
Hypoxia, Hypoxia-Inducible Factors and Liver Fibrosis




 and
and
Abstract
:1. Fibrogenesis and Fibrosis in Chronic Liver Disease Progression: Introductory Remarks
2. Hypoxia and Hypoxia-Inducible Factors: The Strange Case of the Liver and the Cellular “Hypoxic Response”
2.1. Hypoxia and the Liver: Critical Issues
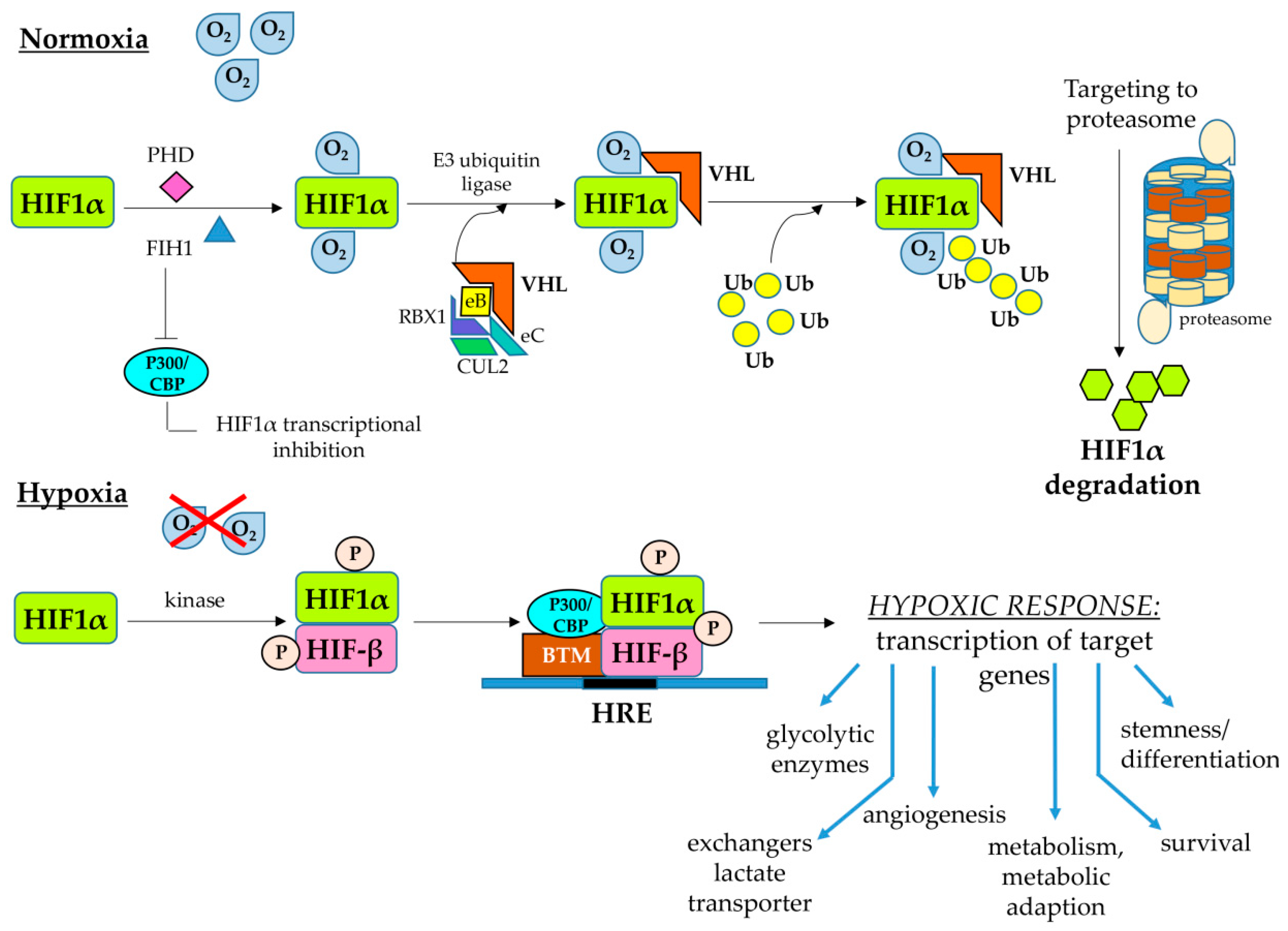
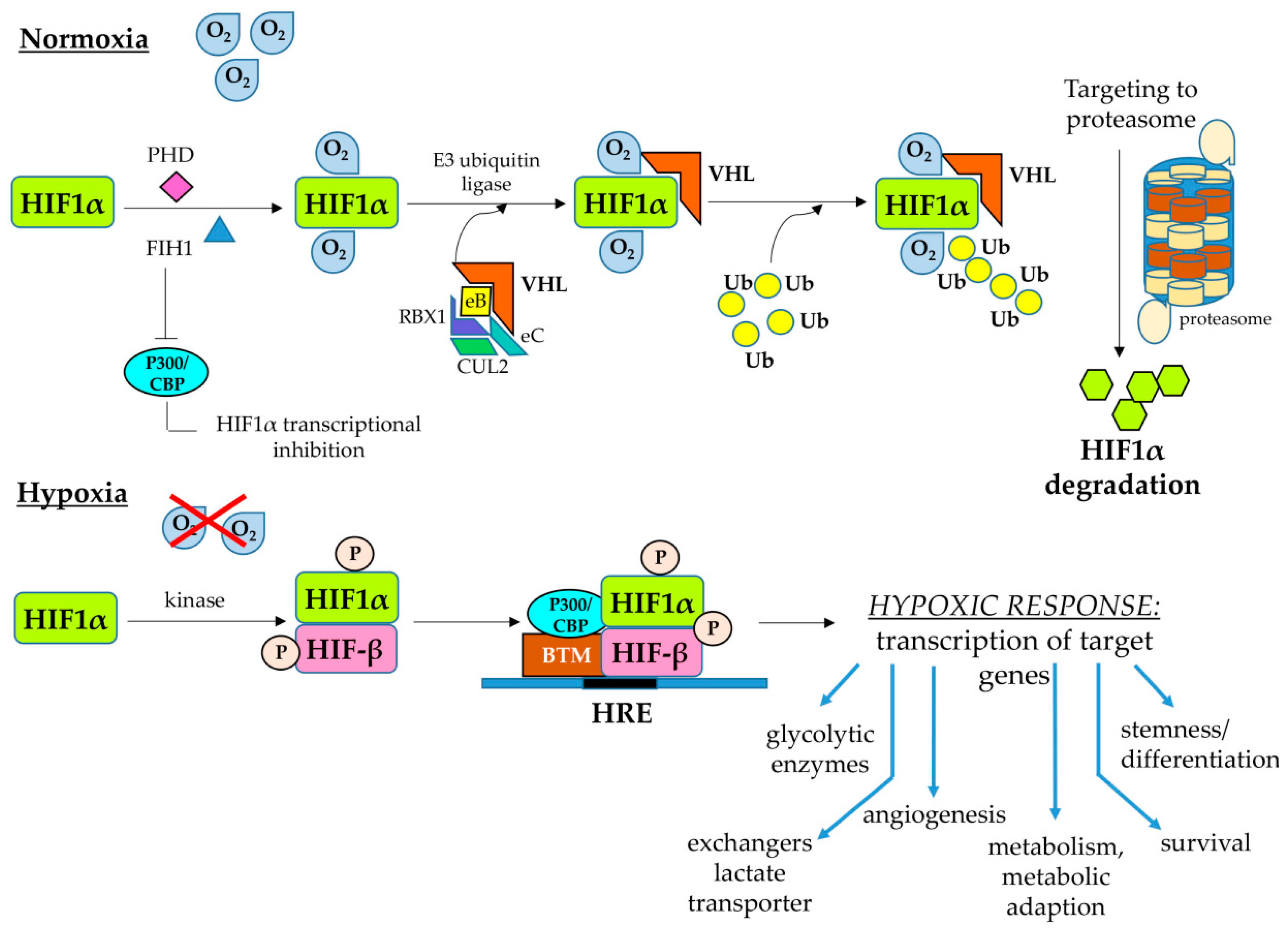
2.2. Hypoxia-Inducible Factors
- (a)
- Genes responding to both HIF1α and HIF2α, in which one can include the genes encoding for (i) VEGF-A; (ii) glucose transporter 1 (GLUT1); (iii) carbonic anhydrase IX and XII; (iv) the inflammatory response-related ones encoding for chemokine receptor CXCR4, the chemokine CXCL12 or IL-1β; (v) the anti-apoptotic protein Bcl2; and (vi) the transcription factor Twist involved in epithelial–mesenchymal transition (EMT).
- (b)
- Genes responding to HIF1α, including those encoding for (i) key enzymes of the glycolytic pathway; (ii) factors involved in autophagy, such as BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 or BNIP3 and the related ligand BNIP3L; (iii) ECM remodeling enzymes, such as lisyl oxidase 2 or LOX2; (iv) the inhibitor of mammalian-target of rapamycin (mTOR) signaling pathway REDD1 (regulated in development and DNA damage responses 1); and (v) inducible NO synthase (iNOs).
- (c)
- Genes responding to HIF2α, including those encoding for (i) factors involved in cell cycle and proliferation or stemness, such as cyclin D1, transforming growth factor-α (TGFα) and Octamer 4; (ii) ECM remodeling enzymes, such as matrix metalloprotease 13 (MMP13); (iii) antioxidant enzymes, such as superoxide dismutase 2 (SOD2); (iv) the delta-like ligand 4 (DLL4) involved in Notch signaling; and (v) the marker for M2 macrophage polarization arginase 1.
2.3. HIFs and the Cellular Response to Hypoxic Conditions: A Focus on Maintenance of Redox Ho-Meostasis
- (a)
- Under hypoxic conditions, HIF1 can modulate the equilibrium between oxidative phosphorylation and glycolytic metabolism, and then ROS generation, mainly by regulating the expression of the LDHA and PDK1 genes encoding for lactate dehydrogenase A (i.e., the enzyme converting pyruvate to lactate) and pyruvate dehydrogenase kinase 1 (i.e., the enzyme which inactivates pyruvate dehydrogenase preventing conversion of pyruvate into acetyl-CoA). Accordingly, deletion of HIF1α in genetically manipulated cells prevented the hypoxia-related switch from oxidative to glycolytic metabolism and maintained high levels of ATP. However, HIF1α-deleted cells can die for an excess of intracellular ROS generation, also revealing the critical role of mitochondrial autophagy for the maintenance of redox homeostasis and the survival of hypoxic cells [61,62].
- (b)
- Another way to modulate ROS generation is by controlling fatty acid oxidation. HIF-1α, and then HIF1, under hypoxic conditions can suppress, through a pathway involving downregulation of C-MYC and then of peroxisome proliferator-activated receptor-gamma (PPAR-gamma) coactivator-1 beta (PGC-1β) expression, the transcription of the medium-chain and long-chain acyl-CoA dehydrogenases (MCAD and LCAD) genes, the enzymes that catalyze the first step of fatty acid oxidation in the mitochondria, subsequently decreasing fatty acid oxidation and mitochondrial ROS generation [57,63].
- (c)
- HIF1 can downregulate mitochondrial ROS generation by stimulating mitochondrial-selective autophagy (or mitophagy), through upregulation of BNIP3 in normal cells or of BNIP3L in tumor cells [61,64]. Conceivably, one can assume that under hypoxic conditions the reduction in the cellular content of mitochondria can suppress both glucose and fatty acids oxidation, resulting in a decreased mitochondrial generation of ROS. What is interesting here is that actual knowledge suggests that liver parenchyma is highly dependent on autophagy for maintaining its normal function and to prevent the development of disease states. Indeed, several laboratories have shown that alterations in autophagy represent a mechanism underlying hepatic diseases of different etiology, both acute or chronic, and even hepatocellular carcinoma development [65,66]. In addition, it has been also shown that autophagy can efficiently fuel fibrogenesis [22,66,67,68,69,70] but, paradoxically, few experimental studies have investigated the possible link between HIF, autophagy and CLDs [71,72], and no one was directly related to fibrogenesis.
- (d)
- Under hypoxic conditions HIFs could reduce mitochondrial ROS generation through two additional mechanisms: (i) HIF1 may act by activating the transcription of the gene encoding for NADH dehydrogenase [ubiquinone] 1α subcomplex, 4-like 2 (NDUFA4L2), resulting in a decrease in flux through the electron transport chain and subsequently in ROS generation [73]; (ii) induction of miR-210 expression, which, in turn, represses ISCU, a gene encoding the iron–sulfur cluster assembly factors that are required for the biogenesis of iron–sulfur-dependent enzyme complexes, which are critical for electron transport, such as complex I, complex III and aconitase [74].
- (e)
- HIFs, under hypoxic conditions, can also contribute to mitochondrial redox homeostasis by regulating the genes encoding for enzymes that are involved in the generation of NADPH and glutathione [57].
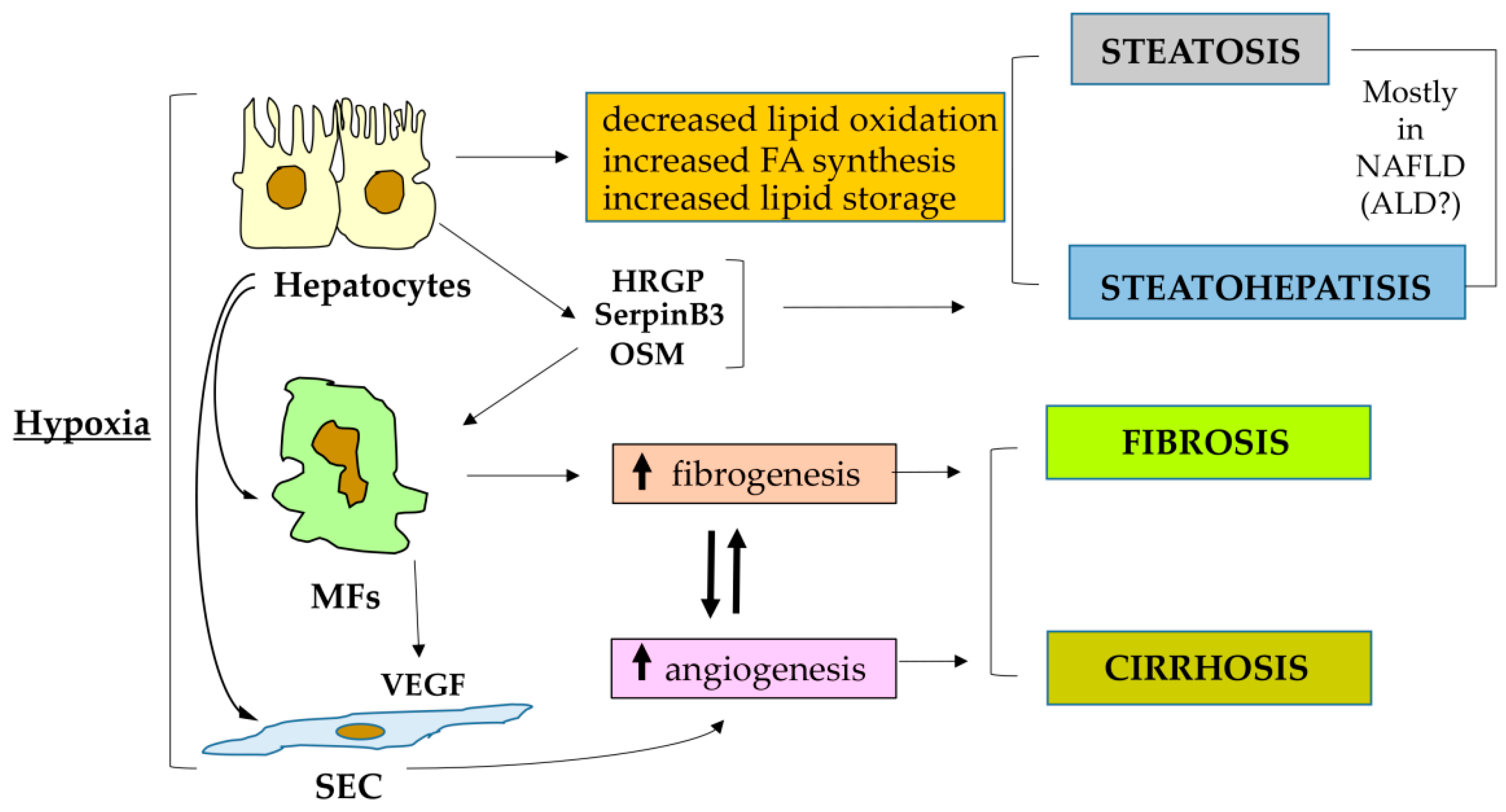
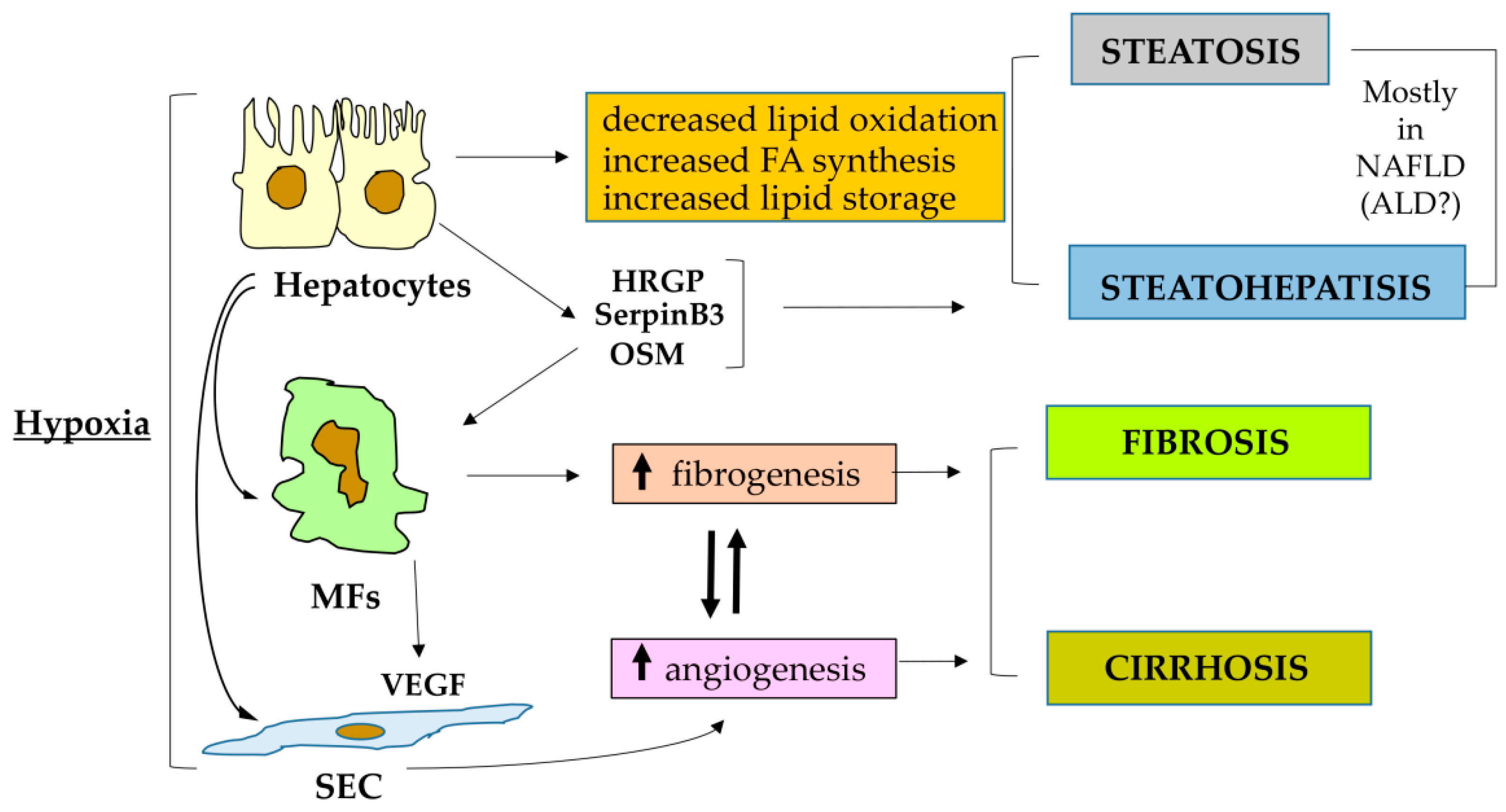
3. Hypoxia, HIFs and Liver Pathological Angiogenesis
4. HIF1α and HIF2α: Two Distinct Critical Players in Fibrogenic CLD Progression
4.1. HIF-1α and HIF1 in Fibrogenic CLD Progression: Of Biliary-Like Fibrosis and Activated Hepatic Stellate Cells
4.2. HIF-1α and HIF1, Metabolic Diseases and Fibrogenic Progression in ALD or NAFLD
4.3. Oncostatin M as a Profibrogenic Mediator Operating through HIF-1α
4.4. HIF-2α, HIF2 and CLD Progression: Relevant for Progressive NAFLD
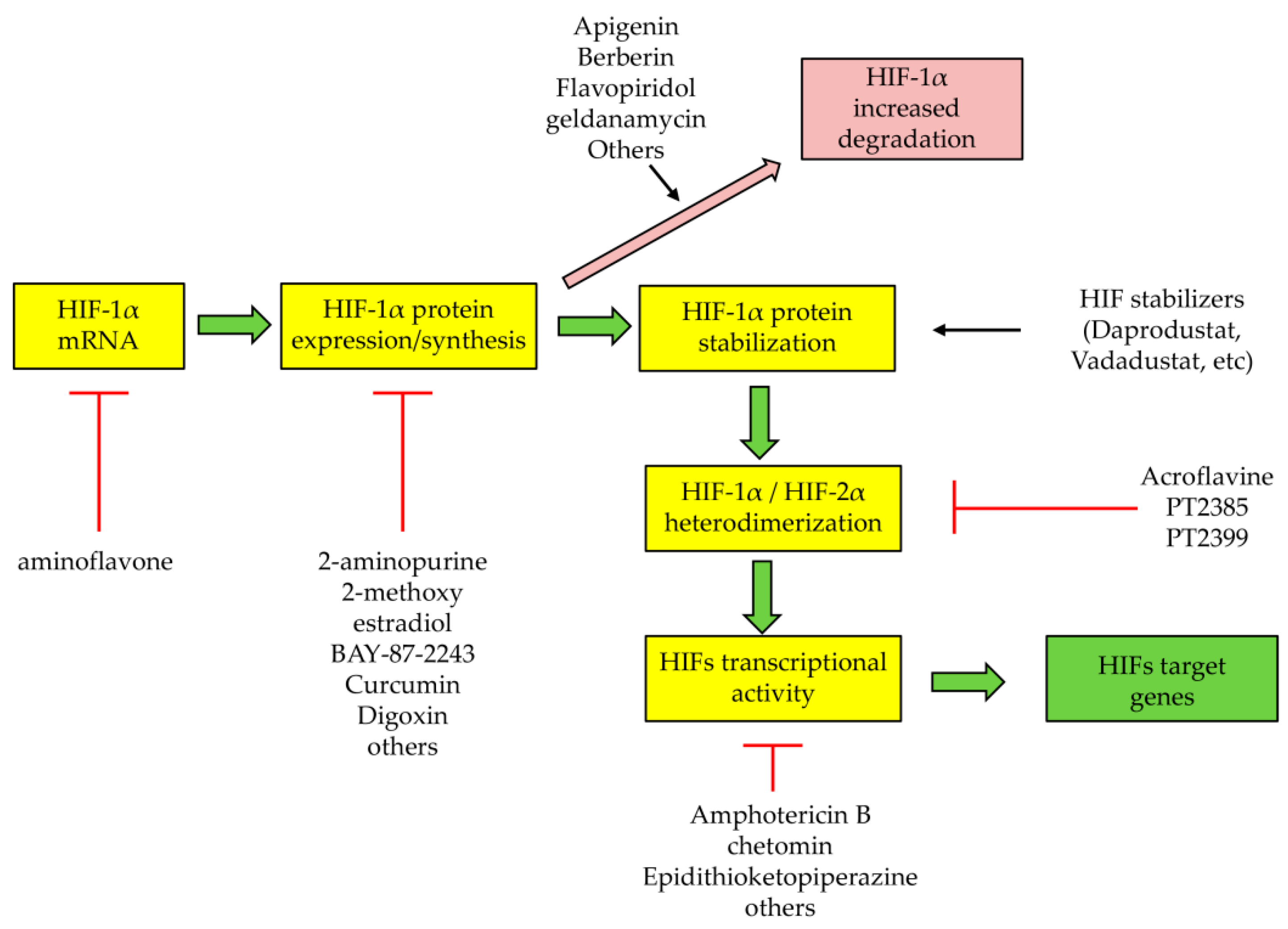
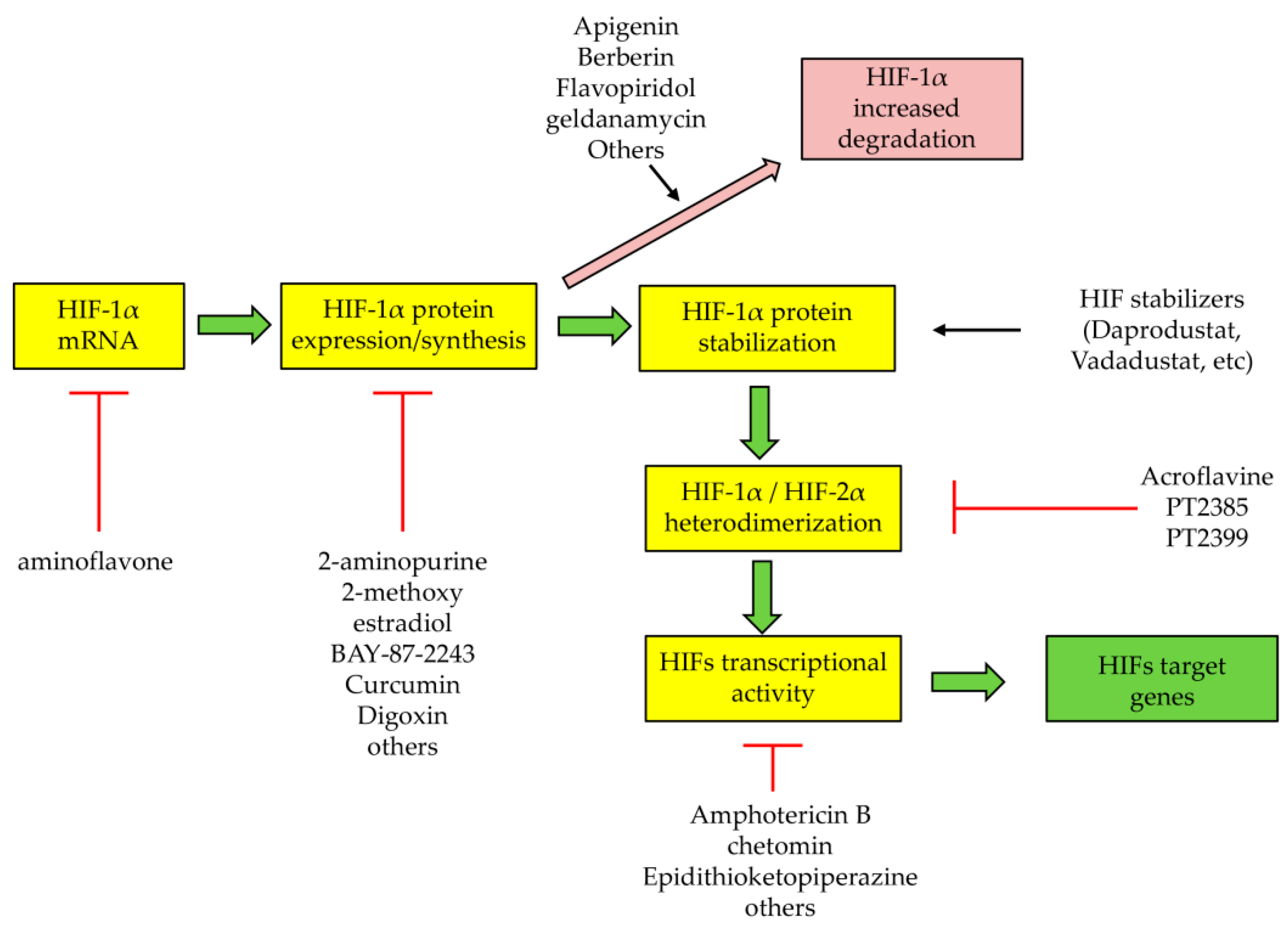
5. Therapeutic Strategies to Target HIFs or HIF-Related Processes and Pathways: Promising but Not Liver Oriented
6. Antifibrotic Therapies for CLD: A Synthetic Overview
6.1. Strategies to Reduce Liver Parenchymal Injury
6.2. Strategies to Target Hepatic Macrophages
6.3. Strategies to Target MFs and to Inhibit Fibrosis through Different Approaches
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Parola, M.; Marra, F.; Pinzani, M. Myofibroblast-like cells and liver fibrogenesis: Emerging concepts in a rapidly moving scenario. Mol. Asp. Med. 2008, 29, 58–66. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef]
- Trautwein, C.; Friedman, S.L.; Schuppan, D.; Pinzani, M. Hepatic fibrosis: Concept to treatment. J. Hepatol. 2015, 62 (Suppl. l), S15–S24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Cannito, S.; Novo, E.; Parola, M. Therapeutic pro-fibrogenic signaling pathways in fibroblasts. Adv. Drug Deliv. Rev. 2017, 121, 57–84. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver fibrosis. Pathophysiology, pathogenetic targets and clinical issues. Mol. Asp. Med. 2019, 65, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Novo, E.; Bocca, C.; Foglia, B.; Protopapa, F.; Maggiora, M.; Parola, M.; Cannito, S. Liver fibrogenesis: Un update on established and emerging basic concepts. Arch. Biochem. Biophys. 2020, 689, 108445. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Guillot, A.; Tacke, F. Liver Macrophages: Old Dogmas and New Insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef] [Green Version]
- Campana, L.; Iredale, J.P. Regression of Liver Fibrosis. Semin. Liver Dis. 2017, 37, 1–10. [Google Scholar] [CrossRef]
- Iredale, J.P.; Thompson, A.; Henderson, N.C. Extracellular matrix degradation in liver fibrosis: Biochemistry and regulation. Biochim. Biophys. Acta 2013, 1832, 876–883. [Google Scholar] [CrossRef] [Green Version]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Rønnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis—Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef]
- Szabo, G.; Momen-Heravi, F. Extracellular vesicles in liver disease and potential as biomarkers and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Olaizola, P.; Lee-Law, P.Y.; Arbelaiz, A.; Lapitz, A.; Perugorria, M.J.; Bujanda, L.; Banales, J.M. MicroRNAs and extracellular vesicles in cholangiopathies. Biochim. Biophys. Acta 2018, 1864, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Urban, S.K.; Mocan, T.; Sänger, H.; Lukacs-Kornek, V.; Kornek, M. Extracellular Vesicles in Liver Diseases: Diagnostic, Prognostic, and Therapeutic Application. Semin. Liver Dis. 2019, 39, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Thoen, L.F.; Guimaraes, E.L.; Dolle, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef]
- Hernández-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Koo, J.H.; Lee, H.J.; Kim, W.; Kim, S.G. Endoplasmic reticulum stress in hepatic stellate cells promotes liver fibrosis via PERK-mediated degradation of HNRNPA1 and up-regulation of SMAD2. Gastroenterology 2016, 150, 181–193.e8. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Seth, D.; Day, C.P. Genetic factors that affect risk of alcoholic and nonalcoholic fatty liver disease. Gastroenterology 2016, 150, 1728–1744. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.; Anstee, Q.M. Genetics of alcoholic liver disease and non-alcoholic steatohepatitis. Clin. Med. 2018, 18 (Suppl. 2), s54–s59. [Google Scholar] [CrossRef] [PubMed]
- Paternostro, C.; David, E.; Novo, E.; Parola, M. Hypoxia, angiogenesis and liver fibrogenesis in the progression of chronic liver diseases. World J. Gastroenterol. 2010, 16, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Nath, B.; Szabo, G. Hypoxia and hypoxia inducible factors: Diverse roles in liver diseases. Hepatology 2012, 55, 622–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocca, C.; Novo, E.; Miglietta, A.; Parola, M. Angiogenesis and Fibrogenesis in Chronic Liver Diseases. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 477–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef]
- Fabris, L.; Spirli, C.; Cadamuro, M.; Fiorotto, R.; Strazzabosco, M. Emerging concepts in biliary repair and fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G102–G116. [Google Scholar] [CrossRef] [Green Version]
- Cannito, S.; Milani, C.; Cappon, A.; Parola, M.; Strazzabosco, M.; Cadamuro, M. Fibroinflammatory Liver Injuries as Preneoplastic Condition in Cholangiopathies. Int. J. Mol. Sci. 2018, 19, 3875. [Google Scholar] [CrossRef] [Green Version]
- Fabris, L.; Fiorotto, R.; Spirli, C.; Cadamuro, M.; Mariotti, V.; Perugorria, M.J.; Banales, J.M.; Strazzabosco, M. Pathobiology of inherited biliary diseases: A roadmap to understand acquired liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Huebert, R.C.; Karlsen, T.; Strazzabosco, M.; LaRusso, N.F.; Gores, G.J. Cholangiocyte pathobiology. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.; Arroyo, A.G.; Sánchez-Madrid, F.; Moreno-Otero, R. Angiogenesis in chronic inflammatory liver disease. Hepatology 2004, 39, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Semela, D.; Bruix, J.; Colle, I.; Pinzani, M.; Bosch, J. Angiogenesis in liver disease. J. Hepatol. 2009, 50, 604–620. [Google Scholar] [CrossRef] [PubMed]
- Rosmorduc, O.; Housset, C. Hypoxia: A link between fibrogenesis, angiogenesis, and carcinogenesis in liver disease. Semin. Liver Dis. 2010, 30, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Lefere, S.; Van Steenkiste, C.; Verhelst, X.; Van Vlierberghe, H.; Devisscher, L.; Geerts, A. Hypoxia-regulated mechanisms in the pathogenesis of obesity and non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2016, 73, 3419–3431. [Google Scholar] [CrossRef]
- Wilson, G.K.; Tennant, A.D.; McKeating, J.A. Hypoxia inducible factors in liver disease and hepatocellular carcinoma: Current understanding and future directions. J. Hepatol. 2014, 61, 1397–1406. [Google Scholar] [CrossRef] [Green Version]
- Ju, C.; Colgan, S.P.; Eltzschig, H.K. Hypoxia-inducible factors as molecular targets for liver diseases. J. Mol. Med. 2016, 94, 613–627. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, F.J.; Xie, C.; Jiang, C. The role of hypoxia-inducible factors in metabolic diseases. Nat. Rev. Endocrinol. 2018, 15, 21–32. [Google Scholar] [CrossRef]
- Kietzmann, T. Liver zonation in health and disease: Hypoxia and hypoxia-inducible transcription factors as concert masters. Int. J. Mol. Sci. 2019, 20, 2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaelin, W.G., Jr.; Ratcliffe, W.J. Oxygen sensing by metazoans: The central role of HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Schödel, J.; Ratcliffe, W.J. Mechanisms of hypoxia signaling: New implications for nephrology. Nat. Rev. Nephrol. 2019, 115, 641–659. [Google Scholar] [CrossRef]
- Wu, D.; Rastinejad, F. Structural characterization of mammalian bHLH-PAS transcription factors. Curr. Opin. Struct. Biol. 2019, 43, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Keith, B.; Adelman, D.M.; Simon, M.C. Targeted mutation of the murine aryl hydrocarbon receptor nuclear translocator 2 (Arnt2) gene reveals partial redundancy with Arnt. Proc. Natl Acad. Sci. USA 2001, 98, 6692–6697. [Google Scholar] [CrossRef] [Green Version]
- Wiesener, M.S.; Turley, H.; Allen, W.E.; Willam, C.; Eckardt, K.U.; Talks, K.L.; Wood, S.M.; Gatter, K.C.; Harris, A.L.; Pugh, C.W.; et al. Induction of endothelial PAS domain protein-1 by hypoxia: Characterization and comparison with hypoxia-inducible factor-1alpha. Blood 1998, 92, 2260–2268. [Google Scholar] [CrossRef]
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, A.; Gradin, K.; et al. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell. 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.E.; Simon, M.C. SnapShot: Hypoxia-Inducible Factors. Cell 2015, 163, 1288–1288.e1. [Google Scholar] [CrossRef] [Green Version]
- Duan, C. Hypoxia-inducible factor 3 biology: Complexities and emerging themes. Am. J. Physiol. Cell. Physiol. 2016, 310, C260–C269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugh, C.W.; Ratcliffe, P.J. New horizons in hypoxia signaling pathways. Exp. Cell Res. 2017, 356, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.A.; Koivunen, P.; Kaelin, W.G., Jr. 2-Oxoglutarate-dependent dioxygenases in cancer. Nat. Rev. Cancer 2020, 20, 710–726. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Semenza, G.L. Maintenance of redox homeostasis by hypoxia-inducible factors. Redox Biol. 2017, 13, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Bellomo, G.; Robino, G.; Barrera, G.; Dianzani, M.U. 4-Hydroxynonenal as a biological signal: Molecular basis and pathophysiological implications. Antioxid. Redox. Signal. 1999, 1, 255–284. [Google Scholar] [CrossRef] [PubMed]
- Robino, G.; Parola, M. Oxidative stress-related molecules and liver fibrosis. J. Hepatol. 2001, 35, 297–306. [Google Scholar] [CrossRef]
- Novo, E.; Parola, M. Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogenes. Tissue Repair. 2008, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 25, 1930–1942. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rautou, P.-E.; Mansouri, A.; Lebrec, D.; Durand, F.; Valla, D.; Moreau, R. Autophagy in liver diseases. J. Hepatol. 2010, 53, 1123–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czaja, M.J.; Ding, W.-X.; Donohue, T.M., Jr.; Friedman, S.L.; Kim, J.-S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; James Ou, J.J.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, P.; Wang, S.; Friedman, S.L. The power of plasticity-metabolic regulation of hepatic stellate cells. Cell Metab. 2021, 33, 242–257. [Google Scholar] [CrossRef]
- Kim, R.S.; Hasegawa, D.; Goossens, N.; Tsuchida, T.; Athwal, V.; Sun, X.; Robinson, C.L.; Bhattacharya, D.; Chou, H.I.; Zhang, D.Y.; et al. The XBP1 Arm of the Unfolded Protein Response Induces Fibrogenic Activity in Hepatic Stellate Cells Through Autophagy. Sci. Rep. 2016, 20, 39342. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Ruart, M.; Chavarria, L.; Campreciós, G.; Suárez-Herrera, N.; Montironi, C.; Guixé-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagán, J.C.; Hernández-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef]
- Ni, H.M.; Bhakta, A.; Wang, S.; Li, Z.; Manley, S.; Huang, H.; Copple, B.; Ding, W.X. Role of hypoxia inducing factor-1beta in alcohol-induced autophagy, steatosis and liver injury in mice. PLoS ONE 2014, 9, e115849. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; de Carvalho Ribeiro, M.; Iracheta-Vellve, A.; Lowe, P.; Ambade, A.; Satishchandran, A.; Bukong, T.; Catalano, D.; Kodys, K.; Szabo, G. Macrophage-Specific Hypoxia-Inducible Factor-1α Contributes to Impaired Autophagic Flux in Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 545–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordóñez, Á.; Corral-Escariz, M.; Soro, I.; López-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting complex I activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Novo, E.; Cannito, S.; Zamara, E.; Valfrè di Bonzo, L.; Caligiuri, A.; Cravanzola, C.; Compagnone, A.; Colombatto, S.; Marra, F.; Pinzani, M.; et al. Proangiogenic cytokines as hypoxia-dependent factors stimulating migration of human hepatic stellate cells. Am. J. Pathol. 2007, 170, 1942–1953. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Q.; Luk, J.M.; Ikeda, K.; Man, K.; Chu, A.C.; Kaneda, K.; Fan, S.T. Regulatory role of vHL/HIF-1α in hypoxia-induced VEGF production in hepatic stellate cells. Biochem. Biophys. Res. Commun. 2004, 317, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Copple, B.L.; Bai, S.; Burgoon, L.D.; Moon, J.O. Hypoxia-inducible factor-1α regulates the expression of genes in hypoxic hepatic stellate cells important for collagen deposition and angiogenesis. Liver Int. 2011, 31, 230–244. [Google Scholar] [CrossRef] [Green Version]
- Aleffi, S.; Navari, N.; Delogu, W.; Galastri, S.; Novo, E.; Rombouts, K.; Pinzani, M.; Parola, M.; Marra, F. Mammalian target of rapamycin mediates the angiogenic effects of leptin in human hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G210–G219. [Google Scholar] [CrossRef] [PubMed]
- Novo, E.; Busletta, C.; di Bonzo, L.V.; Povero, D.; Paternostro, C.; Mareschi, K.; Ferrero, I.; David, E.; Bertolani, C.; Caligiuri, A.; et al. Intracellular reactive oxygen species are required for directional migration of resident and bone marrow-derived hepatic pro-fibrogenic cells. J. Hepatol. 2011, 54, 964–974. [Google Scholar] [CrossRef]
- Novo, E.; Povero, D.; Busletta, C.; Paternostro, C.; di Bonzo, L.V.; Cannito, S.; Compagnone, A.; Bandino, A.; Marra, F.; Colombatto, S.; et al. The biphasic nature of hypoxia-induced directional migration of activated human hepatic stellate cells. J. Pathol. 2012, 226, 588–597. [Google Scholar] [CrossRef]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammoutene, A.; Rautou, P.E. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, 1278–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.O.; Welch, T.P.; Gonzalez, F.J.; Copple, B.L. Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G582–G592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, K.J.; Copple, B.L. Role of Hypoxia-Inducible Factors in the Development of Liver Fibrosis. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Copple, B.L.; Kaska, S.; Wentling, C. Hypoxia-inducible factor activation in myeloid cells contributes to the development of liver fibrosis in cholestatic mice. J. Pharm. Exp. Ther. 2012, 341, 307–316. [Google Scholar] [CrossRef]
- Lee, T.Y.; Leu, Y.L.; Wen, C.K. Modulation of HIF-1α and STAT3 signaling contributes to anti-angiogenic effect of YC-1 in mice with liver fibrosis. Oncotarget 2017, 8, 86206–86216. [Google Scholar] [CrossRef] [Green Version]
- Strickland, J.; Garrison, D.; Copple, B.L. Hypoxia upregulates Cxcl12 in hepatocytes by a complex mechanism involving hypoxia-inducible factors and transforming growth factor-β. Cytokine 2020, 127, 154986. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Y.; Guan, F.; Xiao, Y.; Deng, J.; Chen, H.; Chen, X.; Li, J.; Huang, H.; Shi, C. Hypoxia-inducible factor-1alpha and MAPK co-regulate activation of hepatic stellate cells upon hypoxia stimulation. PLoS ONE 2013, 8, e74051. [Google Scholar] [CrossRef] [Green Version]
- Mochizuki, A.; Pace, A.; Rockwell, C.E.; Roth, K.J.; Chow, A.; O’Brien, K.M.; Albee, R.; Kelly, K.; Towery, K.; Luyendyk, J.P.; et al. Hepatic stellate cells orchestrate clearance of necrotic cells in a hypoxia-inducible factor-1alpha-dependent manner by modulating macrophage phenotype in mice. J. Immunol. 2014, 192, 3847–3857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Huang, Q.; Wang, Y.; Shen, P.; Guan, F.; Li, J.; Huang, H.; Shi, C. Hypoxia-inducible factor-1alpha regulates autophagy to activate hepatic stellate cells. Biochem. Biophys. Res. Commun. 2014, 454, 328–334. [Google Scholar] [CrossRef]
- Liu, J.; Xie, Y.; Cui, Z.; Xia, T.; Wan, L.; Zhou, H.; Zhang, P.; Zhang, Y.; Guan, F.; Liu, W.; et al. Bnip3 interacts with vimentin, an intermediate filament protein, and regulates autophagy of hepatic stellate cells. Aging 2020, 13, 957–972. [Google Scholar] [CrossRef]
- Li, G.; Li, J.; Li, C.; Qi, H.; Dong, P.; Zheng, J.; Yu, F. MicroRNA-125a-5p contributes to hepatic stellate cell activation through targeting FIH1. Cell. Physiol. Biochem. 2016, 38, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Hu, D.; Yu, H.; Xu, W.; Fu, R. Hypoxia-inducible factor 1alpha and ROCK1 regulate proliferation and collagen synthesis in hepatic stellate cells under hypoxia. Mol. Med. Rep. 2018, 18, 3997–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Yang, X.; Kai, J.; Wang, F.; Wang, Z.; Shao, J.; Tan, S.; Chen, A.; Zhang, F.; Wang, S.; et al. HIF-1alpha-upregulated lncRNA-H19 regulates lipid droplet metabolism through the AMPKalpha pathway in hepatic stellate cells. Life Sci. 2020, 255, 117818. [Google Scholar] [CrossRef] [PubMed]
- Hernández, A.; Reyes, D.; Geng, Y.; Arab, J.P.; Cabrera, D.; Sepulveda, R.; Solis, N.; Buist-Homan, M.; Arrese, M.; Moshage, H. Extracellular vesicles derived from fat-laden hepatocytes undergoing chemical hypoxia promote a pro-fibrotic phenotype in hepatic stellate cells. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165857. [Google Scholar] [CrossRef] [PubMed]
- Ba, H.Z.; Liang, Z.H.; Kim, H.S.; Cao, W. TGF-beta1 can be regulated by NDRG2 via the NF-kappaB pathway in hypoxia-induced liver fibrosis. Ann. Transl. Med. 2021, 9, 505. [Google Scholar] [CrossRef]
- Chen, C.; Lou, T. Hypoxia inducible factors in hepatocellular carcinoma. Oncotarget 2017, 8, 46691–46703. [Google Scholar] [CrossRef] [Green Version]
- Kantari-Mimoun, C.; Krzywinska, E.; Castells, M.; Milien, C.; Klose, R.; Meinecke, A.K.; Lemberger, U.; Mathivet, T.; Gojkovic, M.; Schrödter, K.; et al. Boosting the hypoxic response in myeloid cells accelerates resolution of fibrosis and regeneration of the liver in mice. Oncotarget 2017, 8, 15085–15100. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.; Stokes, R.; Cha, K.M.; Clouston, A.; Eslam, M.; Metwally, M.; Swarbrick, M.M.; George, J.; Gunton, J.E. Myeloid cell deletion of Aryl hydrocarbon Receptor Nuclear Translocator (ARNT) induces non-alcoholic steatohepatitis. PLoS ONE 2019, 14, e0225332. [Google Scholar] [CrossRef]
- Gunton, J.E.; Kulkarni, R.N.; Yim, S.; Okada, T.; Hawthorne, W.J.; Tseng, Y.H.; Roberson, R.S.; Ricordi, C.; O’Connell, P.J.; Gonzalez, F.J.; et al. Loss of ARNT/HIF1β mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell 2005, 122, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.; Ho, K.; Stokes, R.; Scott, C.; Lau, S.M.; Hawthorne, W.J.; O’Connell, P.J.; Loudovaris, T.; Kay, T.W.; Kulkarni, R.N.; et al. Hypoxia-inducible factor-1α regulates βcell function in mouse and human islets. J. Clin. Investig. 2010, 120, 2171–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantena, S.K.; Vaughn, D.P.; Andringa, K.K.; Eccleston, H.B.; King, A.L.; Abrams, G.A.; Doeller, J.E.; Kraus, D.W.; Darley-Usmar, V.M.; Bailey, S.M. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem. J. 2009, 417, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arteel, G.E.; Iimuro, Y.; Yin, M.; Raleigh, J.A.; Thurman, R.G. Chronic enteral ethanol treatment causes hypoxia in rat liver tissue in vivo. Hepatology 1997, 25, 920–926. [Google Scholar] [CrossRef]
- Kucejova, B.; Sunny, N.E.; Nguyen, A.D.; Hallac, R.; Fu, X.; Peña-Llopis, S.; Mason, R.P.; Deberardinis, R.J.; Xie, X.J.; Debose-Boyd, R.; et al. Uncoupling hypoxia signaling from oxygen sensing in the liver results in hypoketotic hypoglycemic death. Oncogene 2011, 30, 2147–2160. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Xiang, D.; Wu, D.; Liu, C.; Fang, Y.; Chen, P.; Hu, Y.-P. Hepatic PHD2/HIF-1α axis is involved in postexercise systemic energy homeostasis. FASEB J. 2018, 32, 4670–4680. [Google Scholar] [CrossRef] [Green Version]
- Minamishima, Y.A.; Moslehi, J.; Padera, R.F.; Bronson, R.T.; Liao, R.; Kaelin, W.G., Jr. A feedback loop involving the Phd3 prolyl hydroxylase tunes the mammalian hypoxic response in vivo. Mol. Cell. Biol. 2009, 29, 5729–5741. [Google Scholar] [CrossRef] [Green Version]
- Nath, B.I.; Levin, I.; Csak, T.; Petrasek, J.; Mueller, C.; Kodys, K.; Catalano, D.; Mandrekar, P.; Szabo, G. Hepatocyte-specific hypoxia-inducible factor-1α is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology 2011, 53, 1526–1537. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, Y.; Goda, N.; Kanai, M.; Niwa, D.; Osanai, K.; Yamamoto, Y.; Senoo-Matsuda, N.; Johnson, R.S.; Miura, S.; Kabe, Y.; et al. HIF-1α induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J. Hepatol. 2012, 56, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Mesarwi, O.A.; Shin, M.K.; Bevans-Fonti, S.; Schlesinger, C.; Shaw, J.; Polotsky, V.Y. Hepatocyte Hypoxia Inducible Factor-1 Mediates the Development of Liver Fibrosis in a Mouse Model of Nonalcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0168572. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; He, Y.; Zhao, H.; Xu, X. Hypoxia inducible factor-1 promotes liver fibrosis in nonalcoholic fatty liver disease by activating PTEN/p65 signaling pathway. J. Cell. Biochem. 2019, 120, 14735–14744. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Tanaka, M.; Goda, N. HIF-1-dependent lipin1 induction prevents excessive lipid accumulation in choline-deficient diet-induced fatty liver. Sci. Rep. 2018, 8, 14230. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yang, W.; Gan, L.; Liu, S.; Ni, Q.; Bi, Y.; Han, T.; Liu, Q.; Chen, H.; Hu, Y.; et al. Silencing HIF-1alpha aggravates non-alcoholic fatty liver disease in vitro through inhibiting PPAR-alpha/ANGPTL4 singling pathway. Gastroenterol. Hepatol. 2021, 44, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Tsurusaki, S.; Miyata, N.; Saijou, E.; Okochi, H.; Miyajima, A.; Tanaka, M. Oncostatin M causes liver fibrosis by regulating cooperation between hepatic stellate cells and macrophages in mice. Hepatology 2017, 67, 296–312. [Google Scholar] [CrossRef] [Green Version]
- Foglia, B.; Sutti, S.; Pedicini, D.; Cannito, S.; Bocca, C.; Maggiora, M.; Bevacqua, M.R.; Rosso, C.; Bugianesi, E.; Albano, E.; et al. Oncostatin M, a profibrogenic mediator overexpressed in non-alcoholic fatty liver disease, stimulates migration of hepatic myofibroblasts. Cells 2019, 9, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Miyajima, A. Oncostatin M, a multifunctional cytokine. Rev. Physiol. Biochem. Pharm. 2003, 149, 39–52. [Google Scholar] [CrossRef]
- Elks, C.M.; Stephens, J.M. Oncostatin m modulation of lipid storage. Biology 2015, 4, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Miyajima, A.; Kinoshita, T.; Tanaka, M.; Kamiya, A.; Mukouyama, Y.; Hara, T. Role of oncostatin M in hematopoiesis and liver development. Cytokine Growth Factor Rev. 2000, 11, 177–183. [Google Scholar] [CrossRef]
- Wallace, P.M.; MacMaster, J.F.; Rouleau, K.A.; Brown, T.J.; Loy, J.K.; Donaldson, K.L.; Wahl, A.F. Regulation of inflammatory responses by oncostatin M. J. Immunol. 1999, 162, 5547–5555. [Google Scholar] [CrossRef]
- Znoyko, I.; Sohara, N.; Spicer, S.; Trojanowska, M.; Reuben, A. Expression of oncostatin M and its receptors in normal and cirrhotic human liver. J. Hepatol. 2005, 43, 893–900. [Google Scholar] [CrossRef]
- Levy, M.T.; Trojanowska, M.; Reuben, A. Oncostatin M: A cytokine upregulated in human cirrhosis, increases collagen production by human hepatic stellate cells. J. Hepatol. 2000, 32, 218–226. [Google Scholar] [CrossRef]
- Henkel, J.; Gartner, D.; Dorn, C.; Hellerbrand, C.; Schanze, N.; Elz, S.R.; Püschel, G.P. Oncostatin M produced in kupffer cells in response to pge2: Possible contributor to hepatic insulin resistance and steatosis. Lab. Investig. 2011, 91, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, S.; Kappler, V.; Kaczor, J.; Flügel, D.; Rolvering, C.; Kato, N.; Kietzmann, T.; Behrmann, I.; Haan, C. Hypoxia-inducible factor 1alpha is up-regulated by oncostatin M and participates in oncostatin M signaling. Hepatology 2009, 50, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.K.; Shah, Y.M. A central role for hypoxia-inducible factor (HIF)-2α in hepatic glucose homeostasis. Nutr. Healthy Aging 2017, 4, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Rha, J.; Selak, M.A.; Unger, T.L.; Keith, B.; Liu, Q.; Haase, V.H. Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol. Cell. Biol. 2009, 29, 4527–4538. [Google Scholar] [CrossRef] [Green Version]
- Qu, A.; Taylor, M.; Xue, X.; Matsubara, T.; Metzger, D.; Chambon, P.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-inducible transcription factor 2α promotes steatohepatitis through augmenting lipid accumulation, inflammation, and fibrosis. Hepatology 2011, 54, 472–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morello, E.; Sutti, S.; Foglia, B.; Novo, E.; Cannito, S.; Bocca, C.; Rajsky, M.; Bruzzì, S.; Abate, M.L.; Rosso, C.; et al. Hypoxia-inducible factor 2α drives nonalcoholic fatty liver progression by triggering hepatocyte release of histidine-rich glycoprotein. Hepatology 2018, 67, 2196–2214. [Google Scholar] [CrossRef] [Green Version]
- Bartneck, M.; Fech, V.; Ehling, J.; Govaere, O.; Warzecha, K.T.; Hittatiya, K.; Vucur, M.; Gautheron, J.; Luedde, T.; Trautwein, C.; et al. Histidine-rich glycoprotein promotes macrophage activation and inflammation in chronic liver disease. Hepatology 2016, 63, 1310–1324. [Google Scholar] [CrossRef]
- Cai, H.; Bai, Z.; Ge, R.L. Hypoxia-inducible factor-2 promotes liver fibrosis in non-alcoholic steatohepatitis liver disease via the NF-kappaB signalling pathway. Biochem. Biophys. Res. Commun. 2021, 540, 67–74. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.; Fu, H.; Li, Y.; Wang, L.; Luo, S.; Lu, H. Hypoxia exacerbates nonalcoholic fatty liver disease via the HIF-2alpha/PPARalpha pathway. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E710–E722. [Google Scholar] [CrossRef]
- Turato, C.; Calabrese, F.; Biasiolo, A.; Quarta, S.; Ruvoletto, M.; Tono, N.; Paccagnella, D.; Fassina, G.; Merkel, C.; Harrison, T.J.; et al. SERPINB3 modulates TGF-beta expression in chronic liver disease. Lab. Investig. 2010, 90, 1016–1023. [Google Scholar] [CrossRef] [Green Version]
- Cannito, S.; Turato, C.; Paternostro, C.; Biasiolo, A.; Colombatto, S.; Cambieri, I.; Quarta, S.; Novo, E.; Morello, E.; Villano, G.; et al. Hypoxia up-regulates SERPINB3 through HIF-2α in human liver cancer cells. Oncotarget 2015, 10, 2206–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novo, E.; Villano, G.; Turato, C.; Cannito, S.; Paternostro, C.; Busletta, C.; Biasiolo, A.; Quarta, S.; Morello, E.; Bocca, C.; et al. SerpinB3 Promotes Pro-fibrogenic Responses in Activated Hepatic Stellate Cells. Sci. Rep. 2017, 7, 3420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderborght, B.; De Muynck, K.; Lefere, S.; Geerts, A.; Degroote, H.; Verhelst, X.; Van Vlierberghe, H.; Devisscher, L. Effect of isoform-specific HIF-1alpha and HIF-2alpha antisense oligonucleotides on tumorigenesis, inflammation and fibrosis in a hepatocellular carcinoma mouse model. Oncotarget 2020, 11, 4504–4520. [Google Scholar] [CrossRef]
- Ramakrishnan, S.K.; Zhang, H.; Takahashi, S.; Centofanti, B.; Periyasamy, S.; Weisz, K.; Chen, Z.; Uhler, M.D.; Rui, L.; Gonzalez, F.J.; et al. HIF2α is an essential molecular brake for postprandial hepatic glucagon response independent of insulin signaling. Cell Metab. 2016, 23, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, C.M.; Finger, E.C.; Krieg, A.J.; Wu, C.; Diep, A.N.; LaGory, E.L.; Wei, K.; McGinnis, L.M.; Yuan, J.; Kuo, C.J.; et al. Cross-talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat. Med. 2013, 19, 1325–1330. [Google Scholar] [CrossRef] [Green Version]
- Wei, K.; Piecewicz, S.M.; McGinnis, L.M.; Taniguchi, C.M.; Wiegand, S.J.; Anderson, K.; Chan, C. W-M.; Mulligan, K.X.; Kuo, D.; Yuan, J.; et al. A liver Hif-2α-Irs2 pathway sensitizes hepatic insulin signaling and is modulated by Vegf inhibition. Nat. Med. 2013, 19, 1331–1337. [Google Scholar] [CrossRef]
- Xie, C.; Yagai, T.; Luo, Y.; Liang, X.; Chen, T.; Wang, Q.; Sun, D.; Zhao, J.; Ramakrishnan, S.K.; Sun, L.; et al. Activation of intestinal hypoxia-inducible factor 2α during obesity contributes to hepatic steatosis. Nat. Med. 2017, 23, 1298–1308. [Google Scholar] [CrossRef]
- Cho, H.; Kaelin, W.G. Targeting HIF2 in Clear Cell Renal Cell Carcinoma. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Pharmacologic targeting of hypoxia-inducible factors. Ann. Rev. Pharmacol. Toxicol. 2019, 59, 379–403. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, S. Daprodustat: First approval. Drugs 2020, 80, 1491–1497. [Google Scholar] [CrossRef]
- Markham, A. Vadadustat: First approval. Drugs 2020, 80, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [Green Version]
- Choueiri, T.K.; Kaelin, W.G., Jr. Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat. Med. 2020, 26, 1519–1530. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Lemoinne, S.; Friedman, S.L. New and emerging anti-fibrotic therapeutics entering or already in clinical trials in chronic liver diseases. Curr. Opin. Pharm. 2019, 49, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef]
- Huisman, T.M.; Dieterich, D.T.; Friedman, S.L. Experimental and Investigational Targeted Therapies for the Management of Fibrosis in NASH: An Update. J. Exp. Pharm. 2021, 13, 329–338. [Google Scholar] [CrossRef]
- Weiskirchen, R. Hepatoprotective and antifibrotic agents: It’s time to take the next step. Front. Pharm. 2016, 6, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luangmonkong, T.; Suriguga, S.; Mutsaers, H.A.M.; Groothuis, G.M.M.; Olinga, P.; Boersema, M. Targeting Oxidative Stress for the Treatment of Liver Fibrosis. Rev. Physiol. Biochem. Pharm. 2018, 175, 71–102. [Google Scholar] [CrossRef]
- Tacke, F.; Weiskirchen, R. Non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH)-related liver fibrosis: Mechanisms, treatment and prevention. Ann. Transl. Med. 2021, 9, 729. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Type of Chronic Injury | Agent/Condition | Disease | Pattern of Fibrosis |
|---|---|---|---|
| Parenchymal: viral infection | HBV | ||
| HCV | CVH | Bridging fibrosis, post-necrotic | |
| HBV + HDV | |||
| Parenchymal: altered metabolism | Obesity/T2DM/MS | NAFLD | Pericellular/perisinusoidal fibrosis |
| Parenchymal: toxic | Ethanol | ALD | Pericellular/perisinusoidal fibrosis |
| Parenchymal: immune-mediated | Autoimmune injury to hepatocytes | AH1 | Bridging fibrosis, |
| AH2 | post-necrotic | ||
| Parenchymal: genetically related | α1AT | ||
| Hereditary | WD HH | Bridging fibrosis, post-necrotic | |
| Cholangiopathies: genetically related | Alagille syndrome | ||
| Hereditary | Caroli syndrome ABCB4 deficiency Cystic fibrosis Polycystic disease | Biliary-like fibrosis | |
| Cholangiopathies: immune-mediated | Autoimmune injury to cholangiocytes | PBC PSC | Biliary-like fibrosis |
| Cholangiopathies: unknown | Idiopathic cholangiopathies | Biliary atresia Sarcoidosis | Biliary-like fibrosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foglia, B.; Novo, E.; Protopapa, F.; Maggiora, M.; Bocca, C.; Cannito, S.; Parola, M. Hypoxia, Hypoxia-Inducible Factors and Liver Fibrosis. Cells 2021, 10, 1764. https://doi.org/10.3390/cells10071764
Foglia B, Novo E, Protopapa F, Maggiora M, Bocca C, Cannito S, Parola M. Hypoxia, Hypoxia-Inducible Factors and Liver Fibrosis. Cells. 2021; 10(7):1764. https://doi.org/10.3390/cells10071764
Chicago/Turabian StyleFoglia, Beatrice, Erica Novo, Francesca Protopapa, Marina Maggiora, Claudia Bocca, Stefania Cannito, and Maurizio Parola. 2021. "Hypoxia, Hypoxia-Inducible Factors and Liver Fibrosis" Cells 10, no. 7: 1764. https://doi.org/10.3390/cells10071764
APA StyleFoglia, B., Novo, E., Protopapa, F., Maggiora, M., Bocca, C., Cannito, S., & Parola, M. (2021). Hypoxia, Hypoxia-Inducible Factors and Liver Fibrosis. Cells, 10(7), 1764. https://doi.org/10.3390/cells10071764