
Silibinin Regulates Tumor Progression and Tumorsphere Formation by Suppressing PD-L1 Expression in Non-Small Cell Lung Cancer (NSCLC) Cells


 ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Cell Culture Reagents
2.2. Cell Culture and Treatment
2.3. Cell Viability Assay
2.4. 4′,6-diamidino-2-phenylindole (DAPI) Staining and Morphological Analysis
2.5. Western Blotting Analysis
2.6. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
2.7. Cell Cycle Analysis
2.8. Apoptosis Analysis
2.9. In Vitro Angiogenesis Assay
2.10. Matrigel Invasion Assay
2.11. Wound Healing Assay
2.12. Tumorsphere Formation Assay
2.13. STAT5 and AKT1/2 Inhibitors
2.14. Chromatin Immunoprecipitation (ChIP) Assay
2.15. Statistical Analyses
3. Results
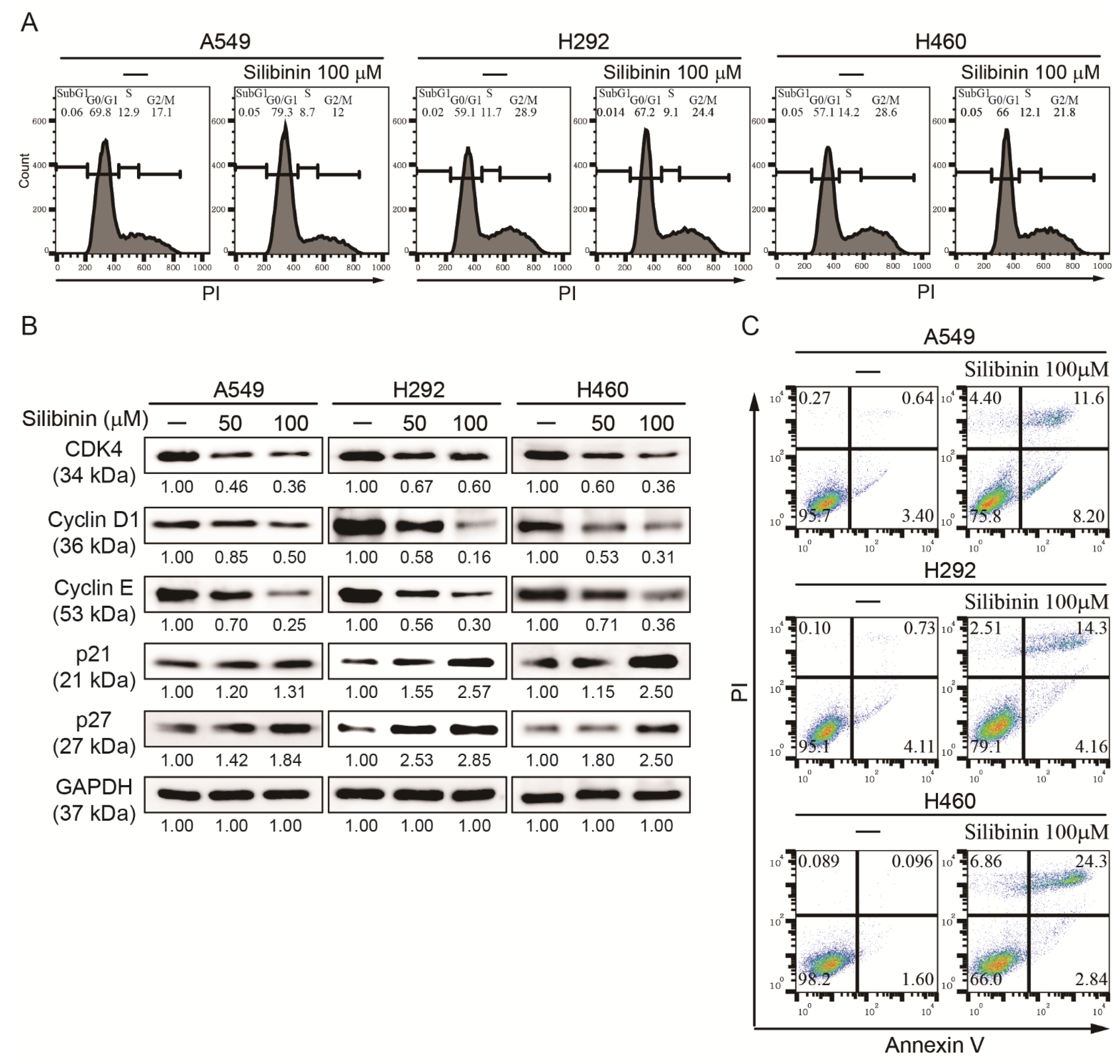
3.1. Silibinin Prevents NSCLC Cell Proliferation and Induces Cell Cycle Arrest and Apoptosis
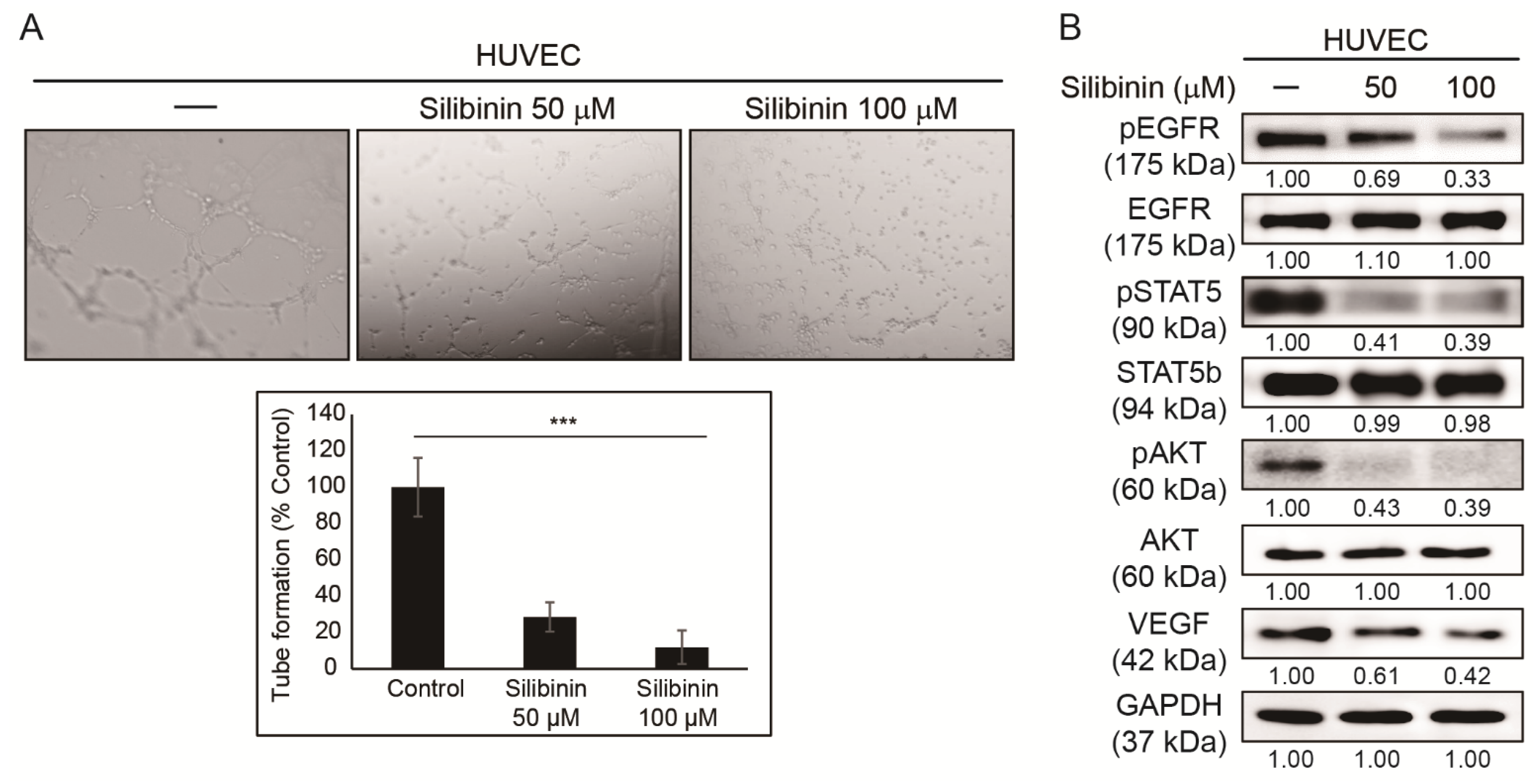
3.2. Silibinin Inhibits Angiogenesis of HUVEC Cells
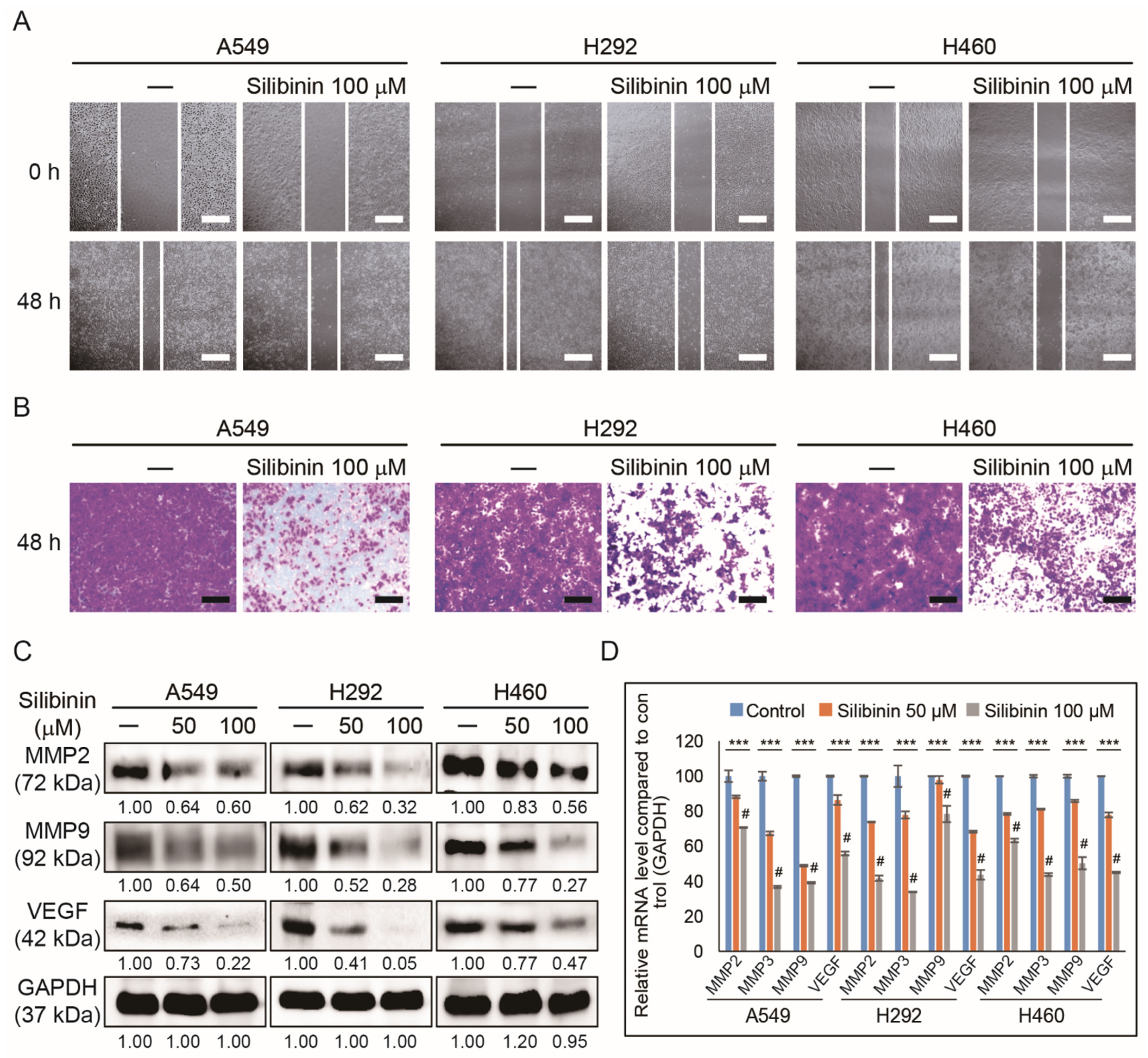
3.3. Silibinin Inhibits NSCLC Cell Migration and Invasion
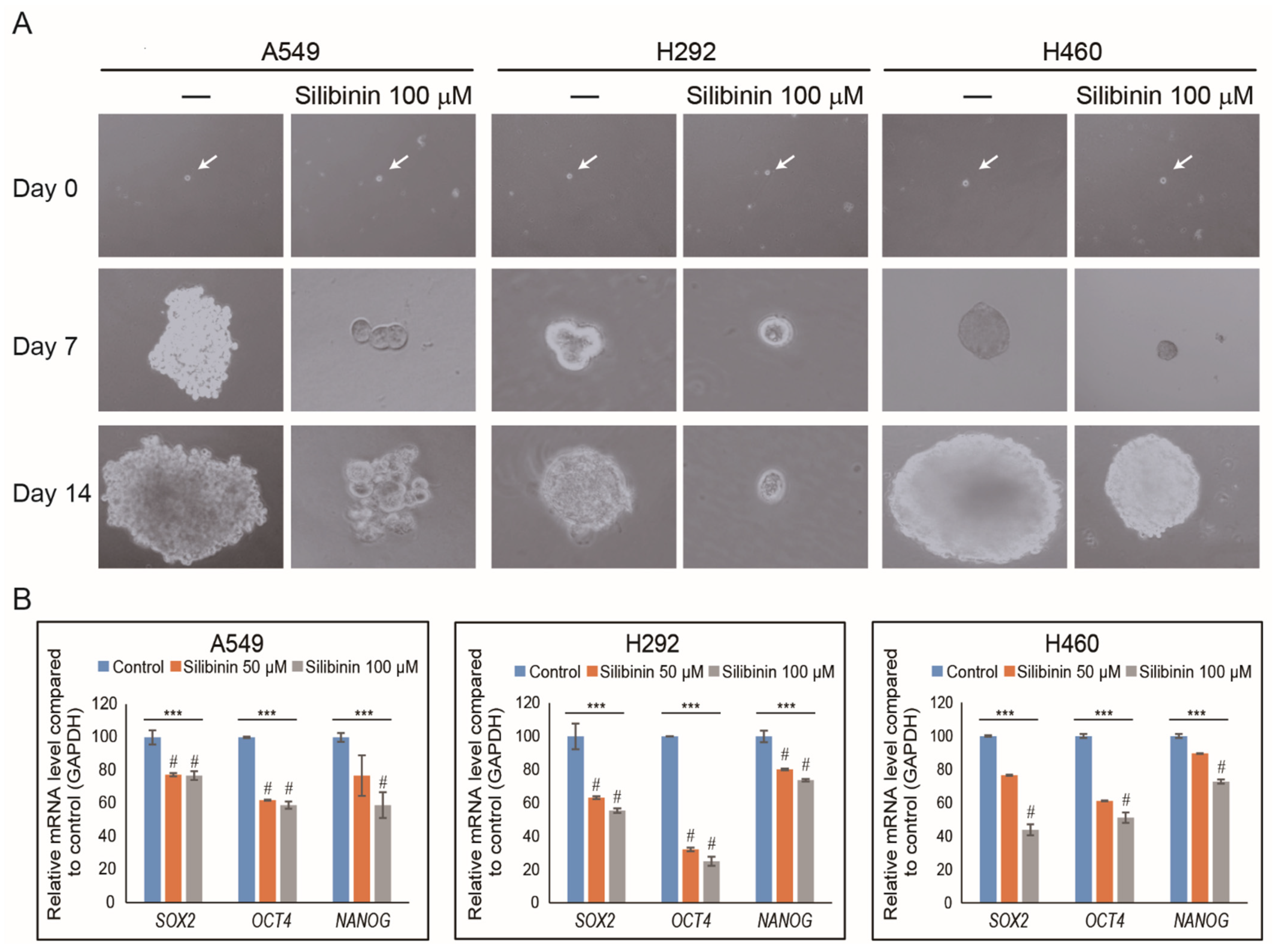
3.4. Silibinin Suppresses Cancer Stemness by Tumorsphere Inhibition in NSCLC Cells
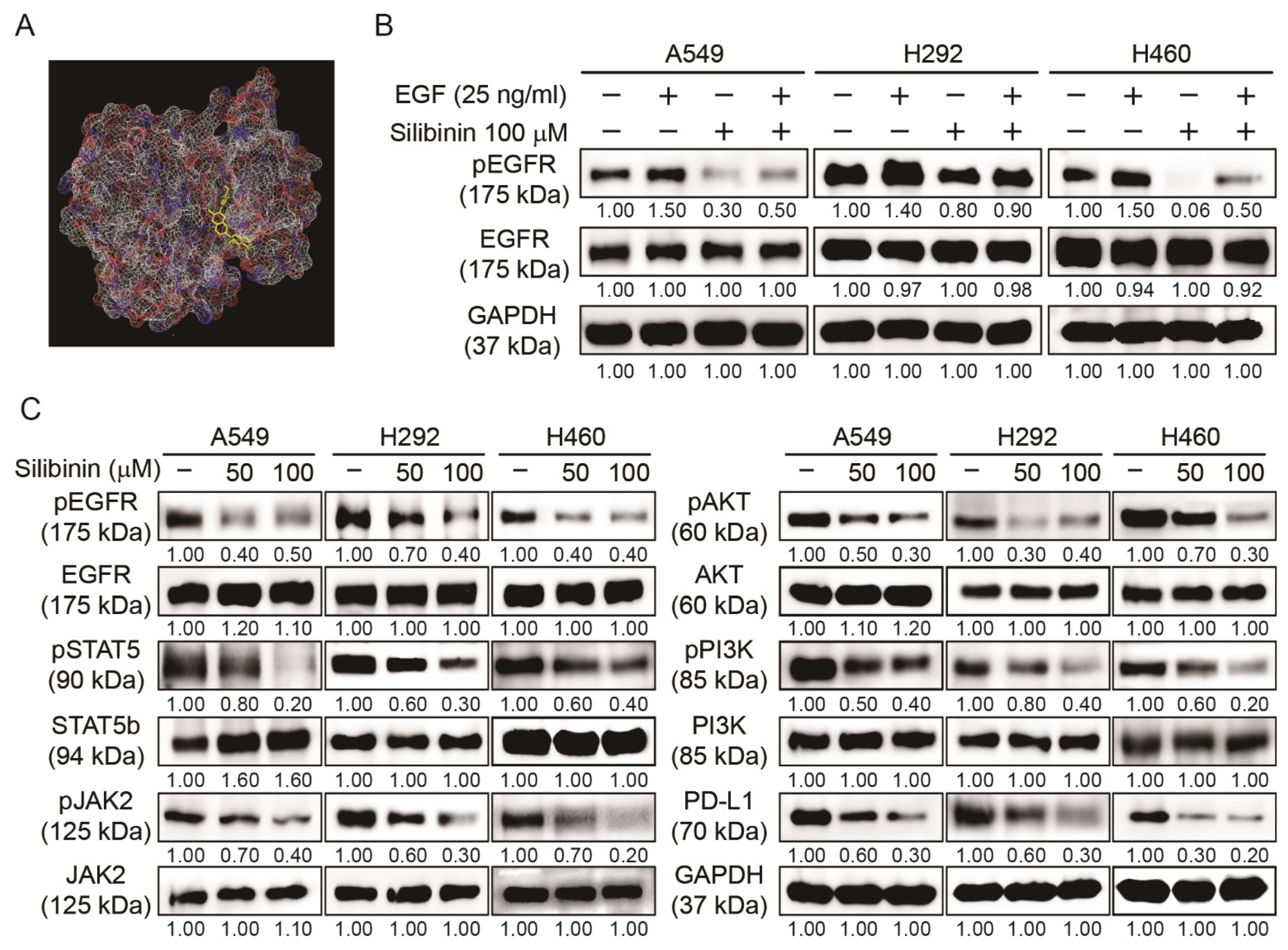
3.5. Silibinin Inhibited EGFR Pathway and PD-L1 Expression in NSCLC Cells
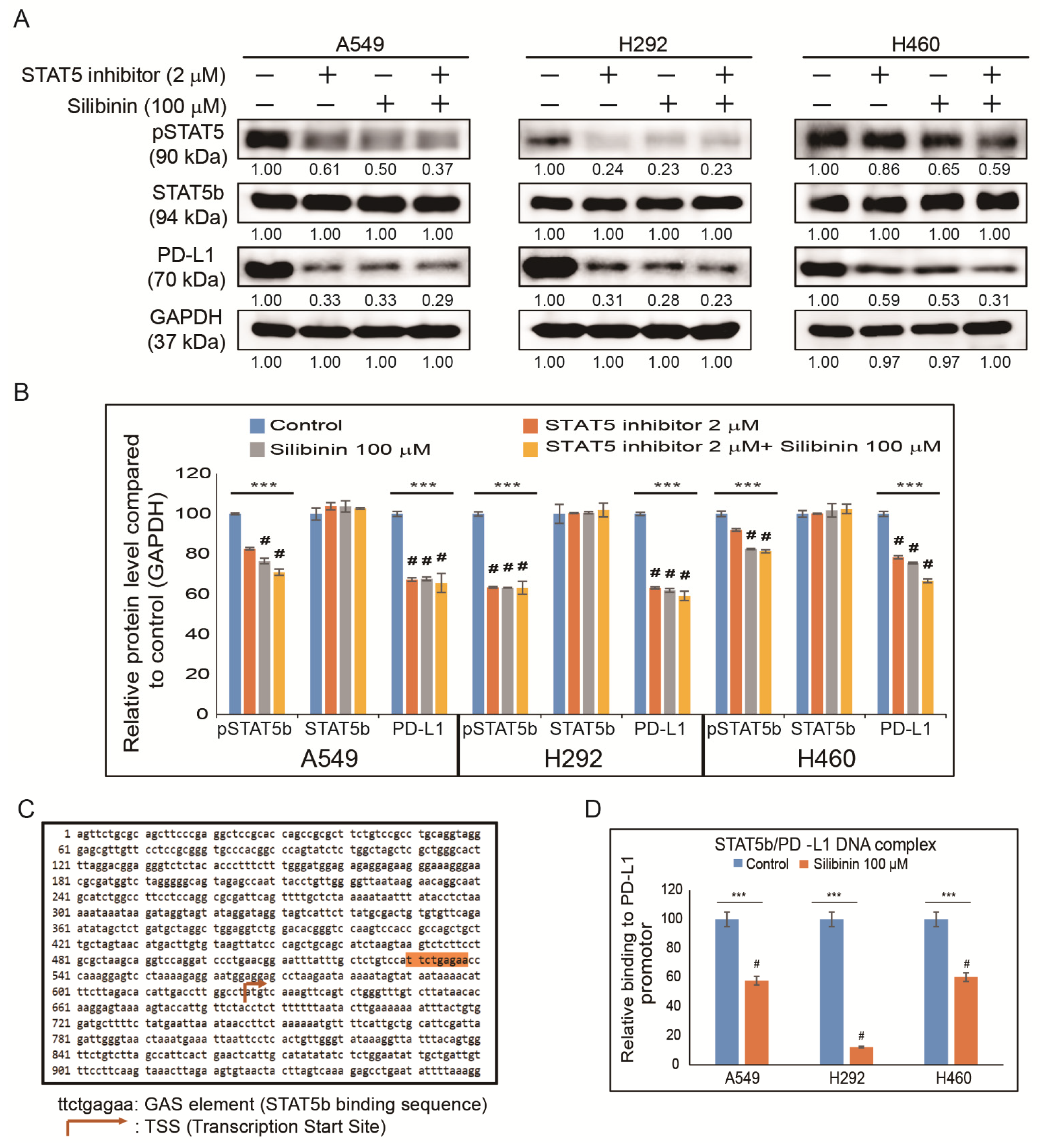
3.6. Silibinin Impaired STAT5-Dependent PD-L1 Expression and STAT5 Binding to the PD-L1 Promoter Region
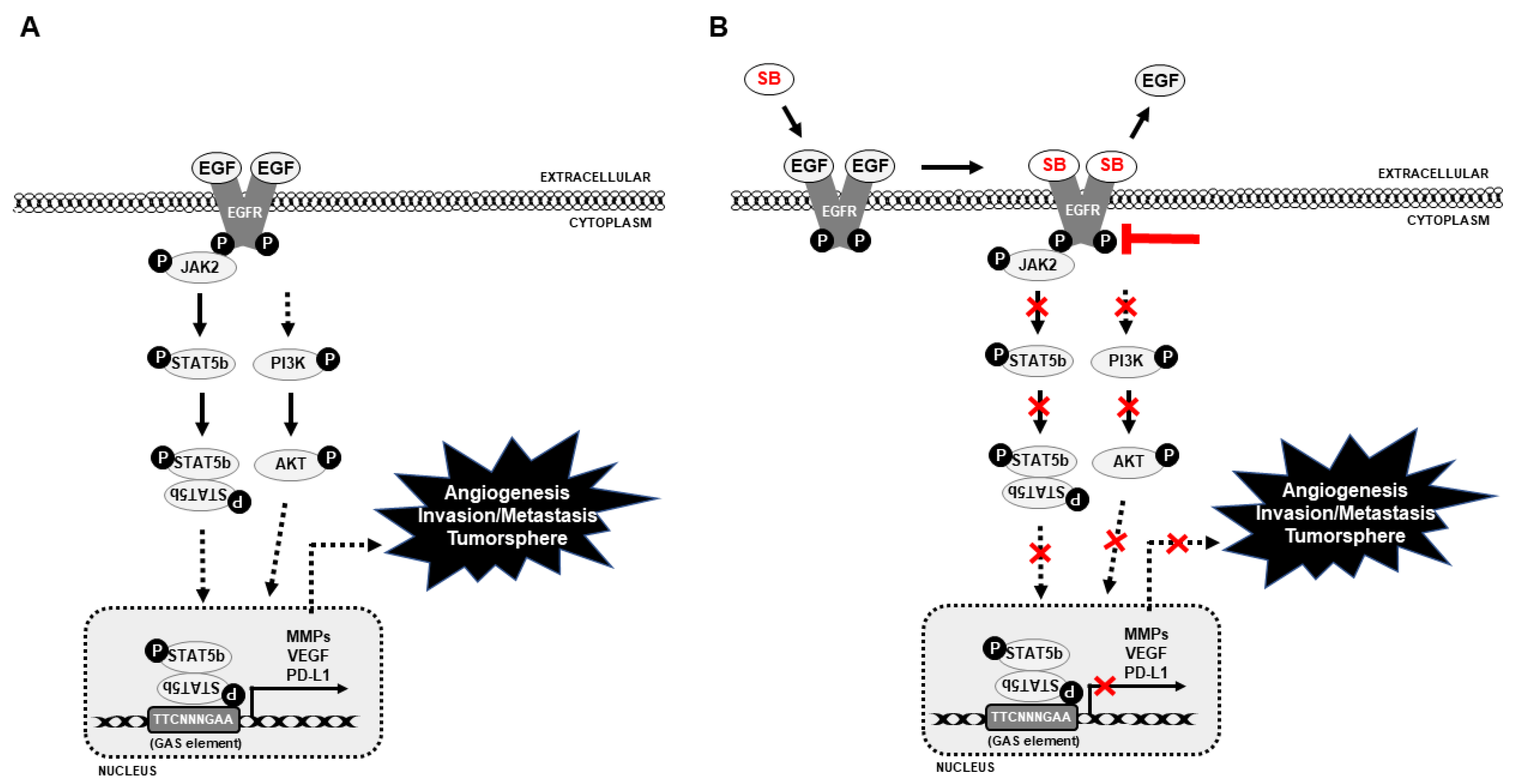
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nowell, P.C. Tumor progression: A brief historical perspective. Semin. Cancer Biol. 2002, 12, 261–266. [Google Scholar] [CrossRef]
- Fidler, I. Cancer Invasion and Metastasis: Biologic and Therapeutic Aspects; Nicolson, G.L., Milas, L., Eds.; Raven Press: New York, NY, USA, 1984; pp. 5–26. [Google Scholar]
- Rajappa, M.; Saxena, P.; Kaur, J. Ocular angiogenesis: Mechanisms and recent advances in therapy. Adv. Clin. Chem. 2010, 50, 103–121. [Google Scholar] [PubMed]
- Zhang, Y.; Xu, W.; Guo, H.; Zhang, Y.; He, Y.; Lee, S.H.; Song, X.; Li, X.; Guo, Y.; Zhao, Y.; et al. NOTCH1 Signaling Regulates Self-Renewal and Platinum Chemoresistance of Cancer Stem-like Cells in Human Non-Small Cell Lung Cancer. Cancer Res. 2017, 77, 3082–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastrana, E.; Silva-Vargas, V.; Doetsch, F. Eyes wide open: A critical review of sphere-formation as an assay for stem cells. Cell Stem Cell 2011, 8, 486–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, A.; Fako, V.; Dang, H.; Forgues, M.; Yu, Z.; Budhu, A.; Wang, X.W. Three-dimensional organotypic culture models of human hepatocellular carcinoma. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Xie, Y.; Shi, X.; Sheng, K.; Han, G.; Li, W.; Zhao, Q.; Jiang, B.; Feng, J.; Li, J.; Gu, Y. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol. Med. Rep. 2019, 19, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Keppler-Noreuil, K.M.; Parker, V.E.; Darling, T.N.; Martinez-Agosto, J.A. Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am. J. Med. Genet. C Semin Med. Genet. 2016, 172, 402–421. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.S.; Wang, Q.; Fu, X.H.; Huang, X.H.; Chen, X.L.; Cao, L.Q.; Chen, L.Z.; Tan, H.X.; Li, W.; Bi, J.; et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol Res. 2009, 39, 177–186. [Google Scholar] [CrossRef]
- Li, B.; Xu, W.W.; Lam, A.K.Y.; Wang, Y.; Hu, H.F.; Guan, X.Y.; Qin, Y.R.; Saremi, N.; Tsao, S.W.; He, Q.Y.; et al. Significance of PI3K/AKT signaling pathway in metastasis of esophageal squamous cell carcinoma and its potential as a target for anti-metastasis therapy. Oncotarget 2017, 8, 38755–38766. [Google Scholar] [CrossRef] [Green Version]
- Chetty, C.; Lakka, S.S.; Bhoopathi, P.; Rao, J.S. MMP-2 alters VEGF expression via alphaVbeta3 integrin-mediated PI3K/AKT signaling in A549 lung cancer cells. Int J. Cancer 2010, 127, 1081–1095. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nipin, S.P.; Darvin, P.; Yoo, Y.B.; Joung, Y.H.; Kang, D.Y.; Kim, D.N.; Hwang, T.S.; Kim, S.Y.; Kim, W.S.; Lee, H.K.; et al. The combination of methylsulfonylmethane and tamoxifen inhibits the Jak2/STAT5b pathway and synergistically inhibits tumor growth and metastasis in ER-positive breast cancer xenografts. BMC Cancer 2015, 15, 474. [Google Scholar] [CrossRef] [Green Version]
- Rundhaug, J.E. Matrix metalloproteinases, angiogenesis, and cancer: Commentary re: A. C. Lockhart et al., Reduction of wound angiogenesis in patients treated with BMS-275291, a broad spectrum matrix metalloproteinase inhibitor. Clin. Cancer Res. 2003, 9, 551–554. [Google Scholar]
- Verma, R.P.; Hansch, C. Matrix metalloproteinases (MMPs): Chemical–biological functions and (Q) SARs. Bioorg. Med. Chem. 2007, 15, 2223–2268. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, A.H. Apoptosis and carcinogenesis. Eur. J. Cell Biol. 1997, 73, 189–197. [Google Scholar] [PubMed]
- Shimoji, M.; Shimizu, S.; Sato, K.; Suda, K.; Kobayashi, Y.; Tomizawa, K.; Takemoto, T.; Mitsudomi, T. Clinical and pathologic features of lung cancer expressing programmed cell death ligand 1 (PD-L1). Lung Cancer 2016, 98, 69–75. [Google Scholar] [CrossRef]
- Kang, D.Y.; Sp, N.; Jo, E.S.; Rugamba, A.; Hong, D.Y.; Lee, H.G.; Yoo, J.S.; Liu, Q.; Jang, K.J.; Yang, Y.M. The Inhibitory Mechanisms of Tumor PD-L1 Expression by Natural Bioactive Gallic Acid in Non-Small-Cell Lung Cancer (NSCLC) Cells. Cancers 2020, 12, 727. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.Y.; Sp, N.; Lee, J.M.; Jang, K.J. Antitumor Effects of Ursolic Acid through Mediating the Inhibition of STAT3/PD-L1 Signaling in Non-Small Cell Lung Cancer Cells. Biomedicines 2021, 9, 297. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974. [Google Scholar] [CrossRef] [Green Version]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.L.; Spigel, D.; Kelly, K.; Chandler, J.C.; Rajan, A.; Hassan, R.; Wong, D.J.L.; Leach, J.; Edenfield, W.J.; Wang, D. Avelumab (MSB0010718C), an anti-PD-L1 antibody, in advanced NSCLC patients: A phase 1b, open-label expansion trial in patients progressing after platinum-based chemotherapy. J. Clin. Oncol. 2015, 33, 8034. [Google Scholar] [CrossRef]
- Yuan, H.; Ma, Q.; Ye, L.; Piao, G. The Traditional Medicine and Modern Medicine from Natural Products. Molecules 2016, 21, 559. [Google Scholar] [CrossRef] [Green Version]
- Cheung, C.W.; Gibbons, N.; Johnson, D.W.; Nicol, D.L. Silibinin—A promising new treatment for cancer. Anticancer Agents Med. Chem. 2010, 10, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Mateen, S.; Raina, K.; Agarwal, R. Chemopreventive and anti-cancer efficacy of silibinin against growth and progression of lung cancer. Nutr. Cancer 2013, 65 (Suppl. 1), 3–11. [Google Scholar] [CrossRef]
- Mirzaaghaei, S.; Foroughmand, A.M.; Saki, G.; Shafiei, M. Combination of Epigallocatechin-3-gallate and Silibinin: A Novel Approach for Targeting Both Tumor and Endothelial Cells. ACS Omega 2019, 4, 8421–8430. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Ning, Z.; Zeng, J.; Fan, J.; Zhou, J.; Zhang, T.; Zhang, L.; Chen, Y.; Gao, Y.; Wang, B.; et al. Silibinin inhibits beta-catenin/ZEB1 signaling and suppresses bladder cancer metastasis via dual-blocking epithelial-mesenchymal transition and stemness. Cell Signal. 2013, 25, 2625–2633. [Google Scholar] [CrossRef]
- Hou, X.; Du, H.; Quan, X.; Shi, L.; Zhang, Q.; Wu, Y.; Liu, Y.; Xiao, J.; Li, Y.; Lu, L.; et al. Silibinin Inhibits NSCLC Metastasis by Targeting the EGFR/LOX Pathway. Front. Pharm. 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Sp, N.; Kang, D.Y.; Jo, E.S.; Rugamba, A.; Kim, W.S.; Park, Y.M.; Hwang, D.Y.; Yoo, J.S.; Liu, Q.; Jang, K.J.; et al. Tannic Acid Promotes TRAIL-Induced Extrinsic Apoptosis by Regulating Mitochondrial ROS in Human Embryonic Carcinoma Cells. Cells 2020, 9, 282. [Google Scholar] [CrossRef] [Green Version]
- Nipin, S.P.; Kang, D.Y.; Kim, B.J.; Joung, Y.H.; Darvin, P.; Byun, H.J.; Kim, J.G.; Park, J.U.; Yang, Y.M. Methylsulfonylmethane Induces G1 Arrest and Mitochondrial Apoptosis in YD-38 Gingival Cancer Cells. Anticancer Res. 2017, 37, 1637–1646. [Google Scholar] [CrossRef] [Green Version]
- Byun, H.J.; Darvin, P.; Kang, D.Y.; Sp, N.; Joung, Y.H.; Park, J.H.; Kim, S.J.; Yang, Y.M. Silibinin downregulates MMP2 expression via Jak2/STAT3 pathway and inhibits the migration and invasive potential in MDA-MB-231 cells. Oncol. Rep. 2017, 37, 3270–3278. [Google Scholar] [CrossRef] [Green Version]
- Bosch-Barrera, J.; Menendez, J.A. Silibinin and STAT3: A natural way of targeting transcription factors for cancer therapy. Cancer Treat. Rev. 2015, 41, 540–546. [Google Scholar] [CrossRef]
- Sellam, L.S.; Zappasodi, R.; Chettibi, F.; Djennaoui, D.; Yahi-Ait Mesbah, N.; Amir-Tidadini, Z.C.; Touil-Boukoffa, C.; Ouahioune, W.; Merghoub, T.; Bourouba, M. Silibinin down-regulates PD-L1 expression in nasopharyngeal carcinoma by interfering with tumor cell glycolytic metabolism. Arch. Biochem. Biophys. 2020, 690, 108479. [Google Scholar] [CrossRef]
- Guo, S.; Bai, X.; Liu, Y.; Shi, S.; Wang, X.; Zhan, Y.; Kang, X.; Chen, Y.; An, H. Inhibition of TMEM16A by Natural Product Silibinin: Potential Lead Compounds for Treatment of Lung Adenocarcinoma. Front. Pharm. 2021, 12, 643489. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, H.; Wang, A.; Ma, Y.; Gan, Y.; Li, G. Silibinin suppresses epithelial-mesenchymal transition in human non-small cell lung cancer cells by restraining RHBDD1. Cell Mol. Biol. Lett. 2020, 25, 36. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Tumorigenesis as a process of gradual loss of original cell identity and gain of properties of neural precursor/progenitor cells. Cell Biosci. 2017, 7, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Zhang, Y.; Chen, Y.; Li, Q.; Chen, J.; Dong, Y.; Shi, W. Silibinin causes apoptosis and cell cycle arrest in some human pancreatic cancer cells. Int. J. Mol. Sci. 2011, 12, 4861–4871. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, J.; Zhang, P.; Dai, L.; Wu, Z.; Wang, L.; Cao, M.; Jiang, J. Silibinin induces G1 arrest, apoptosis and JNK/SAPK upregulation in SW1990 human pancreatic cancer cells. Oncol. Lett. 2018, 15, 9868–9876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [Green Version]
- van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef]
- Dudley, A.C.; Thomas, D.; Best, J.; Jenkins, A. A VEGF/JAK2/STAT5 axis may partially mediate endothelial cell tolerance to hypoxia. Biochem. J. 2005, 390, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiou, S.-H.; Yu, C.-C.; Huang, C.-Y.; Lin, S.-C.; Liu, C.-J.; Tsai, T.-H.; Chou, S.-H.; Chien, C.-S.; Ku, H.-H.; Lo, J.-F. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin. Cancer Res. 2008, 14, 4085–4095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y. Effects of salinomycin on cancer stem cell in human lung adenocarcinoma A549 cells. Med. Chem. 2011, 7, 106–111. [Google Scholar] [CrossRef]
- Wu, L.; Guo, L.; Liang, Y.; Liu, X.; Jiang, L.; Wang, L. Curcumin suppresses stem-like traits of lung cancer cells via inhibiting the JAK2/STAT3 signaling pathway. Oncol. Rep. 2015, 34, 3311–3317. [Google Scholar] [CrossRef] [Green Version]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar] [PubMed]
- Bole-Feysot, C.; Goffin, V.; Edery, M.; Binart, N.; Kelly, P.A. Prolactin (PRL) and its receptor: Actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr. Rev. 1998, 19, 225–268. [Google Scholar] [CrossRef]
- Brooks, A.J.; Putoczki, T. JAK-STAT Signalling Pathway in Cancer. Cancers 2020, 12, 1971. [Google Scholar] [CrossRef]
- Saxena, N.K.; Sharma, D.; Ding, X.; Lin, S.; Marra, F.; Merlin, D.; Anania, F.A. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007, 67, 2497–2507. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.B.; Yao, H.; Li, C.S.; Liang, L.X.; Zhang, Y.; Chen, Y.X.; Fang, J.Y.; Xu, J. Rise of PD-L1 expression during metastasis of colorectal cancer: Implications for immunotherapy. J. Dig. Dis. 2017, 18, 574–581. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rugamba, A.; Kang, D.Y.; Sp, N.; Jo, E.S.; Lee, J.-M.; Bae, S.W.; Jang, K.-J. Silibinin Regulates Tumor Progression and Tumorsphere Formation by Suppressing PD-L1 Expression in Non-Small Cell Lung Cancer (NSCLC) Cells. Cells 2021, 10, 1632. https://doi.org/10.3390/cells10071632
Rugamba A, Kang DY, Sp N, Jo ES, Lee J-M, Bae SW, Jang K-J. Silibinin Regulates Tumor Progression and Tumorsphere Formation by Suppressing PD-L1 Expression in Non-Small Cell Lung Cancer (NSCLC) Cells. Cells. 2021; 10(7):1632. https://doi.org/10.3390/cells10071632
Chicago/Turabian StyleRugamba, Alexis, Dong Young Kang, Nipin Sp, Eun Seong Jo, Jin-Moo Lee, Se Won Bae, and Kyoung-Jin Jang. 2021. "Silibinin Regulates Tumor Progression and Tumorsphere Formation by Suppressing PD-L1 Expression in Non-Small Cell Lung Cancer (NSCLC) Cells" Cells 10, no. 7: 1632. https://doi.org/10.3390/cells10071632
APA StyleRugamba, A., Kang, D. Y., Sp, N., Jo, E. S., Lee, J.-M., Bae, S. W., & Jang, K.-J. (2021). Silibinin Regulates Tumor Progression and Tumorsphere Formation by Suppressing PD-L1 Expression in Non-Small Cell Lung Cancer (NSCLC) Cells. Cells, 10(7), 1632. https://doi.org/10.3390/cells10071632