
sRNA Profiler: A User-Focused Interface for Small RNA Mapping and Profiling

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Setting the Parameters for Pattern Matching
- Set of N strings, where each string has length , sampled from ;
- String of length m, sampled from the alphabet ; and,
- Functionthat maps strings to to identify a matching between two strings, as follows. For two equal-length strings and , if and only if and match.
2.2. Finding the Occurrence of in
2.3. Profiling of vd-sRNA on Both the Viroid Genomic and Antigenomic Strands
2.4. Viroid Small RNA Data
2.5. Software Accessibility and Instructions
3. Results
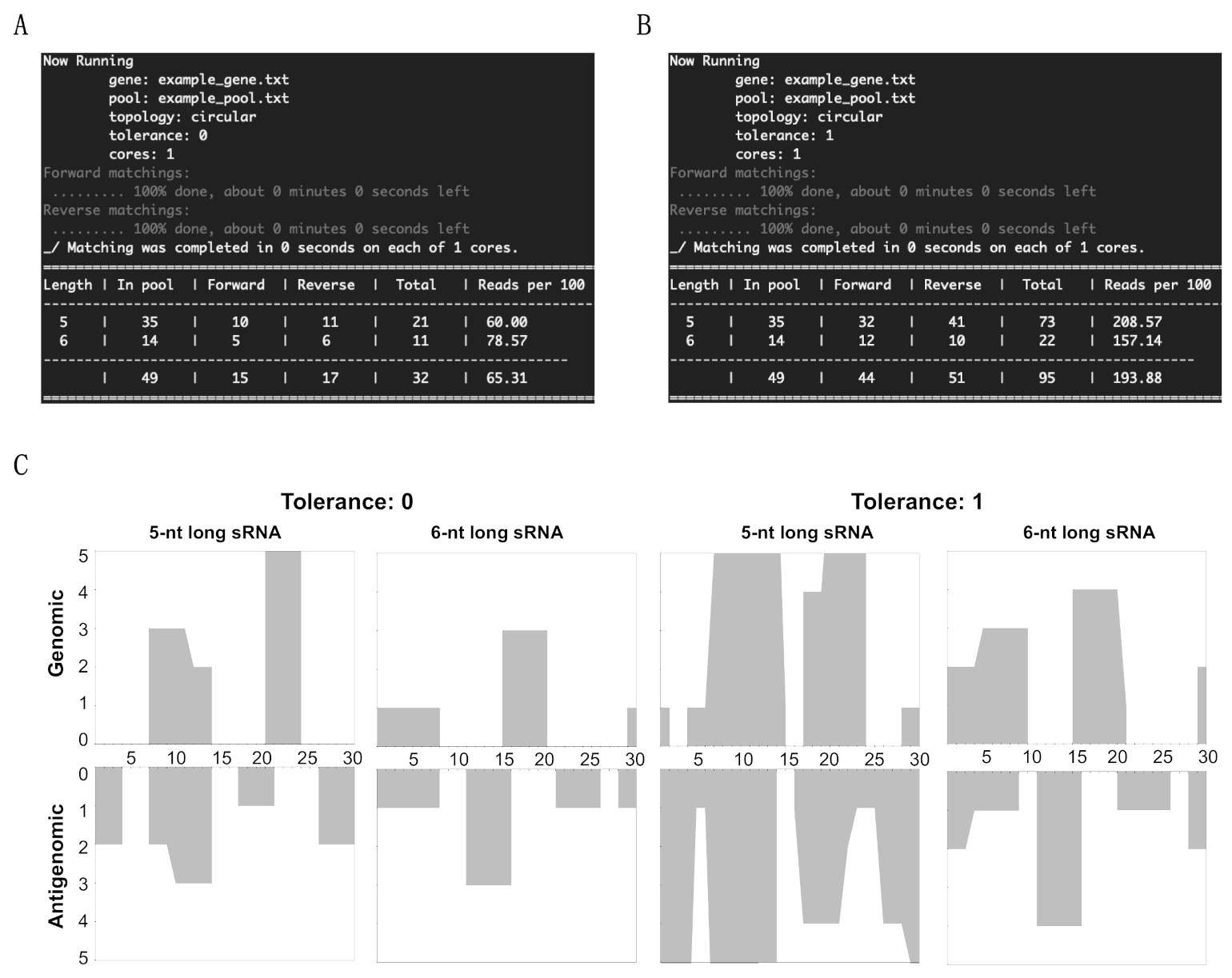
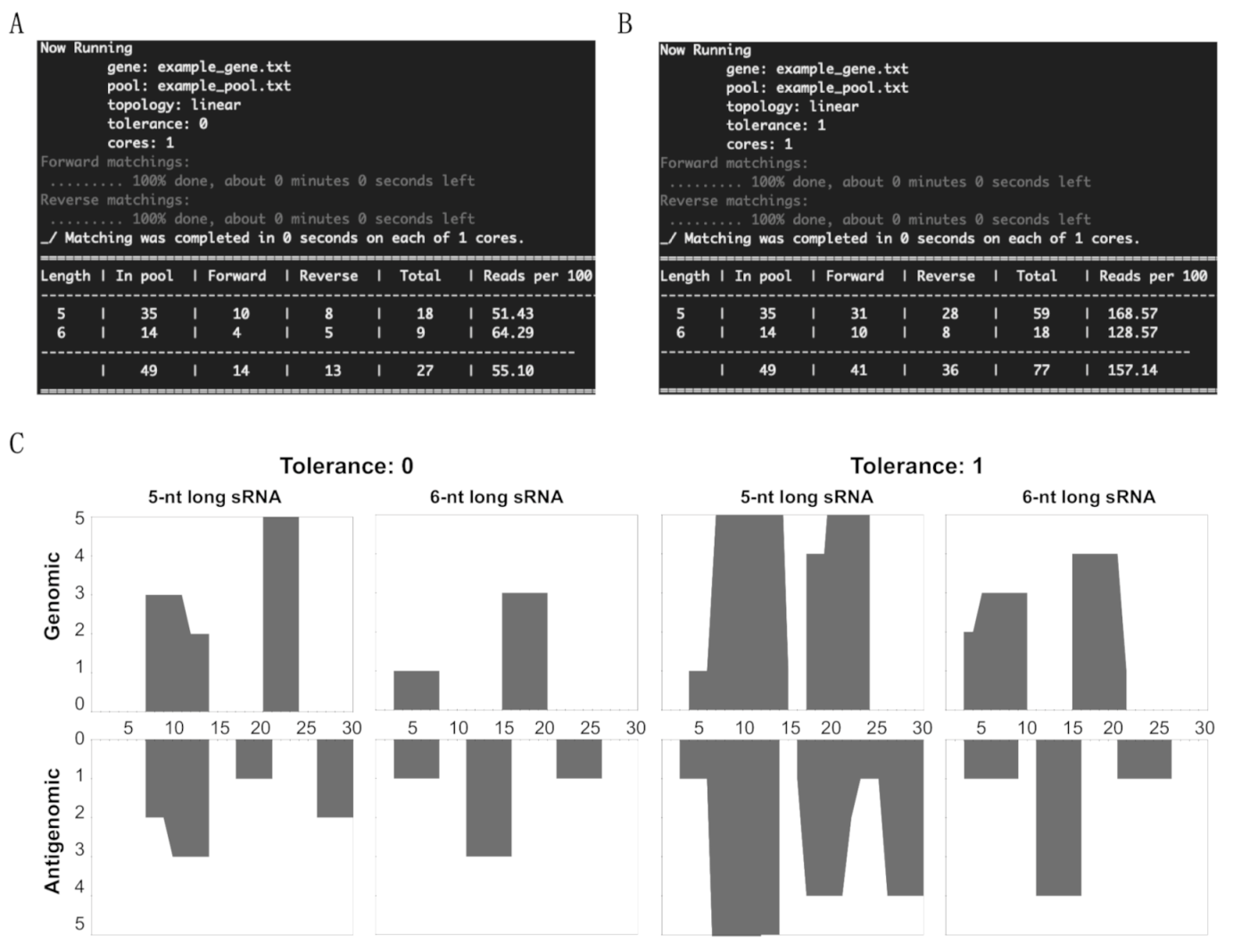
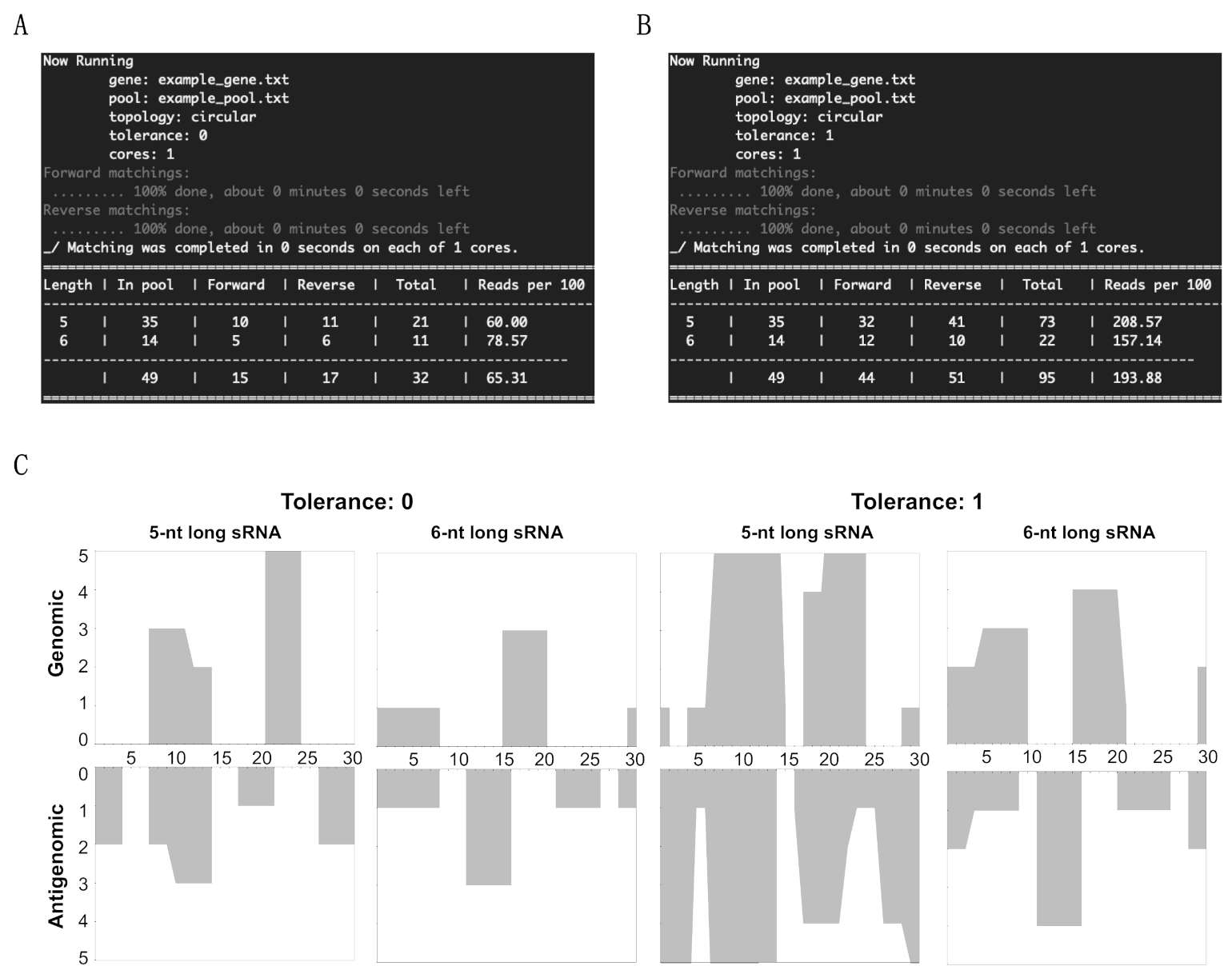
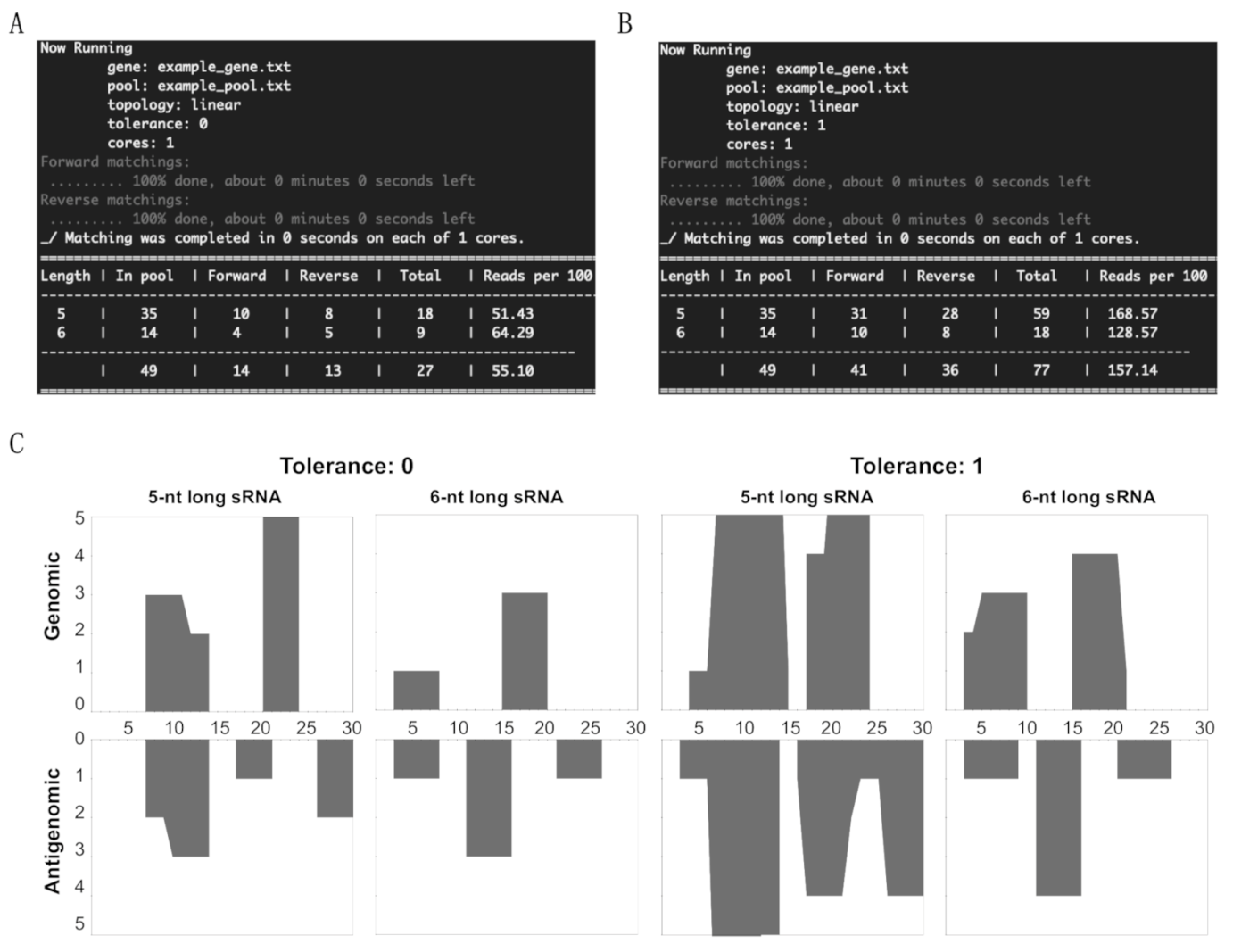
3.1. Overview of sRNA Mapping and Profiling
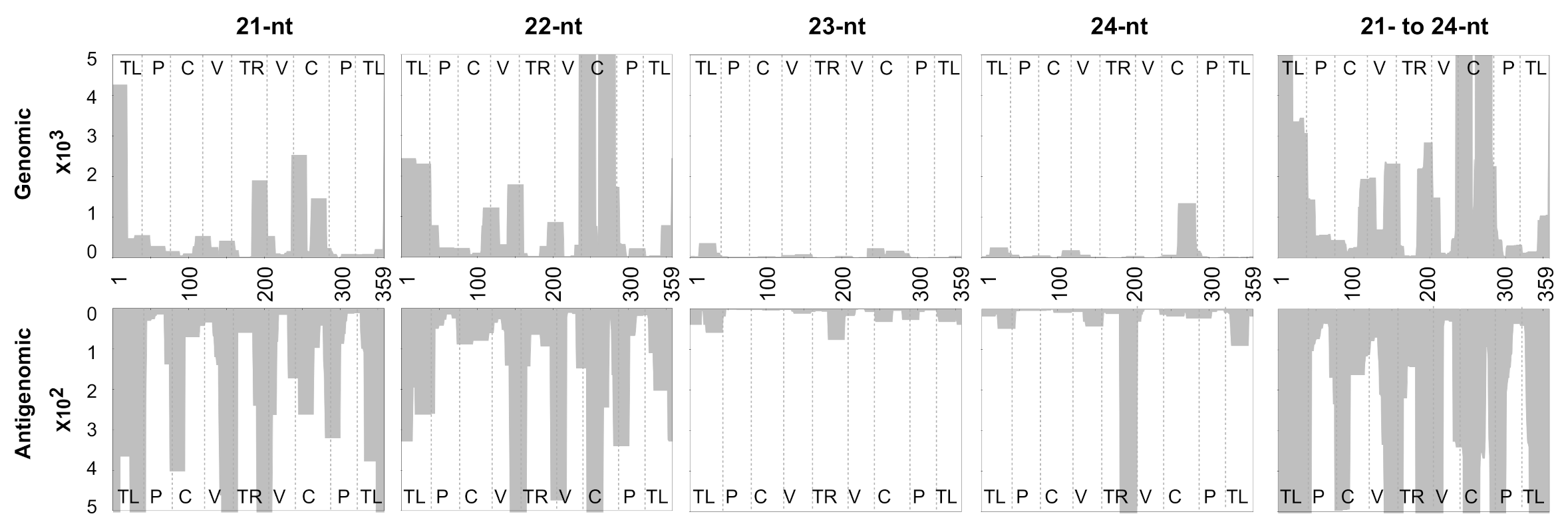
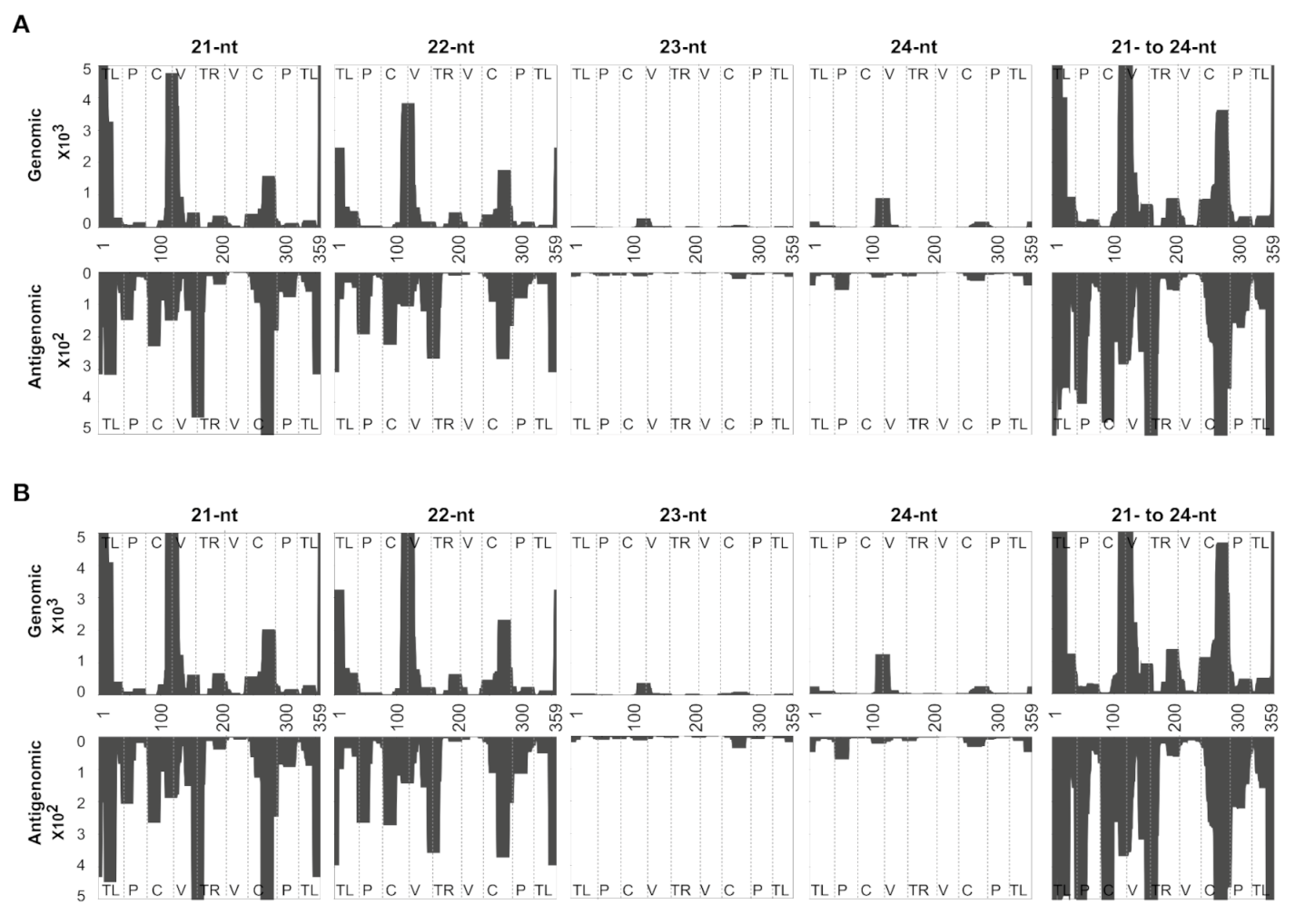
3.2. Mapping of sRNAs Obtained from PSTVd-I-Infected Plants
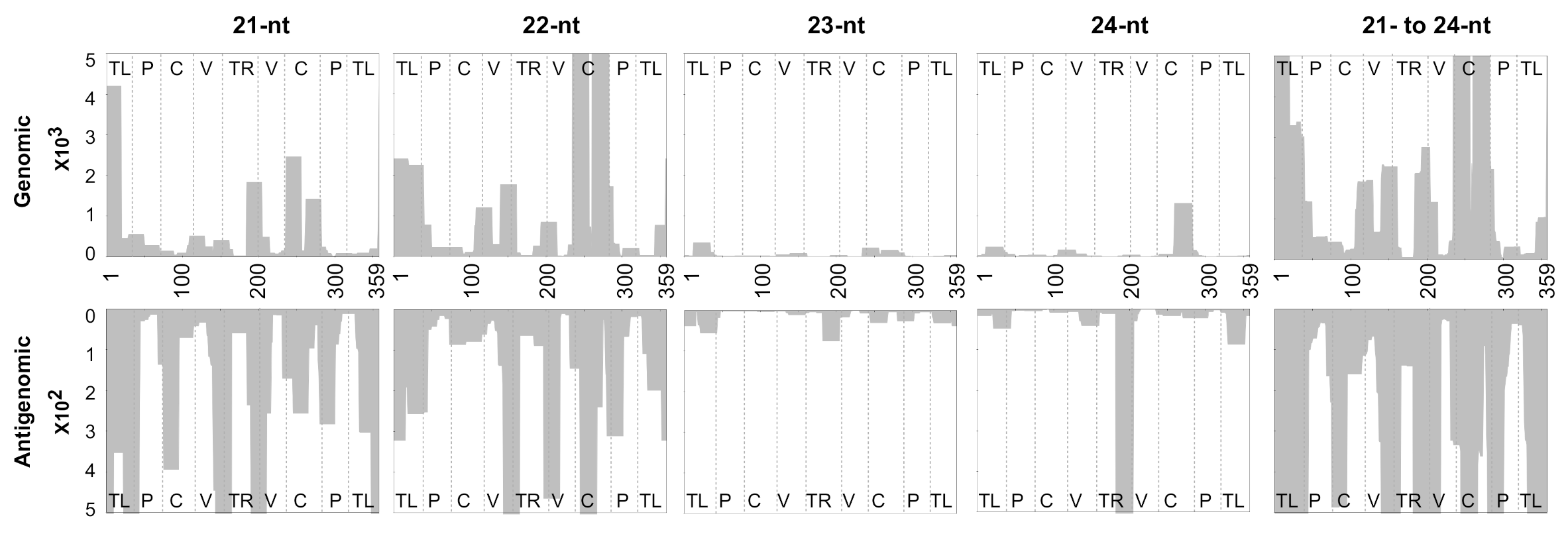
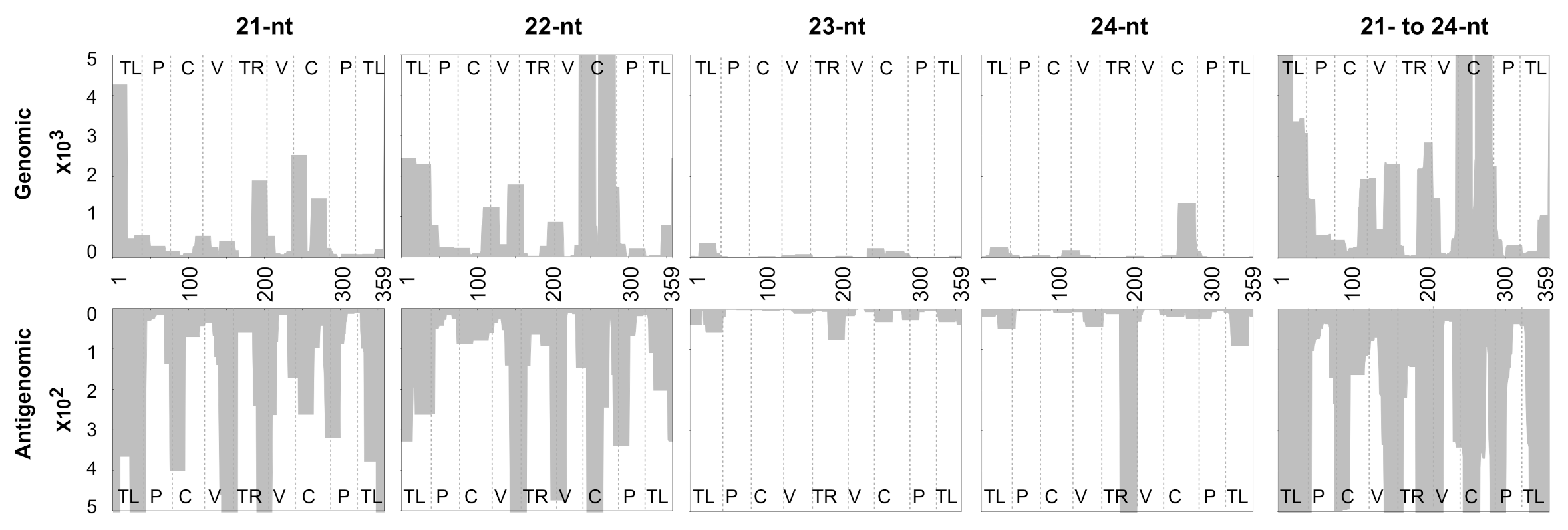
3.3. Profiling of the Mapped sRNAs on PSTVd-I
3.4. Accommodating vd-sRNA of the PSTVd-I Quasi-Species
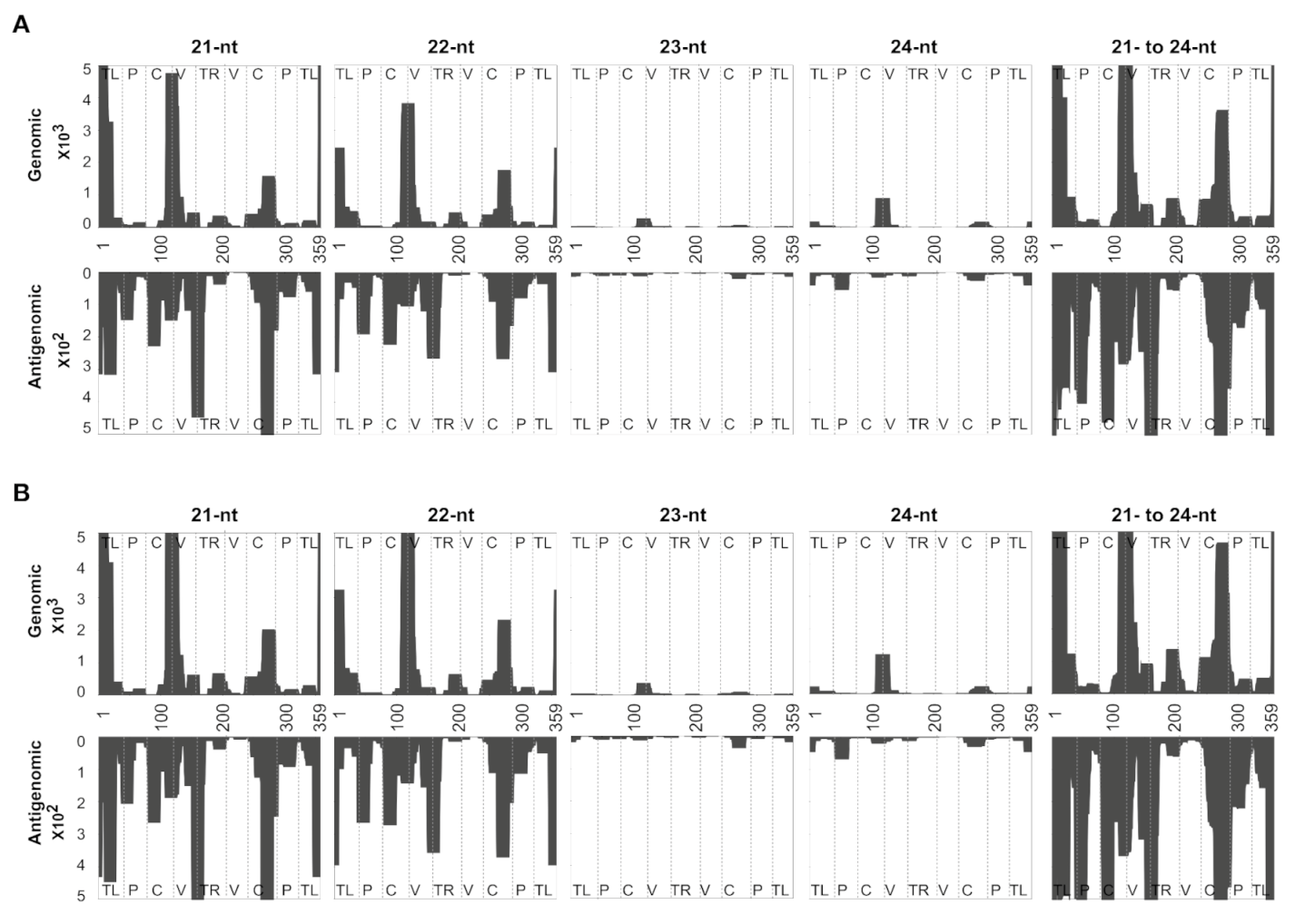
3.5. Evaluating the Mapping Tool Using the vd-sRNAs Obtained from PSTVd-RG1-Infected Plants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kitabayashi, S.; Tsushima, D.; Adkar-Purushothama, C.R.; Sano, T. Identification and molecular mechanisms of key nucleotides causing attenuation in pathogenicity of Dahlia isolate of potato spindle tuber viroid. Int. J. Mol. Sci. 2020, 21, 7352. [Google Scholar] [CrossRef] [PubMed]
- Ding, B. The biology of viroid-host interactions. Annu. Rev. Phytopathol. 2009, 47, 105–131. [Google Scholar] [CrossRef] [PubMed]
- Tsagris, E.M.; Martínez de Alba, A.E.; Gozmanova, M.; Kalantidis, K. Viroids. Cell. Microbiol. 2008, 10, 2168–2179. [Google Scholar] [CrossRef]
- Flores, R.; Hernández, C.; de Alba, A.E.M.; Daròs, J.-A.; Serio, F. Di Viroids and Viroid-Host Interactions. Annu. Rev. Phytopathol. 2005, 43, 117–139. [Google Scholar] [CrossRef]
- Pallas, V.; Martinez, G.; Gomez, G. The interaction between plant viroid-induced symptoms and RNA silencing. Methods Mol. Biol. 2012, 894, 323–343. [Google Scholar]
- Ding, B. Viroids: Self-replicating, mobile, and fast-evolving noncoding regulatory RNAs. Wiley Interdiscip. Rev. RNA 2010, 1, 362–375. [Google Scholar] [CrossRef]
- Dadami, E.; Boutla, A.; Vrettos, N.; Tzortzakaki, S.; Karakasilioti, I.; Kalantidis, K. DICER-LIKE 4 But Not DICER-LIKE 2 may have a positive effect on potato spindle tuber viroid accumulation in nicotiana benthamiana. Mol. Plant 2013, 6, 232–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itaya, A.; Folimonov, A.; Matsuda, Y.; Nelson, R.S.; Ding, B. Potato spindle tuber viroid as inducer of RNA silencing in infected tomato. Mol. Plant. Microbe. Interact. 2001, 14, 1332–1334. [Google Scholar] [CrossRef] [Green Version]
- Papaefthimiou, I.; Hamilton, A.; Denti, M.; Baulcombe, D.; Tsagris, M.; Tabler, M. Replicating potato spindle tuber viroid RNA is accompanied by short RNA fragments that are characteristic of post-transcriptional gene silencing. Nucleic Acids Res. 2001, 29, 2395–2400. [Google Scholar] [CrossRef] [Green Version]
- Bolduc, F.; Hoareau, C.; St-Pierre, P.; Perreault, J.-P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC Mol. Biol. 2010, 11, 16. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, D.; Milev, I.; Vachev, T.; Baev, V.; Yahubyan, G.; Minkov, G.; Gozmanova, M. Small RNA analysis of potato spindle tuber viroid infected phelipanche ramosa. Plant Physiol. Biochem. 2014, 74, 276–282. [Google Scholar] [CrossRef]
- Navarro, B.; Pantaleo, V.; Gisel, A.; Moxon, S.; Dalmay, T.; Bisztray, G.; Di Serio, F.; Burgyán, J. Deep sequencing of viroid-derived small RNAs from grapevine provides new insights on the role of RNA silencing in plant-viroid interaction. PLoS ONE 2009, 4, e7686. [Google Scholar] [CrossRef] [Green Version]
- Tsushima, D.; Adkar-Purushothama, C.R.; Taneda, A.; Sano, T. Changes in relative expression levels of viroid-specific small RNAs and microRNAs in tomato plants infected with severe and mild isolates of Potato spindle tuber viroid. J. Gen. Plant Pathol 2015, 81, 49–62. [Google Scholar] [CrossRef]
- Navarro, B.; Gisel, A.; Rodio, M.E.; Delgado, S.; Flores, R.; Di Serio, F. Small RNAs containing the pathogenic determinant of a chloroplast-replicating viroid guide the degradation of a host mRNA as predicted by RNA silencing. Plant J. 2012, 70, 991–1003. [Google Scholar] [CrossRef]
- Eamens, A.L.; Smith, N.A.; Dennis, E.S.; Wassenegger, M.; Wang, M.B. In Nicotiana species, an artificial microRNA corresponding to the virulence modulating region of Potato spindle tuber viroid directs RNA silencing of a soluble inorganic pyrophosphatase gene and the development of abnormal phenotypes. Virology 2014, 450–451, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Adkar-Purushothama, C.R.; Brosseau, C.; Giguère, T.; Sano, T.; Moffett, P.; Perreault, J.-P. Small RNA Derived from the Virulence Modulating Region of the Potato spindle tuber viroid Silences callose synthase Genes of Tomato Plants. Plant Cell 2015, 27, 2178–2194. [Google Scholar] [CrossRef] [Green Version]
- Adkar-Purushothama, C.R.; Sano, T.; Perreault, J.-P. Viroid derived small RNA induces early flowering in tomato plants by RNA silencing. Mol. Plant Pathol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Adkar-Purushothama, C.R.; Perreault, J.-P. Alterations of the viroid regions that interact with the host defense genes attenuate viroid infection in host plant. RNA Biol. 2018, 15, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Adkar-Purushothama, C.R.; Perreault, J. Current overview on viroid-host interactions. Wiley Interdiscip. Rev. RNA 2020, 11, e1570. [Google Scholar] [CrossRef]
- Adkar-Purushothama, C.R.; Perreault, J.P. Impact of nucleic acid sequencing on viroid biology. Int. J. Mol. Sci. 2020, 21, 5532. [Google Scholar] [CrossRef]
- Diermann, N.; Matoušek, J.; Junge, M.; Riesner, D.; Steger, G. Characterization of plant miRNAs and small RNAs derived from potato spindle tuber viroid (PSTVd) in infected tomato. Biol. Chem. 2010, 391, 1379–1390. [Google Scholar] [CrossRef]
- Brass, J.R.J.; Owens, R.A.; Matoušek, J.; Steger, G. Viroid quasispecies revealed by deep sequencing. RNA Biol. 2017, 14, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Adkar-Purushothama, C.R.; Bolduc, F.; Bru, P.; Perreault, J.-P. Insights Into Potato Spindle Tuber Viroid Quasi-Species From Infection to Disease. Front. Microbiol. 2020, 11, 1235. [Google Scholar] [CrossRef]
- Adkar-Purushothama, C.R.; Kasai, A.; Sugawara, K.; Yamamoto, H.; Yamazaki, Y.; He, Y.-H.; Takada, N.; Goto, H.; Shindo, S.; Harada, T.; et al. RNAi mediated inhibition of viroid infection in transgenic plants expressing viroid-specific small RNAs derived from various functional domains. Sci. Rep. 2015, 5, 17949. [Google Scholar] [CrossRef]
- Kovalskaya, N.; Hammond, R.W. Molecular biology of viroid–host interactions and disease control strategies. Plant Sci. 2014, 228, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Knuth, D.E.; Morris, J.H., Jr.; Pratt, V.R. Fast Pattern Matching in Strings. SIAM J. Comput. 1977, 6, 323–350. [Google Scholar] [CrossRef] [Green Version]
- Gonnet, G.H. Some string matching problems from Bioinformatics which still need better solutions. J. Discret. Algorithms 2004, 2, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Vildhøj, H.W. Topics in Combinatorial Pattern Matching. Ph.D. thesis, Technical University of Denmark, Copenhagen, Denmark, 2015. [Google Scholar]
- Chang, W.I.; Lampe, J. Theoretical and empirical comparisons of approximate string matching algorithms. In Combinatorial Pattern Matching; Apostolico, A., Crochemore, M., Galil, Z., Manber, U., Eds.; Springer: Berlin/Heidelberg, Germany, 1992; pp. 175–184. [Google Scholar]
- Deng, F.; Wang, L.; Liu, X. An efficient algorithm for the blocked pattern matching problem. Bioinformatics 2015, 31, 532–538. [Google Scholar] [CrossRef] [Green Version]
- Bafna, V.; Muthukrishnan, S.; Ravi, R. Computing similarity between RNA strings. In Combinatorial Pattern Matching; Springer: Berlin/Heidelberg, Germany, 1995; pp. 1–16. [Google Scholar]
- Le Gall, F. Faster Algorithms for Rectangular Matrix Multiplication. In Proceedings of the 2012 IEEE 53rd Annual Symposium on Foundations of Computer Science, New Brunswick, NJ, USA, 20–23 October 2012; pp. 514–523. [Google Scholar]
- Adkar-Purushothama, C.R.; Iyer, P.S.; Perreault, J.-P. Potato spindle tuber viroid infection triggers degradation of chloride channel protein CLC-b-like and Ribosomal protein S3a-like mRNAs in tomato plants. Sci. Rep. 2017, 7, 8341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchins, C.J.; Keese, P.; Visvader, J.E.; Rathjen, P.D.; McInnes, J.L.; Symons, R.H. Comparison of multimeric plus and minus forms of viroids and virusoids. Plant Mol. Biol. 1985, 4, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Gago, S.; Elena, S.F.; Flores, R.; Sanjuán, R. Extremely high mutation rate of a hammerhead viroid. Science 2009, 323, 1308. [Google Scholar] [CrossRef] [Green Version]
- López-Carrasco, A.; Ballesteros, C.; Sentandreu, V.; Delgado, S.; Gago-Zachert, S.; Flores, R.; Sanjuán, R. Different rates of spontaneous mutation of chloroplastic and nuclear viroids as determined by high-fidelity ultra-deep sequencing. PLoS Pathog. 2017, 13, e1006547. [Google Scholar] [CrossRef]
- Tessitori, M.; Rizza, S.; Reina, A.; Causarano, G.; Di Serio, F. The genetic diversity of Citrus dwarfing viroid populations is mainly dependent on the infected host species. J. Gen. Virol. 2013, 94, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Owens, R.A.; Sun, Q.; Song, H.; Liu, Y.; Eamens, A.L.; Feng, H.; Tian, H.; Wang, M.-B.; Zhang, R. Silencing of transcription factor encoding gene StTCP23 by small RNAs derived from the virulence modulating region of potato spindle tuber viroid is associated with symptom development in potato. PLoS Pathog. 2019, 15, e1008110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibaut, O.; Claude, B. Innate immunity activation and RNAi interplay in citrus exocortis viroid-tomato pathosystem. Viruses 2018, 10, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aviña-Padilla, K.; Rivera-Bustamante, R.; Kovalskaya, N.Y.; Hammond, R.W. Pospiviroid infection of tomato regulates the expression of genes involved in flower and fruit development. Viruses 2018, 10, 516. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| sRNA Length | Tolerance: 0 | Tolerance: 1 | Non-Matching sRNA | ||
|---|---|---|---|---|---|
| Forward Matching | Reverse Matching | Forward Matching | Reverse Matching | ||
| 5-nt | GGGAT | ACCAC | ACCCG | AGATT | AGGGA |
| ATCCC | CCACG | GGATC | CACGA | CAAGT | |
| AAACC | CCGTG * | TTGAC * | CCCTG * | GGGGC | |
| 6-nt | TTCAGG | AGTTCC | ACAAAA | CAGAGG | ACGCAG |
| CGGGGA | GTGAAC * | GGCTCA | GTGGAC * | GTAGAT | |
| CGGCTT * | CCGTGA * | CACCGA * | CCGCGA * | CGGAAA | |
| sRNA Length | Total sRNA in the Pool | Tolerance: 0 | Normalized Reads (Per Million Reads) | (+):(−) vd-sRNA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (+) vd-sRNA | (−) vd-sRNA | Total vd-sRNA | Total sRNA | (+) vd-sRNA | (−) vd-sRNA | total vd-sRNA | ||||||
| No. of Reads | % | No. of Reads | % | No. of Reads | % | |||||||
| 21-nt | 700,131 | 105,294 | 55,304 | 160,598 | 162,197 | 24,393 | 15.0 | 12,812 | 7.9 | 37,205 | 22.9 | 1.9 |
| 22-nt | 839,033 | 245,515 | 42,665 | 288,180 | 194,376 | 56,878 | 29.3 | 9884 | 5.1 | 66,762 | 34.3 | 5.8 |
| 23-nt | 622,373 | 10,064 | 3154 | 13,218 | 144,183 | 2331 | 1.6 | 731 | 0.5 | 3062 | 2.1 | 3.2 |
| 24-nt | 2,155,006 | 19,858 | 6322 | 26,180 | 499,243 | 4600 | 0.9 | 1465 | 0.3 | 6065 | 1.2 | 3.1 |
| 21–24-nt | 4,316,543 | 380,731 | 107,445 | 488,176 | 1,000,000 | 88,203 | 8.8 | 24,891 | 2.5 | 113,094 | 11.3 | 3.5 |
| sRNA Length | Total sRNA in the Pool | Tolerance: 1 | Normalized Reads (Per Million Reads) | (+):(−) vd-sRNA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (+) vd-sRNA | (−) vd-sRNA | Total vd-sRNA | Total sRNA | (+) vd-sRNA | (−) vd-sRNA | total vd-sRNA | ||||||
| No. of Reads | % | No. of reads | % | No. of Reads | % | |||||||
| 21-nt | 700,131 | 109,114 | 57,707 | 166,821 | 162,197 | 25,278 | 15.6 | 13,369 | 8.2 | 38,647 | 23.8 | 1.9 |
| 22-nt | 839,033 | 251,128 | 45,522 | 296,650 | 194,376 | 58,178 | 29.9 | 10,546 | 5.4 | 68,724 | 35.4 | 5.5 |
| 23-nt | 622,373 | 10,984 | 3511 | 14,495 | 144,183 | 2545 | 1.8 | 813 | 0.6 | 3358 | 2.3 | 3.1 |
| 24-nt | 2,155,006 | 20,556 | 6561 | 27,117 | 499,243 | 4762 | 1.0 | 1520 | 0.3 | 6282 | 1.3 | 3.1 |
| 21–24-nt | 4,316,543 | 391,782 | 113,301 | 505,083 | 1,000,000 | 90,763 | 9.1 | 26,248 | 2.6 | 117,011 | 11.7 | 3.5 |
| sRNA Length | Total sRNA in the Pool | Tolerance: 0 | Normalized Reads (Per Million Reads) | (+):(−) vd-sRNA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (+) vd-sRNA | (−) vd-sRNA | Total vd-sRNA | Total sRNA | (+) vd-sRNA | (−) vd-sRNA | total vd-sRNA | ||||||
| No. of Reads | % | No. of Reads | % | No. of Reads | % | |||||||
| 21-nt | 160,262 | 35,533 | 5778 | 41,311 | 219,387 | 48,642 | 22.2 | 7910 | 3.6 | 56,552 | 25.8 | 6.1 |
| 22-nt | 145,487 | 24,528 | 3913 | 28,441 | 199,161 | 33,577 | 16.9 | 5357 | 2.7 | 38,934 | 19.5 | 6.3 |
| 23-nt | 124,625 | 1448 | 197 | 1645 | 170,603 | 1982 | 1.2 | 270 | 0.2 | 2252 | 1.3 | 7.4 |
| 24-nt | 300,125 | 3135 | 374 | 3509 | 410,849 | 4292 | 1.0 | 512 | 0.1 | 4804 | 1.1 | 8.4 |
| 21–24-nt | 730,499 | 64,644 | 10,272 | 74,906 | 1,000,000 | 88,493 | 8.8 | 14,062 | 1.4 | 102,555 | 10.3 | 6.3 |
| sRNA Length | Total sRNA in the Pool | Tolerance: 1 | Normalized Reads (Per Million Reads) | (+):(−) vd-sRNA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (+) vd-sRNA | (−) vd-sRNA | Total vd-sRNA | Total sRNA | (+) vd-sRNA | (−) vd-sRNA | total vd-sRNA | ||||||
| No. of Reads | % | No. of Reads | % | No. of Reads | % | |||||||
| 21-nt | 160,262 | 46,752 | 7461 | 54,213 | 219,387 | 64,000 | 29.2 | 10,214 | 4.7 | 74,214 | 33.8 | 6.3 |
| 22-nt | 145,487 | 32,779 | 5121 | 37,900 | 199,161 | 44,872 | 22.5 | 7010 | 3.5 | 51,882 | 26.1 | 6.4 |
| 23-nt | 124,625 | 2049 | 280 | 2329 | 170,603 | 2805 | 1.6 | 383 | 0.2 | 3188 | 1.9 | 7.3 |
| 24-nt | 300,125 | 4162 | 491 | 4653 | 410,849 | 5697 | 1.4 | 672 | 0.2 | 6370 | 1.6 | 8.5 |
| 21–24-nt | 730,499 | 85,742 | 13,353 | 99,095 | 1,000,000 | 117,375 | 11.7 | 18,279 | 1.8 | 135,654 | 13.6 | 6.4 |
| ze of the sRNA Pool | Size of the Viroid Genome | Tolerance | Runtime in Seconds |
|---|---|---|---|
| 5,875,050 | 359 | 0 | 1450 |
| 5,875,050 | 359 | 1 | 1800 |
| 730,499 | 359 | 0 | 128 |
| 730,499 | 359 | 1 | 131 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adkar-Purushothama, C.R.; Iyer, P.S.; Sano, T.; Perreault, J.-P. sRNA Profiler: A User-Focused Interface for Small RNA Mapping and Profiling. Cells 2021, 10, 1771. https://doi.org/10.3390/cells10071771
Adkar-Purushothama CR, Iyer PS, Sano T, Perreault J-P. sRNA Profiler: A User-Focused Interface for Small RNA Mapping and Profiling. Cells. 2021; 10(7):1771. https://doi.org/10.3390/cells10071771
Chicago/Turabian StyleAdkar-Purushothama, Charith Raj, Pavithran Sridharan Iyer, Teruo Sano, and Jean-Pierre Perreault. 2021. "sRNA Profiler: A User-Focused Interface for Small RNA Mapping and Profiling" Cells 10, no. 7: 1771. https://doi.org/10.3390/cells10071771
APA StyleAdkar-Purushothama, C. R., Iyer, P. S., Sano, T., & Perreault, J.-P. (2021). sRNA Profiler: A User-Focused Interface for Small RNA Mapping and Profiling. Cells, 10(7), 1771. https://doi.org/10.3390/cells10071771