxp3 gene and inhibit the Tregs development and leads to the dysregulated activation of the gut immune system [98], which mounts inflammation primarily in the gut. Further, the mutation in the human Foxp3 gene leads to a rare autoimmune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome along with other severe autoimmune diseases including arthritis, diabetes, allergy and IBD. This syndrome was seen due to impaired immune response mediated inflammation [17,99,100]. Furthermore Foxp3 expressing Trges are essential for maintaining the balance at the intestinal mucosal surface because intestinal inflammation gets chronic with the decreasing number of Foxp3+ Tregs [101]. DSS induced colitis in mice showings pontaneous depletion of Foxp3+ Tregs leading to an increase in the disease severity [15]. However, adoptive transfer of Tregs in Treg depleted mice (DEREG) showed a decrease in the severity and improved tissue conditions of experimental colitis [15].
Although patients with IBD showed an increased number of Tregs in the inflamed intestinal mucosa than un-inflamed mucosal part [102,103,104]. The phenotype and function of Tregs present in the inflamed mucosa or periphery of IBD patients or in experimental animals differ from those present in peripheral lymphoid organs of healthy control. Patients with IBD showed an increase in the number of peripheral Th17 cells, and a reduction in the peripheral Trges [105]. While in some cases patients with IBD showed higher expression of Foxp3 along with elevated levels of pro-inflammatory cytokines including IL-17A, IL-1β and IL-6 [105]. Moreover, the highest frequency of Foxp3+ IL-17 T-cells (Th17 and Treg intermediate cells) was seen in the inflamed mucosal tissues of patients with IBD [106,107] (Figure 2).
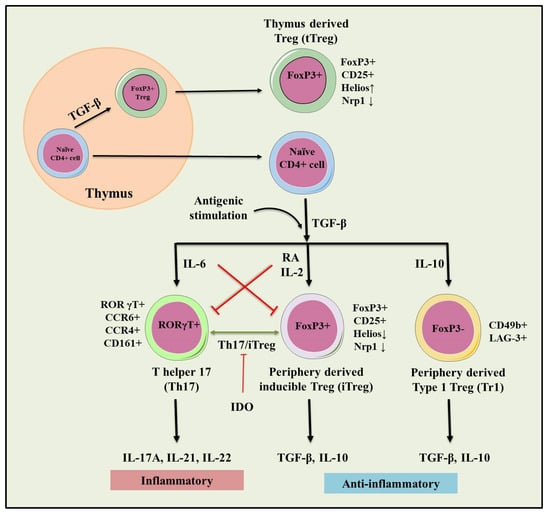
Figure 2.
Role of natural and inducible regulatory T cells (iTregs) involved in the pathogenesis of IBD. Inflammation (IL-6) dependent interconversion of regulatory and effector T cell phenotype and role in dol 2, 3 dioxygenases (IDO) in the generation of inducible Tregs (iTregs). Inflammatory (IL-17A, IL-21, IL-22) and immunosuppressive (TGF-β, IL-10) conditions following antigenic stimulation were seen during the conversion of Th17 to iTreg phenotype. This interconversion plays a crucial role in maintaining tolerance towards IBD.
Foxp3+ Trges express effector T cell-specific transcription factor retinoic acid receptor-related orphan receptor gamma t (RORγt) and differentiate into Th-17 cells. These cells inhibit the immunosuppressive function of Tregs in patients with IBD [108]. Further, Tregs upregulate the expression of T-bet and express pro-inflammatory cytokine IFN-γ. Regulatory T cells are characterized as IFN-γ-expressing Th1 like Tregs and mount intestinal inflammation in patients with IBD. Th1 like Trges evoke inflammation since the IFN-γ-expressing cells accumulate at the site of inflammation in CD and UC and contribute to the IBD pathogenesis [109]. The accumulation of IFN-γ expressing Th1 like Trges is also observed in the inflamed colon in the DSS-induced colitis animal model [109]. The acquisition of pro-inflammatory behavior of Tregs in IBD most likely contributes to the uncontrolled inflammation in vivo. Furthermore, Tregs suppress the colonic inflammation by downregulating Th1 and Th17 responses in the adoptive transfer model of colitis [110,111,112], and passively transferred Trges have shown the ability to control inflammatory lesions in the experimental model of IBD [113].
Trges have been seen to be involved in the tissue repair mechanism of the intestine. Among the different populations of intestinal Treg i.e., Tr1 cells have been shown to mediate repair of the intestinal mucosa that co-expressed Th2 and master transcription factor GATA binding protein 3 (GATA3). Therefore, elevated expression of IL-33R, ST2, and amphiregulin (AREG), an epidermal growth factor receptor ligand was reported [114]. Furthermore, these factors are generally involved in tissue repair and are phenotypically characterized and expressed by GATA3+ Tregs [114]. Tissue repair and protection of gut is rendered by the human Tr1 Trges suppress the proliferation of T effector cells, elicit TNF and IL-1β based innate immune response and secret IL-22 to regulate the repairing of epithelium and promote barrier function [115].
5. Therapeutic Role of Tregs in IBD
Several patients with IBD showed tolerance to the current therapeutic arrangements. Therefore, the need for developing effective, safe and novel therapies for IBD is both attractive and urgent. Newer and effective immunotherapies involving anti-TNF agents (infliximab, adalimumab, and certolizumab) have shown remarkable progress toreducethe need for surgery and hospitalization of IBD patients [116,117]. However, meta-analysis conducted by Ford et al., suggested that usage of anti-TNF agents may increase the risk of getting opportunistic infections in IBD patients [118]. Other recent reports also suggest the higher chance of getting infections such as histoplasmosis, aspergillosis and cytomegalovirus infection [119] and anti-TNF monotherapy was found to be responsible for higher mycobacterial and bacterial infection; however, in a combination therapy with thiopurine will increase the risk of getting serious infections [119,120]. The REFURBISH study was reported that the risk of getting T-cell non-Hodgkin’s lymphoma in IBD patients is higher during combinational therapy with compare to anti-TNF monotherapy [121] whereas the another cohort study delineate that even the ant--TNF monotherapy is associated with lymphoma formation in small number but have the higher statistical significant. And, it put on more on risk during the combinational therapy [122]. Other than this other paradoxical side effects such as psoriasis/psoriasiform skin, development of sarcoidosis-like lesions, late occurrence of arthritis/synovitis and lupus-like syndrome (0.5 to 1% of patients) can also be developed [123,124,125,126,127]. Additionally, novel therapies including JAK inhibitor [128,129,130], anti-MAdCAM-1 [131,132,133,134,135], an anti-SMAD7 antisense oligonucleotide (mongersen) [136,137,138], S1P1 [128,139] and anti-interleukin (IL)-12/23 (ustekinumab) [140,141,142,143,144,145,146] have been under investigation for safety and other purposes.
Recently, cellular therapies have been used as potential therapeutic strategies for IBD patients [147]. The role of Tregs in the preclinical models of colitis has been well understood, and recent investigations and phase 1 clinical trials have proven the safety and efficacy of Trges. A marked difference in the number of Tregs in patients (the inflamed mucosa or peripheral blood) and experimental animal models of IBD have been observed [101,148]. Experimental model of IBD showed an increase in Tregs percentage in the inflamed ileum with a reduced immunosuppressive function and IL-10 production. The dysregulated expression between Th17 cells and Tregs was also observed in UC animal model with downregulated mRNA expression of Foxp3 and IL-10 levels in Trges [149]. In UC patients, a significant increase of IL-17 and Th17 cells and a simultaneous decrease of TGF-β and Trges in serum as compared to healthy control was seen [150]. Th-17 cells and associated cytokines IL-17 and IL-23 were found to be decreased in patients with IBD along with a decrease in the Tregs and associated cytokines IL-10 and TGF-β [151]. IL-10 is known to induce Treg mediated suppression of Th-17 cells in a STAT-3 dependent manner [152]. The improvement in the clinical and histological parameters was observed when Trges were adoptively transferred in Rag−/− or severe combined immunodeficient (SCID) mice [153]. Rapamycin-expanded Trges (Th cells cultured in presence of rapamycin) were shown to suppress colitis in SCID mice [154]. And, ovalbumin (OVA) induced Tregs from DO11.10 mice prevented colitis together with increased TGF-β and IL-10 secretion in SCID-bg mice [155]. The safety and efficacy of OVA-Treg therapy were assessed for refractory CD in an open-labeled multicenter phase I/II clinical trial. This study showed the dose-related efficacy because infusion of ova-specific Tregs treatment was well-tolerated, and 40%patients showed a reduced CD activity on 5 and 8-week post-treatment [156]. In vitro expanded CD45RA+ Tregs cells were shown to express stable Foxp3 locus, which enhanced their suppressive ability and prevented their conversion to Th17 phenotype in the SCID xenotransplant model [24]. Additionally, CD45RA− and CD45RA+ expanded Tregs expressed a high level of gut homing receptor α4β7 integrin, CD62L, and CCR7 to facilitate their intestinal homing [24]. In an active CD mucosa, CD45RA+ Tregs healed the inflammation of lamina propria and mesenteric lymph nodes [24]. Tregs isolated from the lamina propria of active IBD patients and in experimental model (DSS induced colitis) express T-bet and IFN-γ (Th-1 like Tregs) and stimulates the early stages of inflammation. Further, T-bet KO showed the development of less severe colitis with the dysregulated Th1 immune response. It suggests that T-bet expression in Tregs is required for the development of colitis [109]. In the end, Treg immunotherapy with in vitro expanded Tregs (NCT03185000) for treating the Crohn’s disease (TRIBUTE trial) is underway.
6. Animal Models to Study IBD
Numerous animal models have been used to study many immune-effecters such as inflammatory mediators, chemokines and cytokines, pathogenic bacteria and effector Tregs. The experimental animal models of IBD produce the wealth of information to develop further understanding of the pathogenesis of IBD [52,157,158,159]. Genetically modified (GM) mice play an instrumental role to study of gastrointestinal (GI) tract disorders. GM mice including knockout, transgenic and mutant animals have proven a tool to unravel mechanisms underlying the intestinal inflammation and pathogenesis of systemic inflammatory diseases such as colitis and RA. Therefore, experimental induction of colitis in the mouse models has been developed by different chemicals including DSS, TNBS, oxazolone or acetic acid [100,160,161]. Furthermore, adoptive transfer models are being currently used and T-cell deficient mice reconstituted with Trges and depleted naïve T-cells were selected from congenic donor mice (Table 1) [52]. Moreover, few spontaneous and humanized mouse models have been developed to understand the pathogenesis to devising therapeutic treatments against colitis [162].

Table 1.
The different types of animal model induced through various methods are summarized with their implication and role in IBD pathogenesis ([52,162]) (the generation of different genetically engineered mice are reviewed in detailed in [162,163]).
6.1. Bacteria-Infected Models
Studies carried out in the experimental mouse models of IBD suggested sensitivity of Treg cell compartment towards the changes occurred in the microbial environment [19]. Germ-free, pathogen-free, and gnotobiotic mice generally express a lower level of Foxp3 with reduced colonic Tregs compared to the Tregs extracted from wild-type mice. Treg population showed an increase with the decreased bacterial load upon receiving vancomycin treatment [164], and mice infected with Citrobactor rodentium were used to mimic the acute intestinal inflammation. Further, Helicobacter-infected mice are preferred as bacterial infection models over combination models to see the synergistic effect of drugs. Inflammatory signals emanating from bacterial DNA play an important role and inhibit the inducible Tregs, and their differentiation in Toll-like receptor 9 (TLR9) deficient mice showing a higher number of Tregs in small intestine [19,165]. Mice receiving DSS treatment showed a significant increase in the members of Bacteroidaceae and Clostridium spp. from Bacteroides distasonis and Clostridium ramosum families in the intestine [166]. Subsequent studies showed the elevated 16s rRNA gene copy numbers of mucin-degrading Gram-negative bacterium Akkermansia muciniphilia and Enterobacteriaceae to establish a correlation with the disease severity in DSS treated mice [167]. The increased number of microorganisms such as Enterobacteriaceae and adherent-invasive E. coli develops colitis in IL-10 deficient mice and provokes induction of inflammation leading to the cancer development [168,169]. Similarly, an increased number of Bacteroides and Porphyromonas genera in ApcΔ468/IL-10−/− mice mounts inflammation and colon polyposis [170]. Furthermore, increased inflammation with the increased number of Enterobacteriaceae and Bacteriodes was seen in the TNBS induced colitis animals [171].
6.2. Genetically Modified Animal Models
A variety of gene knockout models are available to study the innate and adaptive immune responses elicited by the IBD pathogenesis or other intestinal infections. The modified genetic lines produce phenotypes that investigate the immunological aspects of intestinal infection and inflammation [52]. Commonly knockout (KO) genes used in the murine model of intestinal inflammation are IL-10, IL-23R, CD4+CD25+, NOD2/CARD15, TGF-β1, RAG, ATG16L1, APCmin/+, IL-2, TNF-α, STAT3, NFκB, Muc2, IFN-γ, MyD88 and TLR [163]. A reduced number of Tregs with compromised functional activity was observed due to the genetic modification by knocking out IL-2-/, IL-2R-/, and IL-10-/ genes in some immunodeficient mice to develop IBD models [52,172,173]. Innate or mucosal immunity-related gene-deficient mice such as NOD2-/, myeloid differentiation primary response 88 (MYD88)-/, nuclear factor-kB (NF-kB)−/−, cytokine-deficiency induced colitis susceptibility 1 (CDCS1)-/, multidrug resistance gene 1a (MDR1a)-/, and TLR5-/ showed inflammatory lesions in colon [52,174,175,176,177]. T-cell receptor TCR−/− mice have been used to study various immunobiological phenomena taking place during IBD pathogenesis, and the development of IBD-like lesions are seen due to the over-expression of TNF-α and signal transducer and activator of transcription4 (STAT4) gene in TCR−/− mice [178,179]. Overall, there are various gene-manipulated models wherein epithelial barriers and immune regulation-associated genes are manipulated by knockdown, knock-in, conditional knockout, or transgenic mice [52], and reduced Treg cell number and their impaired functions were observed in these animals. The different animal models used to study IBD pathogenesis is depicted in Figure 3.
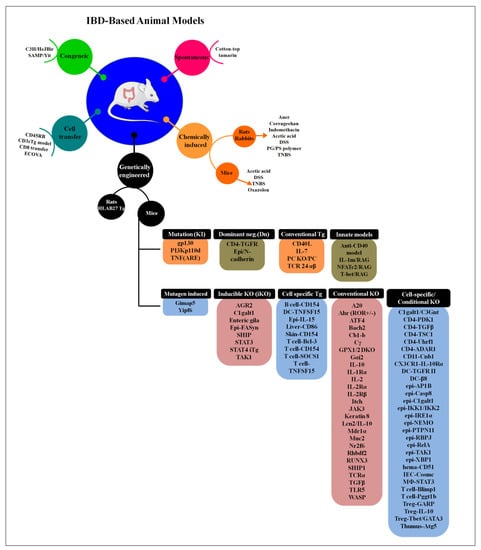
Figure 3.
The different types of animal model used to study IBD pathogenesis. In genetically engineered type, different genes are targeting and according to it they are mainly divided into 9 different groups (the detailed information is reviewd in [162]) (adapted and modified from [157,162]).
6.3. Humanized Mouse Models
Characterization and pre-clinical therapeutic applications are being explored by studying the immune response of patients with IBD in the experimental animal model(s) without putting human life at risk. Evaluation of hematopoietic stem cells (HSCs) with mutations in Foxp3 and bone-marrow-derived CD34+ HSCs received from a patient with mutations and impaired function of IL-10Rshowed severe medical-refractory infantile-onset of IBD.HSCs administration inNOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ(NSG) mice lacking murine MHC-II and expressing HLA-DR1hardly showed the mounted intestinal inflammation as was seen in mice and humans carrying mutations in IL-10 or IL-10R genes [180,181]. The increased frequency of CD19+ B cells was assessed in spleen and mesenteric lymph nodes compared to the control in peripheral lymphoid cells reconstituted mice. Some patients with deleterious IL-10R mutations showed the development of B cell lymphoma and presented a barrier in assessing the potential role of this pathway in the regulation of B cell development [182,183]. Human immune cells recovered from reconstituted NSG mice were found non-responsive to the exogenous IL-10 treatment, and these observations were consistent with the results obtained by using peripheral blood mononuclear cells (PBMCs) from IBD patients [184]. Interestingly, NSG mice harboring transgene encoding human KITLG, GM-CSF, and IL3 (NSG-SGM3) injected with IL-10R1deficient PBMCs were seen susceptible to DSS-induced colitis compared to those receiving healthy control PBMCs. These experimental observations paved the ways to facilitate the development of therapeutic interventional approaches against patients with IL-10R mutations. Fully reconstituted immunodeficient mice with CD34+ HSCs isolated from patients with IL-10R mutations are not suitable for assessing the therapeutic biologics aiming at developing interventional approaches against IBD. Human immune system repopulated mice (humanized mice) would be appropriate to screen gene therapy-based approaches for restoring IL-10R signaling [184,185].
Interleukin-2 is a key cytokine that controls the differentiation, survival and function of Tregs [185,186,187]. Moreover, low dose IL-2 is known to activate Tregs in the peripheral blood and colonic lamina propria isolated from IBD patients in culture as well as HIS mice. And, Tyagi et al., 2021 explored the role of low dose IL-2 in expanding functional Tregs in HSC reconstituted NSG humanized mice [185]. NSG mice reconstituted with healthy donor PBMCs receiving rectal anema with TNBS on day 5 following immune reconstitution to induce colitis. Mice receiving low-dose (10K) IL-2 were shown to reduce the weight loss and histology scores compared to those receiving treatment with a higher (50K) dose of IL-2 [180,185].
Further, the percentage of Foxp3+ IL-10+ TGF-β+ natural Tregs, Foxp3− IL-10+ TGF-β− induced Tregs, CD127− induced Tregs and CD8+ Tregs was measured at different time points in DSS-induced experimental colitis model in murine lamina propria lymphocytes, mesenteric lymph nodes and peripheral blood [148]. %age of Foxp3+ IL-10+ TGF-β+ natural Tregs show a decrease during chronic inflammation induced by IBD in humans and mice and proliferated significantly during remission. The intestinal inflammation exhibited a decrease in the percentage of CD8+ Tregs and remained lower in the remittent stage of human IBD. Only enhanced proliferation of lamina propria lymphocytes derived CD8+ Treg was reported on day 7in DSS-induced murine colitis. Furthermore, results suggest that Foxp3+ IL-10+ TGF-β+ natural Tregs might be crucial for the suppression and protection from immune-related mucosal injury during the chronic stages in IBD [148].
Local delivery of low numbers of human Treg by intradermal injection was shown to prevent skin inflammation in a humanized mouse model (huPBL-SCID-huSkin allograft model) [188]. A dose of only 1 × 105 freshly isolated, non-expanded Tregs injected intradermally close to the transplanted human skin prevented the inflammation induced by the grafted tissue, and intraperitoneal injection of human allogeneic PBMCs and Tregs used were used as 400:1. Inhibition of epidermal thickening sustained Keratin-10 expression, absence of Keratin-16 up-regulation and prevention of human CD3+ T cell influx was observed following the cell administration [188]. Also, concomitant reduction of human T cells was observed in the lymph nodes and spleen of mice. Moreover, injection of Tregs at the contralateral side inhibits the skin inflammation which advocates for the reduction of local and systemic inflammation. In brief, local application of Treg might be an attractive strategy to suppress inflammation in vivo without requiring prior ex-vivo expansion [188]
The simplest way to reconstitute immunodeficient mice with human immune cells is through intravenous or intraperitoneal injection of HSC-CD34 cells, and immunodeficiency is a prerequisite to increase the receptivity of immunodeficient/transgenic mice for human cells or tissues engraftment and repopulation [189]. The extent of immunodeficiency influences survival and function of transplanted human cells, and thus SCID mice lacking T and B cells supported the engraftment followed by repopulation of human immune cells for several weeks [190,191]. However, due to residual innate immunity and leakiness (development of adaptive immune cells in aged mice), SCID mice did not fully support the engraftment of human cells. Reduced natural killer (NK) cells in non-obese diabetic NOD-SCID mice showed significantly improved human cell engraftment [192] due to the reduced production of IFN-γ in NOD/SCID. Profound and long-lasting impairment in adaptive and innate immunity by targeted mutation of IL-2R gamma-chain gene in NSG or BALB/c recombination activating gene (Rag)2−/−IL-2Rγ−/− mice exhibited stable and long-term survival of transplanted human cells and tissues [193,194,195,196,197] (Figure 4).
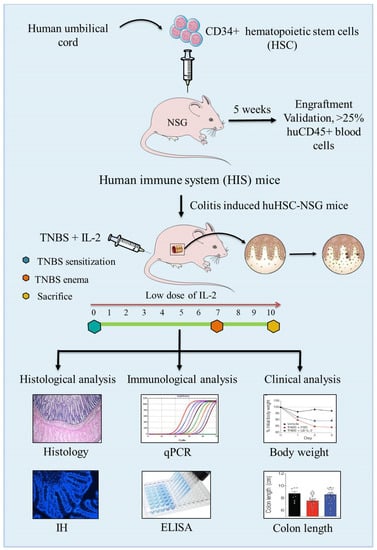
Figure 4.
Schematic presentation of the development of the human immune system (HIS)-repopulated mice by the transplantation of CD34+ human hematopoietic stem cells (HSCs) in NOD.PrkdcscidIl2rg−/− (NSG) immunodeficient mice and the induction of experimental colitis by TNBS to assess the pathology and immunopathogenesis of colitis.
7. Discussion
Different studies concluded that during the progression of IBD, T-lymphocytes exhibit a tremendous role in maintaining intestinal homeostasis and reducing tissue damage by inhibiting immune cell responsiveness with the help of Tregs [198,199]. Furthermore, defects in the number and distribution of functional Tregs and their impaired trafficking ability in the gastrointestinal tract have been examined in patients with IBD [18,104]. Recently, many studies have shown that IBD is ameliorated by restoring anti-inflammatory pathways, which mainly include increased Tregs numbers or maintaining Treg/Th17 balance by suppressing Th1/Th17 cells tipping the immune balance towards the generation of sizeable functional Tregs in IBD [21,200,201,202].
Unmet need for discovery of the novel therapeutic approaches prompt scientists to come up with alternative arrangements since many patients do not respond to treatment with approved drugs [203]. Despite the similarity between mouse and human flora, significant differences make it difficult to study all immune pathways responsible for IBD pathogenesis in one animal model. Therefore, extensive research and seamless efforts to design an ideal animal model aiming at developing a viable therapeutic strategy and selective treatment like small molecules, biologics, traditional and emerging modified therapies with minimal adverse reaction would establish the mouse-human chimeras a better platform to study autoimmune and chronic inflammatory disease. We believe humanized mouse models shall revolutionize the translational biomedical research to study inflammatory diseases.
Chemical induction, adoptive cell transfer, congenital models for IBD have been prominently utilized to study IBD pathogenesis. Further, genetically engineered murine models have been used to experimentally induce colitis to understand the mechanisms underlying intestinal inflammation and preclinical trials for developing novel therapeutic strategies [162]. Nearly 9 groups of IBD mouse models are developed based on gene targeting strategy which includes conventional, cell-specific, inducible, conventional, cell-specific transgenic, dominant-negative, mutagen induced, knock-in and innate [162]. Dysregulated innate and adaptive immune responses drive about 40 different immune-specific KO mice that spontaneously induce intestinal inflammation. The defective mucosal barriers, deregulated necroptosis/apoptosis, antibacterial peptide depletion and endoplasmic reticulum stress induce colitis spontaneously in 18 intestinal epithelial cell-specific KO mice. Although 74 types of genetically engineered mouse strains [163] and 160 IBD susceptible genes in humans indicating complex mechanism of IBD and diverse disease conditions, IBD remains elusive and needs to be studied. Out of 140 susceptible genes responsible for CD and UC, only a few had been identified as NOD2 CD-specific genes [163]. NOD2-KO and Atg16L1 mutant mice comprise of IL-10RA or IL-10RB mutation leading to the severe onset of complicated IBD pathogenesis [204]. Goettel et al. conducted studies on human immune cells reconstituted immunodeficient mice to ameliorate colitis by the expansion of Tregs following low dose IL-2 treatment and test therapeutic molecules [180].
Relevance of efficiency of Tregs as a therapeutic regime to control IBD has been demonstrated in experimental animal models and IBD patients [14,19,22,205]. The induced inflammation and severity of colitis was subsided by the adoptive transfer of Tregs [19], and promising results were observed with the use of ovalbumin (OVA)-specific Tregs in IBD [93,155,156]. The existing therapies to address IBD are based on broad suppression of inflammation that results in variable clinical advantages and unwanted adverse effects. However, passive transfer of Trges is a potential therapeutic regime aiming at promoting immune tolerance in the developed animal models, including humanized model with reconstituted human immune system mice (HIS) that supports the expansion of Tregs. Thus, induction of Tregs generation and re-establishment of immune tolerance leading to immune homeostasis is a potential approach for the long-term treatment of IBD since this regime might minimize the deleterious side effects associated with in-use immunosuppressive approaches.
8. Conclusions
The vital role played in IBD is evident since functional and phenotypic defects along with the compromised numbers and functions of Tregs. These cells significantly subside the intestinal inflammation mounted by the colitis. Therefore, future research concerning IBD progression and its treatment should focus on developing new clinical approaches to increase their regulatory effects including enhancing their homing to inflammation sites, expansion, or enhancing their differentiation, stability, and tissue repair properties. Further, enhancing their survival and anti-inflammatory property to maintain immune homeostasis in the gut is an important aspect. The engineered mouse-human chimera with the repopulated human immune system might be a stepping stone towards studying IBD, its biology and pathogenesis and help developing Tregs-based newer interventional strategies for IBD (Table 2).
Table 2.
Developed humanized mouse models to study IBD pathogenesis.
Author Contributions
Conceptualization: R.K.T. Writing: S.N., K.S., R.K.T. Formatting: S.S. and N.T. Preparation of figures and table(s): S.S. and N.T. Review and editing: R.K.T., R.P.N.M., N.T. and S.S. All authors have read and agreed to the published version of the manuscript.
Funding
Rajeev Tyagi would like to express his gratitude to Ramalingaswami Re-entry Fellowship Project (No. BT/RLF/Re-entry/27/2018), DBT, New Delhi, India for generously supporting this study.
Acknowledgments
Rajeev Tyagi offers his sincere thanks to the central instrumentation facility of CSIR-IMTECH, Chandigarh. He offers his gratitude to Jeremy A. Goettel, VUMC, USA to allow him studying the role of low-dose IL-2 in expanding Tregs in humanized animal model (CD34+ HSC) engrafted NSG mice.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| IBD | Inflammatory bowel disease |
| Tregs | Regulatory T-cells |
| CD | Crohn’s disease |
| UC | Ulcerative colitis |
| Th cells | T-helper cells |
| HIS | Human immune system |
| DCs | Dendritic cells |
| ILCs | Innate lymphoid cells |
| TNF | Tumor necrosis factor (TNF) |
| IFN-γ | Interferon-gamma |
| TGF-β | Transforming growth factor |
| TLA1 | TNF like cytokine 1A |
| DR3 | Death domain receptor 3 |
| TNBS | 2,4,6-trinitrobenzene sulfonic acid |
| DSS | Dextran sulfate sodium |
| NOD2 | Nucleotide-binding oligomerization domain 2 |
| ATG16L1 | Aumiddlehagy-related 16-like 1 |
| CCR | Chemokine receptor |
| RA | Retinoic acid |
| iTregs | Inducible Treg cells |
| Foxp3 | Forkhead box P3 |
| tTregs | Thymus-derived Treg cells |
| pTregs | Peripheral Treg cells |
| Nrp1 | Neuropilin-1 |
| TSDR | Treg specific demethylated region |
| Tr1 | Type 1 T-regulatory cells |
| GATA3 | GATA binding protein 3 |
| GM | Genetically modified |
| TLR | Toll-like receptor |
| HSCs | Hemamiddleoietic stem cells |
References
- Seyedian, S.S.; Nokhostin, F.; Malamir, M.D. A review of the diagnosis, prevention, and treatment methods of inflammatory bowel disease. J. Med. Life 2019, 12, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Yeshi, K.; Ruscher, R.; Hunter, L.; Daly, N.L.; Loukas, A.; Wangchuk, P. Revisiting Inflammatory Bowel Disease: Pathology, Treatments, Challenges and Emerging Therapeutics Including Drug Leads from Natural Products. J. Clin. Med. 2020, 9, 1273. [Google Scholar] [CrossRef]
- de Souza, H.S.P.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef] [Green Version]
- Loddo, I.; Romano, C. Inflammatory Bowel Disease: Genetics, Epigenetics, and Pathogenesis. Front. Immunol. 2015, 6, 551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imam, T.; Park, S.; Kaplan, M.H.; Olson, M.R. Effector T Helper Cell Subsets in Inflammatory Bowel Diseases. Front. Immunol. 2018, 9, 1212. [Google Scholar] [CrossRef] [PubMed]
- Tindemans, I.; Joosse, M.E.; Samsom, J.N. Dissecting the heterogeneity in T-cell mediated inflammation in IBD. Cells 2020, 9, 110. [Google Scholar] [CrossRef] [Green Version]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S. Naturally arising Foxp3− expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat. Immunol. 2005, 6, 345–352. [Google Scholar] [CrossRef]
- Pereira, L.M.S.; Gomes, S.T.M.; Ishak, R.; Vallinoto, A.C.R. Regulatory T Cell and Forkhead Box Protein 3 as Modulators of Immune Homeostasis. Front. Immunol. 2017, 8, 605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Hu, B.; Xu, D.; Liew, F.Y. CD4+CD25+ regulatory T cells cure murine colitis: The role of IL-10, TGF-beta, and CTLA4. J. Immunol. 2003, 171, 5012–5017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wing, K.; Sakaguchi, S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat. Immunol. 2010, 11, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Fanelli, G.; Albany, C.J.; Giganti, G.; Lombardi, G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Mohammadnia-Afrouzi, M.; Hosseini, A.; Khalili, A.; Abediankenari, S.; Hosseini, V.; Maleki, I. Decrease of CD4(+) CD25(+) CD127(low) Foxp3(+) regulatory T cells with impaired suppressive function in untreated ulcerative colitis patients. Autoimmunity 2015, 48, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Boschetti, G.; Kanjarawi, R.; Bardel, E.; Collardeau-Frachon, S.; Duclaux-Loras, R.; Moro-Sibilot, L.; Almeras, T.; Flourié, B.; Nancey, S.; Kaiserlian, D. Gut Inflammation in Mice Triggers Proliferation and Function of Mucosal Foxp3+ Regulatory T Cells but Impairs Their Conversion from CD4+ T Cells. J. Crohns. Colitis. 2016, 11, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sznurkowska, K.; Luty, J.; Bryl, E.; Witkowski, J.M.; Hermann-Okoniewska, B.; Landowski, P.; Kosek, M.; Szlagatys-Sidorkiewicz, A. Enhancement of Circulating and Intestinal T Regulatory Cells and Their Expression of Helios and Neuropilin-1 in Children with Inflammatory Bowel Disease. J. Inflamm. Res. 2020, 13, 995–1005. [Google Scholar] [CrossRef]
- Bacchetta, R.; Barzaghi, F.; Roncarolo, M.-G. From IPEX syndrome to FOXP3 mutation: A lesson on immune dysregulation. Ann. N. Y. Acad. Sci. 2018, 1417, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Pedros, C.; Duguet, F.; Saoudi, A.; Chabod, M. Disrupted regulatory T cell homeostasis in inflammatory bowel diseases. World J. Gastroenterol. 2016, 22, 974–995. [Google Scholar] [CrossRef]
- Mayne, C.G.; Williams, C.B. Induced and natural regulatory T cells in the development of inflammatory bowel disease. Inflamm. Bowel Dis. 2013, 19, 1772–1788. [Google Scholar] [CrossRef] [Green Version]
- Rocamora-Reverte, L.; Tuzlak, S.; von Raffay, L.; Tisch, M.; Fiegl, H.; Drach, M.; Reichardt, H.M.; Villunger, A.; Tischner, D.; Wiegers, G.J. Glucocorticoid receptor-deficient Foxp3+ regulatory T cells fail to control experimental inflammatory bowel disease. Front. Immunol. 2019, 10, 472. [Google Scholar] [CrossRef]
- Giuffrida, P.; Cococcia, S.; Delliponti, M.; Lenti, M.V.; Di Sabatino, A. Controlling gut inflammation by restoring anti-inflammatory pathways in inflammatory bowel disease. Cells 2019, 8, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clough, J.N.; Omer, O.S.; Tasker, S.; Lord, G.M.; Irving, P.M. Regulatory T-cell therapy in Crohn’s disease: Challenges and advances. Gut 2020, 69, 942–952. [Google Scholar] [CrossRef] [Green Version]
- Mottet, C.; Uhlig, H.H.; Powrie, F. Cutting edge: Cure of colitis by CD4+CD25+ regulatory T cells. J. Immunol. 2003, 170, 3939–3943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canavan, J.B.; Scottà, C.; Vossenkämper, A.; Goldberg, R.; Elder, M.J.; Shoval, I.; Marks, E.; Stolarczyk, E.; Lo, J.W.; Powell, N.; et al. Developing in vitro expanded CD45RA+ regulatory T cells as an adoptive cell therapy for Crohn’s disease. Gut 2016, 65, 584–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iftekhar, A.; Sigal, M. Defence and adaptation mechanisms of the intestinal epithelium upon infection. Int. J. Med. Microbiol. 2021, 311, 151486. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.O. Fight them or feed them: How the intestinal mucus layer manages the gut microbiota. Gastroenterol. Rep. 2019, 7, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakshani, C.R.; Morales-Garcia, A.L.; Althaus, M.; Wilcox, M.D.; Pearson, J.P.; Bythell, J.C.; Burgess, J.G. Evolutionary conservation of the antimicrobial function of mucus: A first defence against infection. npj Biofilms Microbiomes 2018, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Zhang, J. Role of intestinal microbiota and metabolites on gut homeostasis and human diseases. Bmc Immunol. 2017, 18, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagg, A.J. Intestinal dendritic cells in health and gut inflammation. Front. Immunol. 2018, 9, 2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denning, T.L.; Wang, Y.C.; Patel, S.R.; Williams, I.R.; Pulendran, B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 2007, 8, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ye, Q.; Zeng, X.; Qiao, S. Functions of macrophages in the maintenance of intestinal homeostasis. J. Immunol. Res. 2019, 2019, 1512969. [Google Scholar] [CrossRef] [Green Version]
- Pandiyan, P.; Bhaskaran, N.; Zou, M.; Schneider, E.; Jayaraman, S.; Huehn, J. Microbiome dependent regulation of Tregs and Th17 cells in mucosa. Front. Immunol. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef] [PubMed]
- Okumura, R.; Takeda, K. Maintenance of gut homeostasis by the mucosal immune system. Proc. Jpn. Acad. Ser. B 2016, 92, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Yue, B.; Luo, X.; Yu, Z.; Mani, S.; Wang, Z.; Dou, W. Inflammatory bowel disease: A potential result from the collusion between gut microbiota and mucosal immune system. Microorganisms 2019, 7, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Chen, Z. Inflammatory bowel disease related innate immunity and adaptive immunity. Am. J. Transl. Res. 2016, 8, 2490–2497. [Google Scholar] [PubMed]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine networks in the pathophysiology of inflammatory bowel disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Guan, Q.; Zhang, J. Recent Advances: The Imbalance of Cytokines in the Pathogenesis of Inflammatory Bowel Disease. Mediat. Inflamm. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, N.; Hisamatsu, T.; Honda, H.; Kobayashi, T.; Chinen, H.; Takayama, T.; Kitazume, M.; Okamoto, S.; Koganei, K.; Sugita, A.; et al. TL1A Produced by Lamina Propria Macrophages Induces Th1 and Th17 Immune Responses in Cooperation with IL-23 in Patients with Crohn’s Disease. Inflamm. Bowel Dis. 2010, 16, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Sidhu-Varma, M.; Shih, D.Q.; Targan, S.R. Differential Levels of Tl1a Affect the Expansion and Function of Regulatory T Cells in Modulating Murine Colitis. Inflamm. Bowel Dis. 2016, 22, 548–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takedatsu, H.; Michelsen, K.S.; Wei, B.; Landers, C.J.; Thomas, L.S.; Dhall, D.; Braun, J.; Targan, S.R. TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T-helper 1 and T-helper 17 activation. Gastroenterology 2008, 135, 552–567. [Google Scholar] [CrossRef] [Green Version]
- Meylan, F.; Song, Y.J.; Fuss, I.; Villarreal, S.; Kahle, E.; Malm, I.J.; Acharya, K.; Ramos, H.L.; Lo, L.; Mentink-Kane, M.M.; et al. The TNF-family cytokine TL1A drives IL-13-dependent small intestinal inflammation. Mucosal. Immunol. 2011, 4, 172–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Kim, N.-M.; Jiang, Y.-W.; Zhang, H.; Zheng, D.; Zhu, F.-X.; Liang, R.; Li, B.; Xu, H.-X. Cambogin suppresses dextran sulphate sodium-induced colitis by enhancing Treg cell stability and function. Br. J. Pharm. 2018, 175, 1085–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, M.H.; Kamada, N.; Warner, N.; Kim, Y.-G.; Nuñez, G. The ever-expanding function of NOD2: Autophagy, viral recognition, and T cell activation. Trends Immunol. 2011, 32, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuballa, P.; Huett, A.; Rioux, J.D.; Daly, M.J.; Xavier, R.J. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS ONE 2008, 3, e3391. [Google Scholar] [CrossRef]
- Cooney, R.; Baker, J.; Brain, O.; Danis, B.; Pichulik, T.; Allan, P.; Ferguson, D.J.; Campbell, B.J.; Jewell, D.; Simmons, A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 2010, 16, 90–97. [Google Scholar] [CrossRef]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.-G.; Magalhães, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Barreau, F.; Meinzer, U.; Chareyre, F.; Berrebi, D.; Niwa-Kawakita, M.; Dussaillant, M.; Foligne, B.; Ollendorff, V.; Heyman, M.; Bonacorsi, S.; et al. CARD15/NOD2 is required for Peyer’s patches homeostasis in mice. PLoS ONE 2007, 2, e523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabat, A.; Harrison, O.; Riffelmacher, T.; Moghaddam, A.E.; Pearson, C.; Laing, A.; Abeler-Dörner, L.; Forman, S.; Grencis, R.; Sattentau, Q.; et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. eLife 2016, 5, e12444. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Arakaki, R.; Saito, M.; Tsunematsu, T.; Kudo, Y.; Ishimaru, N. Role of regulatory T cell in the pathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.A.; Meingassner, J.G.; Lipp, M.; Moore, H.D.; Rot, A. CCR7 is required for the in vivo function of CD4+ CD25+ regulatory T cells. J. Exp. Med. 2007, 204, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Bromley, S.K.; Means, T.K.; Jones, K.J.; Hayashi, F.; Bhan, A.K.; Luster, A.D. CCR4-dependent regulatory T cell function in inflammatory bowel disease. J. Exp. Med. 2007, 204, 1327–1334. [Google Scholar] [CrossRef] [Green Version]
- McNamee, E.N.; Masterson, J.C.; Veny, M.; Collins, C.B.; Jedlicka, P.; Byrne, F.R.; Ng, G.Y.; Rivera-Nieves, J. Chemokine receptor CCR7 regulates the intestinal TH1/TH17/Treg balance during Crohn’s-like murine ileitis. J. Leukoc. Biol. 2015, 97, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Kuk, W.; Rivera-Nieves, J.; Lopez-Ramirez, M.A.; Eckmann, L.; Ginsberg, M.H. β7 Integrin Inhibition Can Increase Intestinal Inflammation by Impairing Homing of CD25(hi)Foxp3(+) Regulatory T Cells. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 369–385. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, D.; Tsun, A.; Li, B. Foxp3+ regulatory T cells and their functional regulation. Cell Mol. Immunol. 2015, 12, 558–565. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription FactorFoxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Putnam, A.L.; Xu-Yu, Z.; Szot, G.L.; Lee, M.R.; Zhu, S.; Gottlieb, P.A.; Kapranov, P.; Gingeras, T.R.; Fazekas de St Groth, B.; et al. CD127 expression inversely correlates with Foxp3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 2006, 203, 1701–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trzonkowski, P.; Bieniaszewska, M.; Juścińska, J.; Dobyszuk, A.; Krzystyniak, A.; Marek, N.; Myśliwska, J.; Hellmann, A. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+ CD25+ CD127−T regulatory cells. Clin. Immunol. 2009, 133, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Haribhai, D.; Williams, J.B.; Jia, S.; Nickerson, D.; Schmitt, E.G.; Edwards, B.; Ziegelbauer, J.; Yassai, M.; Li, S.-H.; Relland, L.M. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity 2011, 35, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Josefowicz, S.Z.; Niec, R.E.; Kim, H.Y.; Treuting, P.; Chinen, T.; Zheng, Y.; Umetsu, D.T.; Rudensky, A.Y. Extrathymically generated regulatory T cells control mucosal TH 2 inflammation. Nature 2012, 482, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Darrigues, J.; van Meerwijk, J.P.; Romagnoli, P. Age-dependent changes in regulatory T lymphocyte development and function: A mini-review. Gerontology 2018, 64, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Fujikado, N.; Kolodin, D.; Benoist, C.; Mathis, D. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 2015, 348, 589–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, W.; Beckett, O.; Ma, Q.; Li, M.O. Transforming growth factor-β signaling curbs thymic negative selection promoting regulatory T cell development. Immunity 2010, 32, 642–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretschmer, K.; Apostolou, I.; Hawiger, D.; Khazaie, K.; Nussenzweig, M.C.; von Boehmer, H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 2005, 6, 1219–1227. [Google Scholar] [CrossRef]
- Knoechel, B.; Lohr, J.; Kahn, E.; Bluestone, J.A.; Abbas, A.K. Sequential development of interleukin 2–dependent effector and regulatory T cells in response to endogenous systemic antigen. J. Exp. Med. 2005, 202, 1375–1386. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.; Bluestone, J.A.; Stephan, S. Peripherally induced tregs–role in immune homeostasis and autoimmunity. Front. Immunol. 2013, 4, 232. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Josefowicz, S.; Chaudhry, A.; Peng, X.P.; Forbush, K.; Rudensky, A.Y. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 2010, 463, 808–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.-j.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+ CD25− naive T cells to CD4+ CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Schlenner, S.M.; Weigmann, B.; Ruan, Q.; Chen, Y.; von Boehmer, H. Smad3 binding to the Foxp3 enhancer is dispensable for the development of regulatory T cells with the exception of the gut. J. Exp. Med. 2012, 209, 1529–1535. [Google Scholar] [CrossRef] [Green Version]
- Tai, X.; Cowan, M.; Feigenbaum, L.; Singer, A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat. Immunol. 2005, 6, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-M.; Hall, J.A.; Blank, R.B.; Bouladoux, N.; Oukka, M.; Mora, J.R.; Belkaid, Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 2007, 204, 1775–1785. [Google Scholar] [CrossRef] [Green Version]
- Benson, M.J.; Pino-Lagos, K.; Rosemblatt, M.; Noelle, R.J. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J. Exp. Med. 2007, 204, 1765–1774. [Google Scholar] [CrossRef] [Green Version]
- Shevach, E.M.; Thornton, A.M. tTregs, pTregs, and iTregs: Similarities and differences. Immunol. Rev. 2014, 259, 88–102. [Google Scholar] [CrossRef] [Green Version]
- Shevyrev, D.; Tereshchenko, V. Treg heterogeneity, function, and homeostasis. Front. Immunol. 2020, 10, 3100. [Google Scholar] [CrossRef] [Green Version]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef]
- Collison, L.W.; Pillai, M.R.; Chaturvedi, V.; Vignali, D.A.A. Regulatory T cell suppression is potentiated by target T cells in a cell contact, IL-35- and IL-10-dependent manner. J. Immunol. 2009, 182, 6121–6128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, W.J.; Verbsky, J.W.; Barchet, W.; Colonna, M.; Atkinson, J.P.; Ley, T.J. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 2004, 21, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Gondek, D.C.; Lu, L.-F.; Quezada, S.A.; Sakaguchi, S.; Noelle, R.J. Cutting Edge: Contact-Mediated Suppression by CD4+CD25+ Regulatory Cells Involves a Granzyme B-Dependent, Perforin-Independent Mechanism. J. Immunol. 2005, 174, 1783–1786. [Google Scholar] [CrossRef] [Green Version]
- Karreci, E.S.; Eskandari, S.K.; Dotiwala, F.; Routray, S.K.; Kurdi, A.T.; Assaker, J.P.; Luckyanchykov, P.; Mihali, A.B.; Maarouf, O.; Borges, T.J. Human regulatory T cells undergo self-inflicted damage via granzyme pathways upon activation. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Lin, X.; Chen, M.; Liu, Y.; Guo, Z.; He, X.; Brand, D.; Zheng, S.G. Advances in distinguishing natural from induced Foxp3(+) regulatory T cells. Int. J. Clin. Exp. Pathol. 2013, 6, 116–123. [Google Scholar]
- Singh, K.; Hjort, M.; Thorvaldson, L.; Sandler, S. Concomitant analysis of Helios and Neuropilin-1 as a marker to detect thymic derived regulatory T cells in naive mice. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgoffe, G.M.; Woo, S.-R.; Turnis, M.E.; Gravano, D.M.; Guy, C.; Overacre, A.E.; Bettini, M.L.; Vogel, P.; Finkelstein, D.; Bonnevier, J.; et al. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature 2013, 501, 252–256. [Google Scholar] [CrossRef]
- Thornton, A.M.; Korty, P.E.; Tran, D.Q.; Wohlfert, E.A.; Murray, P.E.; Belkaid, Y.; Shevach, E.M. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol. 2010, 184, 3433–3441. [Google Scholar] [CrossRef] [Green Version]
- Gottschalk, R.A.; Corse, E.; Allison, J.P. Expression of Helios in peripherally induced Foxp3+ regulatory T cells. J. Immunol. 2012, 188, 976–980. [Google Scholar] [CrossRef] [Green Version]
- Elkord, E. Helios should not be cited as a marker of human thymus-derived Tregs. Commentary: Helios+ and Helios− cells coexist within the natural Foxp3+ T regulatory cell subset in humans. Front. Immunol. 2016, 7, 276. [Google Scholar] [CrossRef] [Green Version]
- Szurek, E.; Cebula, A.; Wojciech, L.; Pietrzak, M.; Rempala, G.; Kisielow, P.; Ignatowicz, L. Differences in expression level of helios and neuropilin-1 do not distinguish thymus-derived from extrathymically-induced CD4+ Foxp3+ regulatory T cells. PLoS ONE 2015, 10, e0141161. [Google Scholar]
- Lee, S.; Park, K.; Kim, J.; Min, H.; Seong, R.H. Foxp3 expression in induced regulatory T cells is stabilized by C/EBP in inflammatory environments. Embo Rep. 2018, 19, e45995. [Google Scholar] [CrossRef] [PubMed]
- Polansky, J.K.; Kretschmer, K.; Freyer, J.; Floess, S.; Garbe, A.; Baron, U.; Olek, S.; Hamann, A.; von Boehmer, H.; Huehn, J. DNA methylation controls Foxp3 gene expression. Eur. J. Immunol. 2008, 38, 1654–1663. [Google Scholar] [CrossRef] [PubMed]
- Opstelten, R.; de Kivit, S.; Slot, M.C.; van den Biggelaar, M.; Iwaszkiewicz-Grześ, D.; Gliwiński, M.; Scott, A.M.; Blom, B.; Trzonkowski, P.; Borst, J. GPA33: A marker to identify stable human regulatory T cells. J. Immunol. 2020, 204, 3139–3148. [Google Scholar] [CrossRef]
- Levings, M.K.; Gregori, S.; Tresoldi, E.; Cazzaniga, S.; Bonini, C.; Roncarolo, M.G. Differentiation of Tr1 cells by immature dendritic cells requires IL-10 but not CD25+CD4+ Tr cells. Blood 2005, 105, 1162–1169. [Google Scholar] [CrossRef] [Green Version]
- Groux, H.; O’Garra, A.; Bigler, M.; Rouleau, M.; Antonenko, S.; De Vries, J.E.; Roncarolo, M.G. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997, 389, 737–742. [Google Scholar] [CrossRef]
- Zeng, H.; Zhang, R.; Jin, B.; Chen, L. Type 1 regulatory T cells: A new mechanism of peripheral immune tolerance. Cell Mol. Immunol. 2015, 12, 566–571. [Google Scholar] [CrossRef] [Green Version]
- Gagliani, N.; Magnani, C.F.; Huber, S.; Gianolini, M.E.; Pala, M.; Licona-Limon, P.; Guo, B.; De’Broski, R.H.; Bulfone, A.; Trentini, F. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med. 2013, 19, 739. [Google Scholar] [CrossRef]
- Barrat, F.J.; Cua, D.J.; Boonstra, A.; Richards, D.F.; Crain, C.; Savelkoul, H.F.; de Waal-Malefyt, R.; Coffman, R.L.; Hawrylowicz, C.M.; O’Garra, A. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J. Exp. Med. 2002, 195, 603–616. [Google Scholar] [CrossRef]
- Levings, M.K.; Sangregorio, R.; Galbiati, F.; Squadrone, S.; de Waal Malefyt, R.; Roncarolo, M.-G. IFN-α and IL-10 induce the differentiation of human type 1 T regulatory cells. J. Immunol. 2001, 166, 5530–5539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wildin, R.S.; Ramsdell, F.; Peake, J.; Faravelli, F.; Casanova, J.L.; Buist, N.; Levy-Lahad, E.; Mazzella, M.; Goulet, O.; Perroni, L.; et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 2001, 27, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef]
- Boehm, F.; Martin, M.; Kesselring, R.; Schiechl, G.; Geissler, E.K.; Schlitt, H.-J.; Fichtner-Feigl, S. Deletion of Foxp3+ regulatory T cells in genetically targeted mice supports development of intestinal inflammation. BMC Gastroenterol. 2012, 12, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maul, J.; Loddenkemper, C.; Mundt, P.; Berg, E.; Giese, T.; Stallmach, A.; Zeitz, M.; Duchmann, R. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology 2005, 128, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Holmén, N.; Lundgren, A.; Lundin, S.; Bergin, A.-M.; Rudin, A.; Sjövall, H.; Ohman, L. Functional CD4+CD25high regulatory T cells are enriched in the colonic mucosa of patients with active ulcerative colitis and increase with disease activity. Inflamm. Bowel Dis. 2006, 12, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Hovhannisyan, Z.; Treatman, J.; Littman, D.R.; Mayer, L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology 2011, 140, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, X.P.; Zhao, Z.B.; Chen, J.H.; Yu, C.G. Expression of CD4+ forkhead box P3 (FOXP3)+ regulatory T cells in inflammatory bowel disease. J. Dig. Dis 2011, 12, 286–294. [Google Scholar] [CrossRef]
- Eastaff-Leung, N.; Mabarrack, N.; Barbour, A.; Cummins, A.; Barry, S. Foxp3+ Regulatory T Cells, Th17 Effector Cells, and Cytokine Environment in Inflammatory Bowel Disease. J. Clin. Immunol. 2009, 30, 80–89. [Google Scholar] [CrossRef]
- Li, J.; Ueno, A.; Iacucci, M.; Fort Gasia, M.; Jijon, H.B.; Panaccione, R.; Kaplan, G.G.; Beck, P.L.; Luider, J.; Barkema, H.W.; et al. Crossover Subsets of CD4+ T Lymphocytes in the Intestinal Lamina Propria of Patients with Crohn’s Disease and Ulcerative Colitis. Dig. Dis. Sci. 2017, 62, 2357–2368. [Google Scholar] [CrossRef]
- Ueno, A.; Jijon, H.; Chan, R.; Ford, K.; Hirota, C.; Kaplan, G.G.; Beck, P.L.; Iacucci, M.; Fort Gasia, M.; Barkema, H.W.; et al. Increased prevalence of circulating novel IL-17 secreting Foxp3 expressing CD4+ T cells and defective suppressive function of circulating Foxp3+ regulatory cells support plasticity between Th17 and regulatory T cells in inflammatory bowel disease patients. Inflamm. Bowel Dis. 2013, 19, 2522–2534. [Google Scholar] [CrossRef]
- Yang, B.H.; Hagemann, S.; Mamareli, P.; Lauer, U.; Hoffmann, U.; Beckstette, M.; Föhse, L.; Prinz, I.; Pezoldt, J.; Suerbaum, S.; et al. Foxp3+ T cells expressing RORγt represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol. 2016, 9, 444–457. [Google Scholar] [CrossRef]
- Di Giovangiulio, M.; Rizzo, A.; Franzè, E.; Caprioli, F.; Facciotti, F.; Onali, S.; Favale, A.; Stolfi, C.; Fehling, H.-J.; Monteleone, G.; et al. Tbet Expression in Regulatory T Cells Is Required to Initiate Th1-Mediated Colitis. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Ogino, H.; Nakamura, K.; Ihara, E.; Akiho, H.; Takayanagi, R. CD4+CD25+ Regulatory T Cells Suppress Th17-Responses in an Experimental Colitis Model. Dig. Dis. Sci. 2011, 56, 376–386. [Google Scholar] [CrossRef]
- Uhlig, H.H.; Coombes, J.; Mottet, C.; Izcue, A.; Thompson, C.; Fanger, A.; Tannapfel, A.; Fontenot, J.D.; Ramsdell, F.; Powrie, F. Characterization of Foxp3+ CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J. Immunol. 2006, 177, 5852–5860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, O.J.; Srinivasan, N.; Pott, J.; Schiering, C.; Krausgruber, T.; Ilott, N.E.; Maloy, K.J. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3+ Treg cell function in the intestine. Mucosal Immunol. 2015, 8, 1226–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, B.; Banz, A.; Bienvenu, B.; Cordier, C.; Dautigny, N.; Bécourt, C.; Lucas, B. Suppression of CD4+ T lymphocyte effector functions by CD4+ CD25+ cells in vivo. J. Immunol. 2004, 172, 3391–3398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin–EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, L.; Stahl, M.; Han, X.; Nazli, A.; MacDonald, K.N.; Wong, M.Q.; Tsai, K.; Dizzell, S.; Jacobson, K.; Bressler, B. Suppressive and gut-reparative functions of human type 1 T regulatory cells. Gastroenterology 2019, 157, 1584–1598. [Google Scholar] [CrossRef] [Green Version]
- Ananthakrishnan, A.N.; Cagan, A.; Cai, T.; Gainer, V.S.; Shaw, S.Y.; Savova, G.; Churchill, S.; Karlson, E.W.; Kohane, I.; Liao, K.P. Comparative effectiveness of infliximab and adalimumab in Crohn’s disease and ulcerative colitis. Inflamm. Bowel Dis. 2016, 22, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Vulliemoz, M.; Brand, S.; Juillerat, P.; Mottet, C.; Ben-Horin, S.; Michetti, P. TNF-Alpha Blockers in Inflammatory Bowel Diseases: Practical Recommendations and a User’s Guide: An Update. Digestion 2020, 101, 20–30. [Google Scholar] [CrossRef]
- Ford, A.C.; Peyrin-Biroulet, L. Opportunistic Infections with Anti-Tumor Necrosis Factor-α Therapy in Inflammatory Bowel Disease: Meta-Analysis of Randomized Controlled Trials. Am. J. Gastroenterol. 2013, 108, 1268–1276. [Google Scholar] [CrossRef]
- Fellermann, K. Adverse events of tumor necrosis factor inhibitors. Dig. Dis. 2013, 31, 374–378. [Google Scholar] [CrossRef]
- Kirchgesner, J.; Lemaitre, M.; Carrat, F.; Zureik, M.; Carbonnel, F.; Dray-Spira, R. Risk of serious and opportunistic infections associated with treatment of inflammatory bowel diseases. Gastroenterology 2018, 155, 337–346.e310. [Google Scholar] [CrossRef] [Green Version]
- Deepak, P.; Sifuentes, H.; Sherid, M.; Stobaugh, D.; Sadozai, Y.; Ehrenpreis, E.D. T-cell non-Hodgkin’s lymphomas reported to the FDA AERS with tumor necrosis factor-alpha (TNF-α) inhibitors: Results of the REFURBISH study. Off. J. Am. Coll. Gastroenterol. ACG 2013, 108, 99–105. [Google Scholar] [CrossRef]
- Lemaitre, M.; Kirchgesner, J.; Rudnichi, A.; Carrat, F.; Zureik, M.; Carbonnel, F.; Dray-Spira, R. Association between use of thiopurines or tumor necrosis factor antagonists alone or in combination and risk of lymphoma in patients with inflammatory bowel disease. JAMA 2017, 318, 1679–1686. [Google Scholar] [CrossRef]
- Eickstaedt, J.B.; Killpack, L.; Tung, J.; Davis, D.; Hand, J.L.; Tollefson, M.M. Psoriasis and psoriasiform eruptions in pediatric patients with inflammatory bowel disease treated with anti–tumor necrosis factor alpha agents. Pediatric Dermatol. 2017, 34, 253–260. [Google Scholar] [CrossRef]
- Decock, A.; Van Assche, G.; Vermeire, S.; Wuyts, W.; Ferrante, M. Sarcoidosis-like lesions: Another paradoxical reaction to anti-TNF therapy? J. Crohns. Colitis 2017, 11, 378–383. [Google Scholar] [CrossRef]
- Alivernini, S.; Pugliese, D.; Tolusso, B.; Bui, L.; Petricca, L.; Guidi, L.; Mirone, L.; Rapaccini, G.L.; Federico, F.; Ferraccioli, G. Paradoxical arthritis occurring during anti-TNF in patients with inflammatory bowel disease: Histological and immunological features of a complex synovitis. Rmd Open 2018, 4, e000667. [Google Scholar] [CrossRef]
- Perše, M.; Unkovič, A. The role of TNF in the pathogenesis of inflammatory bowel disease. In Biological Therapy for Inflammatory Bowel Disease; IntechOpen: London, UK, 2019. [Google Scholar]
- Shovman, O.; Tamar, S.; Amital, H.; Watad, A.; Shoenfeld, Y. Diverse patterns of anti-TNF-α-induced lupus: Case series and review of the literature. Clin. Rheumatol. 2018, 37, 563–568. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Wolf, D.C.; D’Haens, G.; Vermeire, S.; Hanauer, S.B.; Ghosh, S.; Smith, H.; Cravets, M.; Frohna, P.A. Ozanimod induction and maintenance treatment for ulcerative colitis. N. Engl. J. Med. 2016, 374, 1754–1762. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Sands, B.E.; Danese, S.; D’Haens, G.R.; Vermeire, S.; Schreiber, S.; Feagan, B.G.; Reinisch, W.; Friedman, G.; Woodworth, D.A. Efficacy and safety of oral tofacitinib as maintenance therapy in patients with moderate to severe ulcerative colitis: Results from a phase 3 randomised controlled trial. Gastroenterology 2017, 152, S199. [Google Scholar] [CrossRef]
- D’Haens, G.R.; Panaccione, R.; Higgins, P.; Colombel, J.-F.; Feagan, B.G.; Moscariello, M.; Chan, G.; Healey, P.J.; Niezychowski, W.; Wang, W. 856 Efficacy and safety of oral tofacitinib for maintenance therapy in patients with moderate to severe Crohn’s disease: Results of a phase 2B randomized placebo-controlled trial. Gastroenterology 2016, 150, S183. [Google Scholar] [CrossRef]
- Villablanca, E.J.; Cassani, B.; Von Andrian, U.H.; Mora, J.R. Blocking lymphocyte localization to the gastrointestinal mucosa as a therapeutic strategy for inflammatory bowel diseases. Gastroenterology 2011, 140, 1776–1784.e1775. [Google Scholar] [CrossRef] [Green Version]
- Feagan, B.G.; Rutgeerts, P.; Sands, B.E.; Hanauer, S.; Colombel, J.-F.; Sandborn, W.J.; Van Assche, G.; Axler, J.; Kim, H.-J.; Danese, S. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2013, 369, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Sands, B.E.; Feagan, B.G.; Rutgeerts, P.; Colombel, J.-F.; Sandborn, W.J.; Sy, R.; D’Haens, G.; Ben-Horin, S.; Xu, J.; Rosario, M. Effects of vedolizumab induction therapy for patients with Crohn’s disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology 2014, 147, 618–627.e613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombel, J.-F.; Sands, B.E.; Rutgeerts, P.; Sandborn, W.; Danese, S.; D’Haens, G.; Panaccione, R.; Loftus, E.V.; Sankoh, S.; Fox, I. The safety of vedolizumab for ulcerative colitis and Crohn’s disease. Gut 2017, 66, 839–851. [Google Scholar] [CrossRef]
- Loftus Jr, E.V.; Colombel, J.-F.; Feagan, B.G.; Vermeire, S.; Sandborn, W.J.; Sands, B.E.; Danese, S.; D’Haens, G.R.; Kaser, A.; Panaccione, R. Long-term efficacy of vedolizumab for ulcerative colitis. J. Crohns. Colitis 2017, 11, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Monteleone, G.; Neurath, M.F.; Ardizzone, S.; Di Sabatino, A.; Fantini, M.C.; Castiglione, F.; Scribano, M.L.; Armuzzi, A.; Caprioli, F.; Sturniolo, G.C.; et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N. Engl. J. Med. 2015, 372, 1104–1113. [Google Scholar] [CrossRef] [Green Version]
- Marafini, I.; Monteleone, I.; Dinallo, V.; Di Fusco, D.; De Simone, V.; Laudisi, F.; Fantini, M.C.; Di Sabatino, A.; Pallone, F.; Monteleone, G. CCL20 is negatively regulated by TGF-β1 in intestinal epithelial cells and reduced in Crohn’s disease patients with a successful response to Mongersen, a Smad7 antisense oligonucleotide. J. Crohns. Colitis 2017, 11, 603–609. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sands, B.E.; Rossiter, G.; Li, X.; Usiskin, K.; Zhan, X.; Colombel, J.-F. Effects of mongersen (GED-0301) on endoscopic and clinical outcomes in patients with active Crohn’s disease. Gastroenterology 2018, 154, 61–64.e66. [Google Scholar] [CrossRef]
- Danese, S.; Furfaro, F.; Vetrano, S. Targeting S1P in inflammatory bowel disease: New avenues for modulating intestinal leukocyte migration. J. Crohns. Colitis 2018, 12, S678–S686. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, C.L.; Kimball, A.B.; Papp, K.A.; Yeilding, N.; Guzzo, C.; Wang, Y.; Li, S.; Dooley, L.T.; Gordon, K.B.; Investigators, P.S. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008, 371, 1665–1674. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Fedorak, R.N.; Scherl, E.; Fleisher, M.R.; Katz, S.; Johanns, J.; Blank, M.; Rutgeerts, P.; Ustekinumab Crohn’s Disease Study Group. A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn’s disease. Gastroenterology 2008, 135, 1130–1141. [Google Scholar] [CrossRef] [Green Version]
- Sandborn, W.J.; Gasink, C.; Gao, L.-L.; Blank, M.A.; Johanns, J.; Guzzo, C.; Sands, B.E.; Hanauer, S.B.; Targan, S.; Rutgeerts, P. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N. Engl. J. Med. 2012, 367, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Hibi, T.; Imai, Y.; Murata, Y.; Matsushima, N.; Zheng, R.; Gasink, C. Efficacy and safety of ustekinumab in Japanese patients with moderately to severely active Crohn’s disease: A subpopulation analysis of phase 3 induction and maintenance studies. Intest Res. 2017, 15, 475. [Google Scholar] [CrossRef] [Green Version]
- Nigam, G.B.; Limdi, J.K. An update on the role of anti-IL-12/IL23 agents in the management of inflammatory bowel disease. Br. Med. Bull. 2021, 138, 29–40. [Google Scholar] [CrossRef]
- Jefremow, A.; Neurath, M.F. All are Equal, Some are More Equal: Targeting IL 12 and 23 in IBD—A Clinical Perspective. Immunotargets Ther. 2020, 9, 289. [Google Scholar] [CrossRef]
- Kashani, A.; Schwartz, D.A. The Expanding Role of Anti–IL-12 and/or Anti–IL-23 Antibodies in the Treatment of Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2019, 15, 255. [Google Scholar]
- Misselwitz, B.; Juillerat, P.; Sulz, M.C.; Siegmund, B.; Brand, S. Emerging treatment options in inflammatory Bowel disease: Janus Kinases, stem cells, and more. Digestion 2020, 101, 69–82. [Google Scholar] [CrossRef]
- Sun, X.; He, S.; Lv, C.; Sun, X.; Wang, J.; Zheng, W.; Wang, D. Analysis of murine and human Treg subsets in inflammatory bowel disease. Mol. Med. Rep. 2017, 16, 2893–2898. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.-H.; Zhang, J.; Chen, X.; Xie, Y.-F.; Pang, Y.-H.; Liu, X.-J. Increased CD4 + CD45RA - Foxp3 low cells alter the balance between Treg and Th17 cells in colitis mice. World J. Gastroenterol. 2016, 22, 9356. [Google Scholar] [CrossRef]
- Gong, Y.; Lin, Y.; Zhao, N.; He, X.; Lu, A.; Wei, W.; Jiang, M. The Th17/Treg Immune Imbalance in Ulcerative Colitis Disease in a Chinese Han Population. Mediat. Inflamm. 2016, 2016, 7089137. [Google Scholar] [CrossRef] [Green Version]
- Geng, X.; Xue, J. Expression of Treg/Th17 cells as well as related cytokines in patients with inflammatory bowel disease. Pak. J. Med. Sci. 2016, 32, 1164–1168. [Google Scholar] [CrossRef]
- Chaudhry, A.; Samstein, R.M.; Treuting, P.; Liang, Y.; Pils, M.C.; Heinrich, J.-M.; Jack, R.S.; Wunderlich, F.T.; Brüning, J.C.; Müller, W.; et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 2011, 34, 566–578. [Google Scholar] [CrossRef] [Green Version]
- Song-Zhao, G.X.; Maloy, K.J. Experimental mouse models of T cell-dependent inflammatory bowel disease. Methods Mol. Biol. 2014, 1193, 199–211. [Google Scholar] [CrossRef]
- Ogino, H.; Nakamura, K.; Iwasa, T.; Ihara, E.; Akiho, H.; Motomura, Y.; Akahoshi, K.; Igarashi, H.; Kato, M.; Kotoh, K.; et al. Regulatory T cells expanded by rapamycin in vitro suppress colitis in an experimental mouse model. J. Gastroenterol. 2012, 47, 366–376. [Google Scholar] [CrossRef]
- Zhou, P.; Borojevic, R.; Streutker, C.; Snider, D.; Liang, H.; Croitoru, K. Expression of dual TCR on DO11.10 T cells allows for ovalbumin-induced oral tolerance to prevent T cell-mediated colitis directed against unrelated enteric bacterial antigens. J. Immunol. 2004, 172, 1515–1523. [Google Scholar] [CrossRef] [Green Version]
- Desreumaux, P.; Foussat, A.; Allez, M.; Beaugerie, L.; Hébuterne, X.; Bouhnik, Y.; Nachury, M.; Brun, V.; Bastian, H.; Belmonte, N.; et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology 2012, 143, 1207–1217.e1202. [Google Scholar] [CrossRef]
- Mizoguchi, A. Animal models of inflammatory bowel disease. Prog. Mol. Biol. Transl. Sci. 2012, 105, 263–320. [Google Scholar] [CrossRef]
- Cominelli, F.; Arseneau, K.O.; Rodriguez-Palacios, A.; Pizarro, T.T. Uncovering Pathogenic Mechanisms of Inflammatory Bowel Disease Using Mouse Models of Crohn’s Disease-Like Ileitis: What is the Right Model? Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Corridoni, D.; Arseneau, K.O.; Cominelli, F. Inflammatory bowel disease. Immunol. Lett 2014, 161, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 2017, 12, 1295–1309. [Google Scholar] [CrossRef]
- Hernández-Chirlaque, C.; Aranda, C.J.; Ocón, B.; Capitán-Cañadas, F.; Ortega-González, M.; Carrero, J.J.; Suárez, M.D.; Zarzuelo, A.; Sánchez de Medina, F.; Martínez-Augustin, O. Germ-free and Antibiotic-treated Mice are Highly Susceptible to Epithelial Injury in DSS Colitis. J. Crohns. Colitis 2016, 10, 1324–1335. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, E.; Low, D.; Ezaki, Y.; Okada, T. Recent updates on the basic mechanisms and pathogenesis of inflammatory bowel diseases in experimental animal models. Intest. Res. 2020, 18, 16. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, A.; Takeuchi, T.; Himuro, H.; Okada, T.; Mizoguchi, E. Genetically engineered mouse models for studying inflammatory bowel disease. J. Pathol. 2015, 238, 14. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, E.A.; King, K.Y.; Baldridge, M.T. Mouse Microbiota Models: Comparing Germ-Free Mice and Antibiotics Treatment as Tools for Modifying Gut Bacteria. Front. Physiol 2018, 9, 1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.A.; Bouladoux, N.; Sun, C.M.; Wohlfert, E.A.; Blank, R.B.; Zhu, Q.; Grigg, M.E.; Berzofsky, J.A.; Belkaid, Y. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity 2008, 29, 637–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. [Google Scholar] [CrossRef]
- Håkansson, Å.; Tormo-Badia, N.; Baridi, A.; Xu, J.; Molin, G.; Hagslätt, M.L.; Karlsson, C.; Jeppsson, B.; Cilio, C.M.; Ahrné, S. Immunological alteration and changes of gut microbiota after dextran sulfate sodium (DSS) administration in mice. Clin. Exp. Med. 2015, 15, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.C.; Jobin, C. The complex interplay between inflammation, the microbiota and colorectal cancer. Gut Microbes 2013, 4, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Yang, I.; Eibach, D.; Kops, F.; Brenneke, B.; Woltemate, S.; Schulze, J.; Bleich, A.; Gruber, A.D.; Muthupalani, S.; Fox, J.G.; et al. Intestinal microbiota composition of interleukin-10 deficient C57BL/6J mice and susceptibility to Helicobacter hepaticus-induced colitis. PLoS ONE 2013, 8, e70783. [Google Scholar] [CrossRef] [Green Version]
- Dennis, K.L.; Wang, Y.; Blatner, N.R.; Wang, S.; Saadalla, A.; Trudeau, E.; Roers, A.; Weaver, C.T.; Lee, J.J.; Gilbert, J.A.; et al. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10-producing T cells. Cancer Res. 2013, 73, 8. [Google Scholar] [CrossRef] [Green Version]
- Ettreiki, C.; Gadonna-Widehem, P.; Mangin, I.; Coëffier, M.; Delayre-Orthez, C.; Anton, P.M. Juvenile ferric iron prevents microbiota dysbiosis and colitis in adult rodents. World J. Gastroenterol. 2012, 18, 2619–2629. [Google Scholar] [CrossRef]
- Poussier, P.; Ning, T.; Chen, J.; Banerjee, D.; Julius, M. Intestinal inflammation observed in IL-2R/IL-2 mutant mice is associated with impaired intestinal T lymphopoiesis. Gastroenterology 2000, 118, 880–891. [Google Scholar] [CrossRef]
- Long, S.A.; Cerosaletti, K.; Bollyky, P.L.; Tatum, M.; Shilling, H.; Zhang, S.; Zhang, Z.-Y.; Pihoker, C.; Sanda, S.; Greenbaum, C. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4+ CD25+ regulatory T-cells of type 1 diabetic subjects. Diabetes 2010, 59, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Rubino, S.J.; Selvanantham, T.; Girardin, S.E.; Philpott, D.J. Nod-like receptors in the control of intestinal inflammation. Curr. Opin. Immunol. 2012, 24, 398–404. [Google Scholar] [CrossRef]
- Maggio-Price, L.; Shows, D.; Waggie, K.; Burich, A.; Zeng, W.; Escobar, S.; Morrissey, P.; Viney, J.L. Helicobacter bilis infection accelerates and H. hepaticus infection delays the development of colitis in multiple drug resistance-deficient (mdr1a-/-) mice. Am. J. Pathol. 2002, 160, 739–751. [Google Scholar] [CrossRef]
- Hapfelmeier, S.; Stecher, B.; Barthel, M.; Kremer, M.; Müller, A.J.; Heikenwalder, M.; Stallmach, T.; Hensel, M.; Pfeffer, K.; Akira, S. The Salmonella pathogenicity island (SPI)-2 and SPI-1 type III secretion systems allow Salmonella serovar typhimurium to trigger colitis via MyD88-dependent and MyD88-independent mechanisms. J. Immunol. 2005, 174, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Uematsu, S.; Jang, M.H.; Chevrier, N.; Guo, Z.; Kumagai, Y.; Yamamoto, M.; Kato, H.; Sougawa, N.; Matsui, H.; Kuwata, H. Detection of pathogenic intestinal bacteria by Toll-like receptor 5 on intestinal CD11c+ lamina propria cells. Nat. Immunol. 2006, 7, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Kontoyiannis, D.; Pasparakis, M.; Pizarro, T.T.; Cominelli, F.; Kollias, G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: Implications for joint and gut-associated immunopathologies. Immunity 1999, 10, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Burich, A.; Hershberg, R.; Waggie, K.; Zeng, W.; Brabb, T.; Westrich, G.; Viney, J.L.; Maggio-Price, L. Helicobacter-induced inflammatory bowel disease in IL-10-and T cell-deficient mice. Am. J. Physiol.-Gastrointest. Liver Physiol. 2001, 281, G764–G778. [Google Scholar] [CrossRef]
- Goettel, J.A.; Kotlarz, D.; Emani, R.; Canavan, J.B.; Konnikova, L.; Illig, D.; Frei, S.M.; Field, M.; Kowalik, M.; Peng, K.; et al. Low-Dose Interleukin-2 Ameliorates Colitis in a Preclinical Humanized Mouse Model. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 193–195. [Google Scholar] [CrossRef] [Green Version]
- Glocker, E.O.; Kotlarz, D.; Boztug, K.; Gertz, E.M.; Schaffer, A.A.; Noyan, F.; Perro, M.; Diestelhorst, J.; Allroth, A.; Murugan, D.; et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 2009, 361, 2033–2045. [Google Scholar] [CrossRef] [Green Version]
- Neven, B.; Mamessier, E.; Bruneau, J.; Kaltenbach, S.; Kotlarz, D.; Suarez, F.; Masliah-Planchon, J.; Billot, K.; Canioni, D.; Frange, P. A Mendelian predisposition to B-cell lymphoma caused by IL-10R deficiency. Blood J. Am. Soc. Hematol. 2013, 122, 3713–3722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shouval, D.S.; Ebens, C.L.; Murchie, R.; McCann, K.; Rabah, R.; Klein, C.; Muise, A.; Snapper, S.B. Large B-cell lymphoma in an adolescent patient with IL-10 receptor deficiency and history of infantile inflammatory bowel disease. J. Pediatric Gastroenterol. Nutr. 2016, 63, e15. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, R.K.; Li, J.; Jacobse, J.; Snapper, S.B.; Shouval, D.S.; Goettel, J.A. Humanized mouse models of genetic immune disorders and hematological malignancies. Biochem. Pharm. 2020, 174, 113671. [Google Scholar] [CrossRef]
- Tyagi, R.K.; Jacobse, J.; Li, J.; Allaman, M.M.; Otipoby, K.L.; Sampson, E.R.; Wilson, K.T.; Goettel, J.A. HLA-Restriction of Human Treg Cells Is Not Required for Therapeutic Efficacy of Low-Dose IL-2 in Humanized Mice. Front. Immunol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.H.; Cantrell, D.A. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu. Rev. Immunol. 2018, 36, 22. [Google Scholar] [CrossRef]
- Śledzińska, A.; Mucha, M.V.d.; Bergerhoff, K.; Hotblack, A.; Demane, D.F.; Ghorani, E.; Akarca, A.U.; Marzolini, M.A.V.; Solomon, I.; Vargas, F.A.; et al. Regulatory T Cells Restrain Interleukin-2- and Blimp-1-Dependent Acquisition of Cytotoxic Function by CD4 + T Cells. Immunity 2020, 52, 15. [Google Scholar] [CrossRef]
- Landman, S.; de Oliveira, V.L.; van Erp, P.E.J.; Fasse, E.; Bauland, S.C.G.; Joosten, I.; Koenen, H.J.P.M. Intradermal injection of low dose human regulatory T cells inhibits skin inflammation in a humanized mouse model. Sci. Rep. 2018, 8, 10044. [Google Scholar] [CrossRef]
- Zhang, B.; Duan, Z.; Zhao, Y. Mouse models with human immunity and their application in biomedical research. J. Cell. Mol. Med. 2009, 13, 1043–1058. [Google Scholar] [CrossRef] [Green Version]
- McCune, J.M.; Namikawa, R.; Kaneshima, H.; Shultz, L.D.; Lieberman, M.; Weissman, I.L. The SCID-hu mouse: Murine model for the analysis of human hematolymphoid differentiation and function. Science 1988, 241, 1632–1639. [Google Scholar] [CrossRef]
- Mosier, D.E.; Gulizia, R.J.; Baird, S.M.; Wilson, D.B. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature 1988, 335, 256–259. [Google Scholar] [CrossRef]
- Tournoy, K.G.; Depraetere, S.; Pauwels, R.A.; Leroux-Roels, G.G. Mouse strain and conditioning regimen determine survival and function of human leucocytes in immunodeficient mice. Clin. Exp. Immunol. 2000, 119, 9. [Google Scholar] [CrossRef]
- Ito, R.; Takahashi, T.; Katano, I.; Ito, M. Current advances in humanized mouse models. Cell Mol. Immunol. 2012, 9, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Shultz, L.D.; Brehm, M.A.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Pearson, T.; King, M.; Giassi, L.; Carney, L.; Gott, B.; Lyons, B.; Rossini, A.A.; Greiner, D.L. Humanized NOD/LtSz-scid IL2 receptor common gamma chain knockout mice in diabetes research. Ann. N. Y. Acad. Sci. 2007, 1103, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K. NOD/SCID/γ c null mouse: An excellent recipient mouse model for engraftment of human cells. Blood J. Am. Soc. Hematol. 2002, 100, 3175–3182. [Google Scholar]
- King, M.; Pearson, T.; Shultz, L.D.; Leif, J.; Bottino, R.; Trucco, M.; Atkinson, M.A.; Wasserfall, C.; Herold, K.C.; Woodland, R.T. A new Hu-PBL model for the study of human islet alloreactivity based on NOD-scid mice bearing a targeted mutation in the IL-2 receptor gamma chain gene. Clin. Immunol. 2008, 126, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Harrison, O.J.; Powrie, F.M. Regulatory T cells and immune tolerance in the intestine. Cold Spring Harb. Perspect. Biol. 2013, 5, a018341. [Google Scholar] [CrossRef]
- Sharma, A.; Rudra, D. Emerging functions of regulatory T cells in tissue homeostasis. Front. Immunol. 2018, 9, 883. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, L.-Y.; Wu, J.; Xu, L.-F.; Sun, M. The JAK2 inhibitor AG490 regulates the Treg/Th17 balance and alleviates DSS-induced intestinal damage in IBD rats. Clin. Exp. Pharm. Physiol 2020. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Liang, W.; Wang, T.; Sui, J.; Wang, J.; Deng, Z.; Chen, D. Saponins regulate intestinal inflammation in colon cancer and IBD. Pharm. Res. 2019, 144, 66–72. [Google Scholar] [CrossRef]
- Lee, S.H.; Kwon, J.E.; Cho, M.-L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef] [Green Version]
- Hazel, K.; O’Connor, A. Emerging treatments for inflammatory bowel disease. Ther. Adv. Chronic Dis. 2020, 11, 2040622319899297. [Google Scholar] [CrossRef] [PubMed]
- Hitotsumatsu, O.; Ahmad, R.-C.; Tavares, R.; Wang, M.; Philpott, D.; Turer, E.E.; Lee, B.L.; Shiffin, N.; Advincula, R.; Malynn, B.A.; et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity 2008, 28, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantini, M.C.; Monteleone, G. Update on the Therapeutic Efficacy of Tregs in IBD: Thumbs up or Thumbs down? Inflamm. Bowel Dis. 2017, 23, 1682–1688. [Google Scholar] [CrossRef]
- Weigmann, B.; Schughart, N.; Wiebe, C.; Sudowe, S.; Lehr, H.A.; Jonuleit, H.; Vogel, L.; Becker, C.; Neurath, M.F.; Grabbe, S.; et al. Allergen-induced IgE-dependent gut inflammation in a human PBMC-engrafted murine model of allergy. J. Allergy Clin. Immunol. 2012, 129, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Nolte, T.; Zadeh-Khorasani, M.; Safarov, O.; Rueff, F.; Gülberg, V.; Herbach, N.; Wollenberg, A.; Mueller, T.; Siebeck, M.; Wolf, E.; et al. Oxazolone and ethanol induce colitis in non-obese diabetic-severe combined immunodeficiency interleukin-2Rγ(null) mice engrafted with human peripheral blood mononuclear cells. Clin. Exp. Immunol. 2013, 172, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Goettel, J.; Shouval, D.; Lexmond, W.; Muise, A.; Fiebiger, E.; Snapper, S. Development of Novel Humanized Murine Models to Assess Mucosal Homeostasis: Human anti-CD3 Antibody or TNBS Administration Leads to Small and Large Bowel Inflammation Respectively in Immunodeficient Mice Transferred With Human T Cells. Gastroenterology 2013, 144, S-32. [Google Scholar] [CrossRef]
- Goettel, J.; Biswas, B.; Lexmond, W.; Sun, J.; Ouahed, J.; McCann, K.; Shouval, D.; Milford, E.; Fiebiger, E.; Muise, A.; et al. Human Hematopoietic Stem Cells With a Defined Immunodeficiency and Enteropathy Transfer Clinical Phenotype to a Novel Humanized Mouse Strain. Gastroenterology 2014, 146, S-81. [Google Scholar] [CrossRef]
- Goettel, J.A.; Gandhi, R.; Kenison, J.E.; Yeste, A.; Murugaiyan, G.; Sambanthamoorthy, S.; Griffith, A.E.; Patel, B.; Shouval, D.S.; Weiner, H.L.; et al. AHR Activation Is Protective against Colitis Driven by T Cells in Humanized Mice. Cell Rep. 2016, 17, 1318–1329. [Google Scholar] [CrossRef] [Green Version]
- Jodeleit, H.; Winkelmann, P.; Caesar, J.; Sterz, S.; Holdt, L.M.; Beigel, F.; Stallhofer, J.; Breiteneicher, S.; Bartnik, E.; Leeuw, T.; et al. Head-to-head study of oxelumab and adalimumab in a mouse model of ulcerative colitis based on NOD/Scid IL2Rγnull mice reconstituted with human peripheral blood mononuclear cells. Dis. Model. Mech. 2021, 14. [Google Scholar] [CrossRef]
- Harshe, R.P.; Xie, A.; Vuerich, M.; Frank, L.A.; Gromova, B.; Zhang, H.; Robles, R.J.; Mukherjee, S.; Csizmadia, E.; Kokkotou, E.; et al. Endogenous antisense RNA curbs CD39 expression in Crohn’s disease. Nat. Commun. 2020, 11, 5894. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).