
Cockayne Syndrome-Associated CSA and CSB Mutations Impair Ribosome Biogenesis, Ribosomal Protein Stability, and Global Protein Folding

 ,
,
Abstract
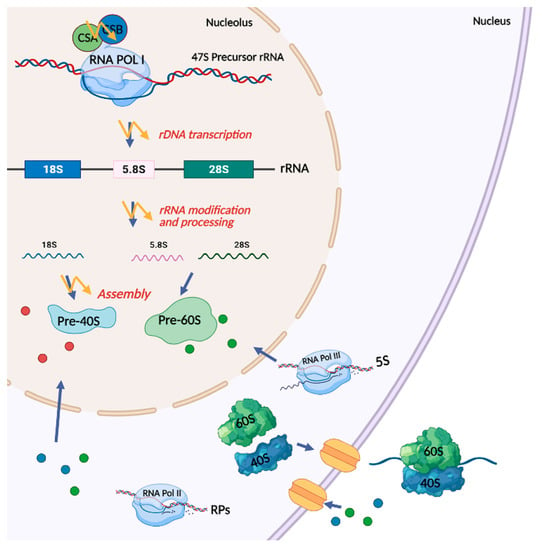
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Cell Culture
2.2. Antibodies
2.3. RNA Extraction and qRT-PCR
2.3.1. RNA Extraction
2.3.2. qRT-PCR
2.4. Ribosome Isolation
2.5. Protein Folding and Stability
2.6. Heat Sensitivity Assay
2.7. Cell Growth
2.8. Western Blot
2.9. Northern Blot
2.10. Mass Spectrometry
2.11. Statistical Analysis
3. Results
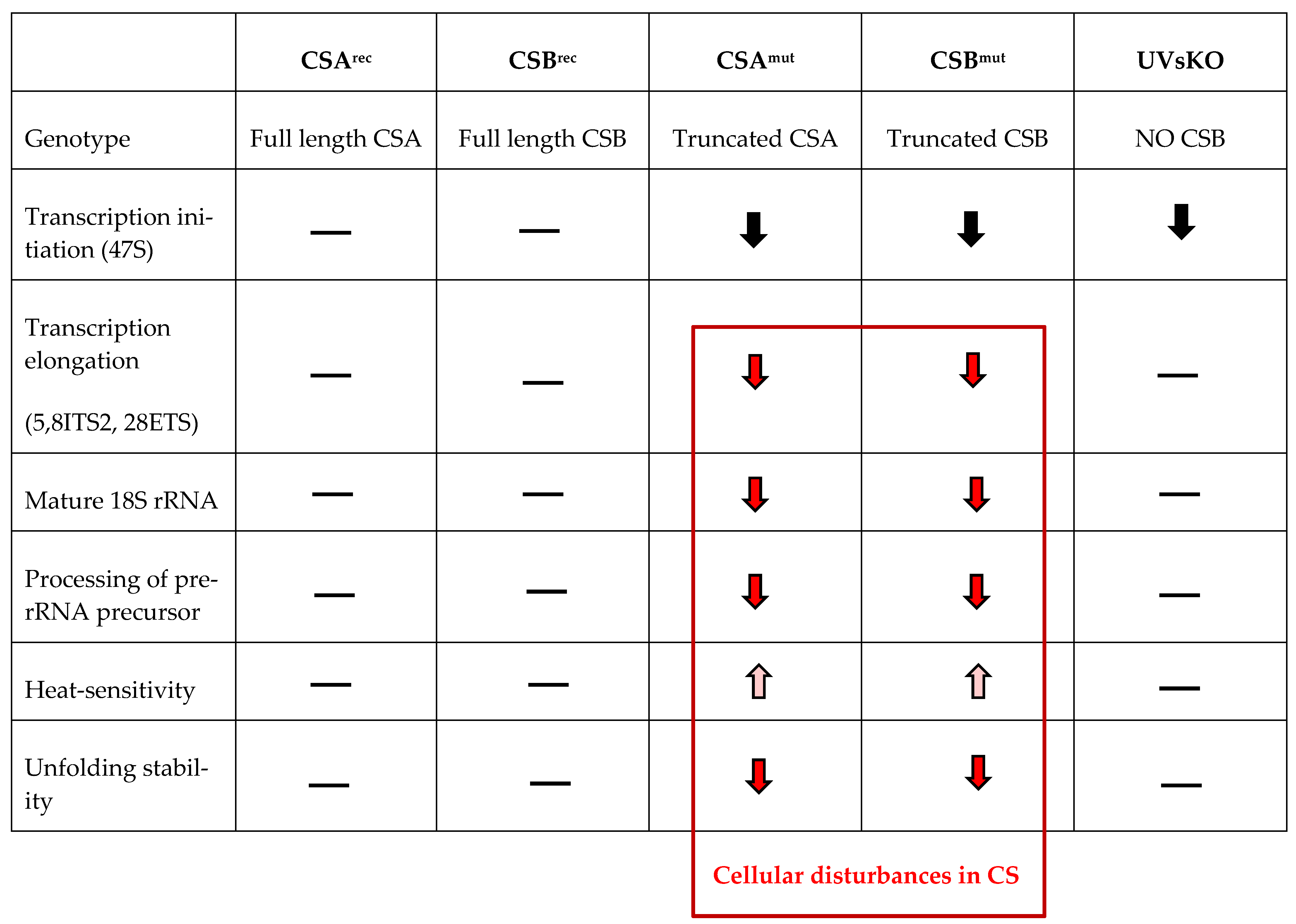
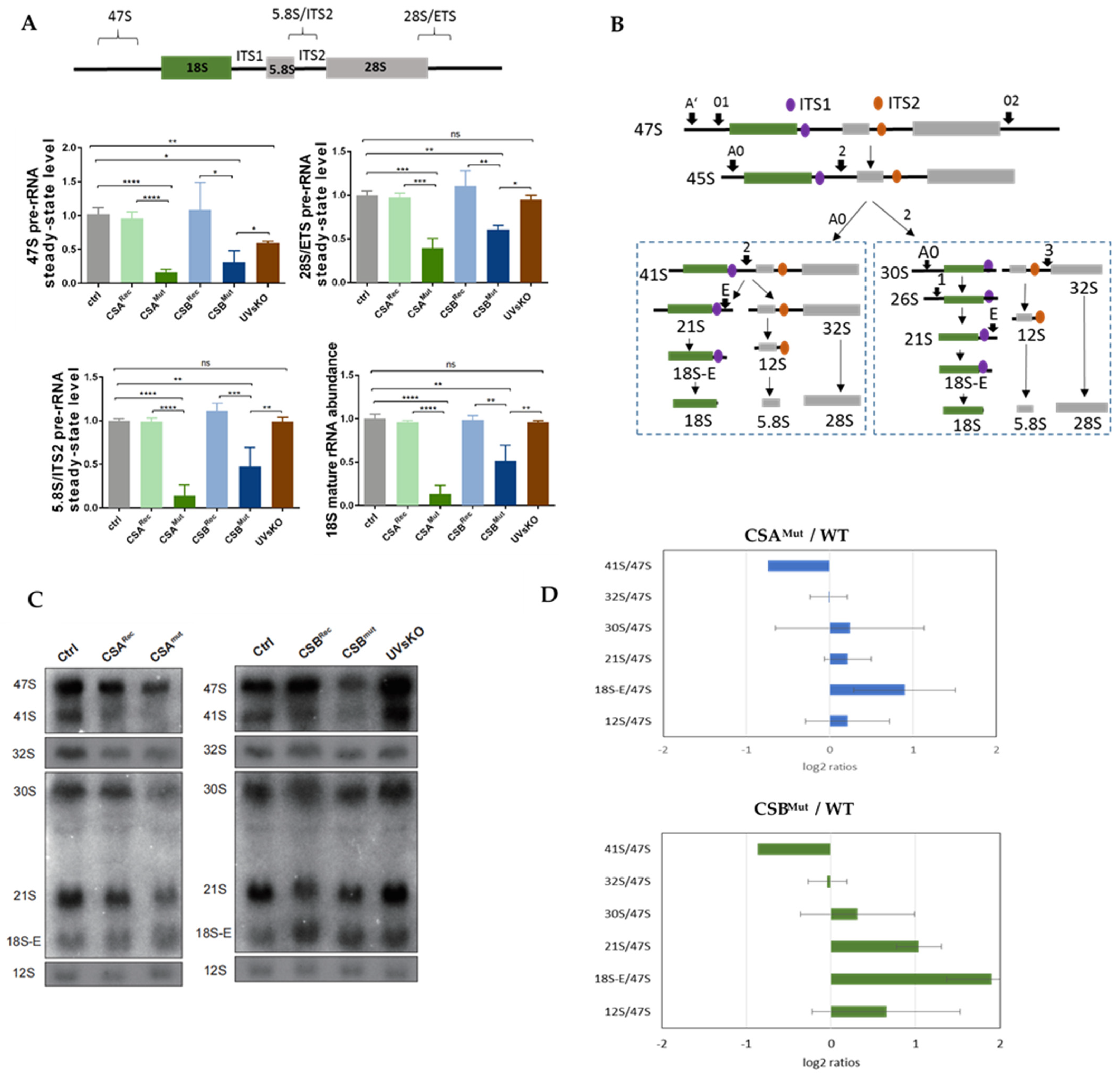
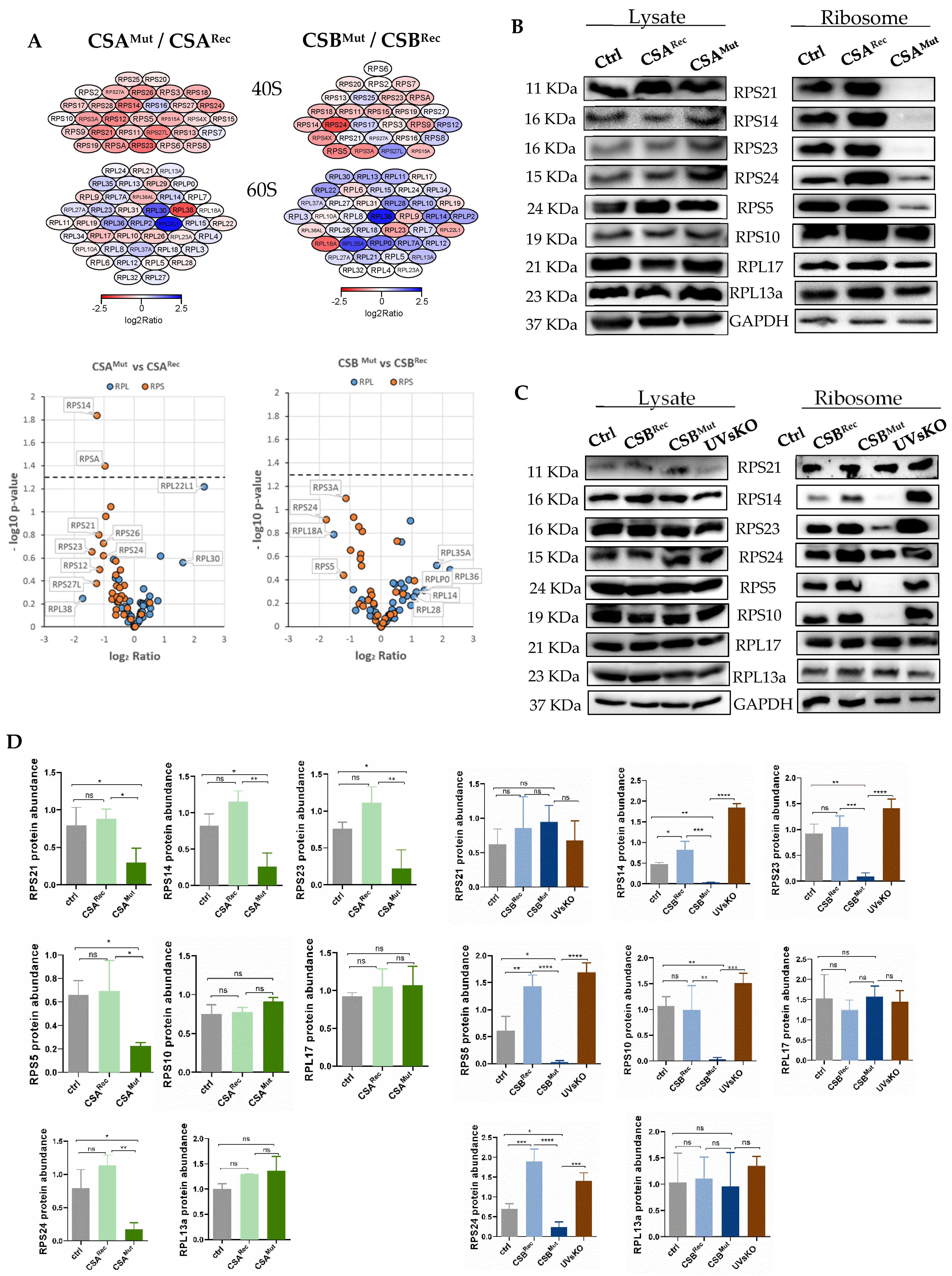
3.1. RNA Polymerase I Transcription and Processing Disturbances in CS Cells
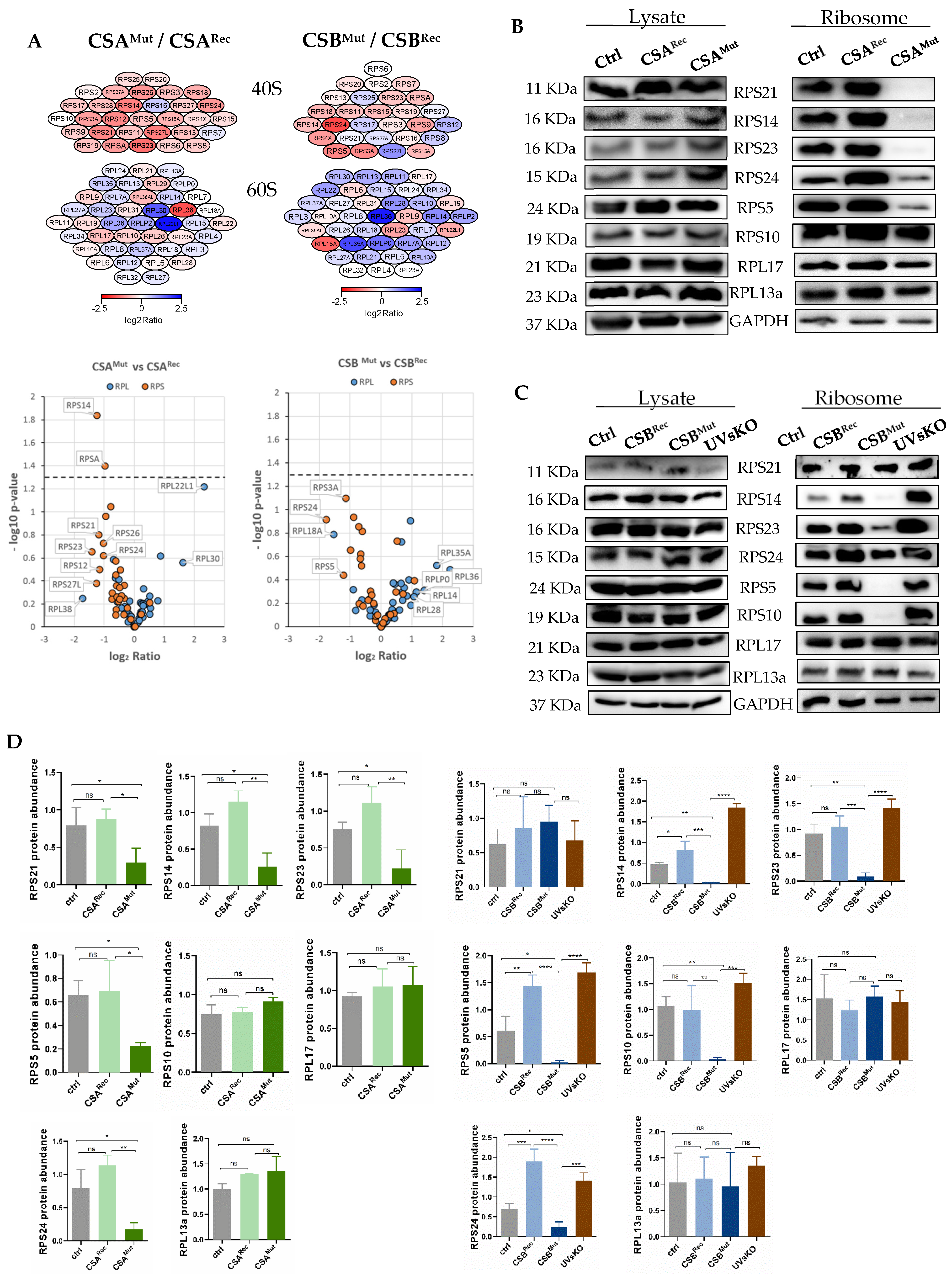
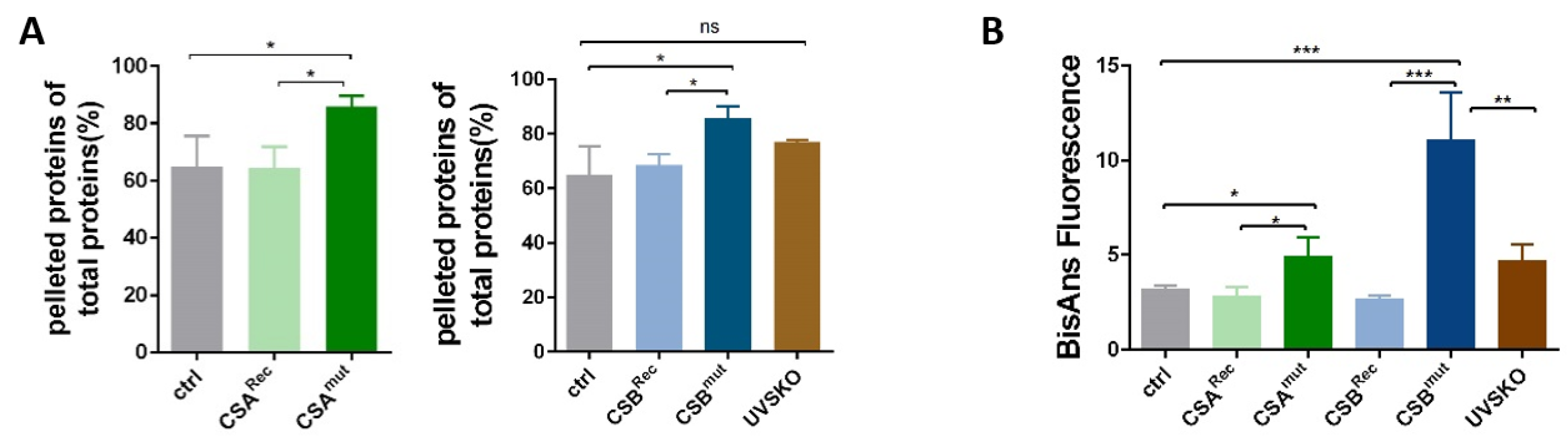
3.2. Isolated Ribosomes from CS Cells Display Stability Defects
3.3. Reduced Heat and Unfolding Stability in CS cells
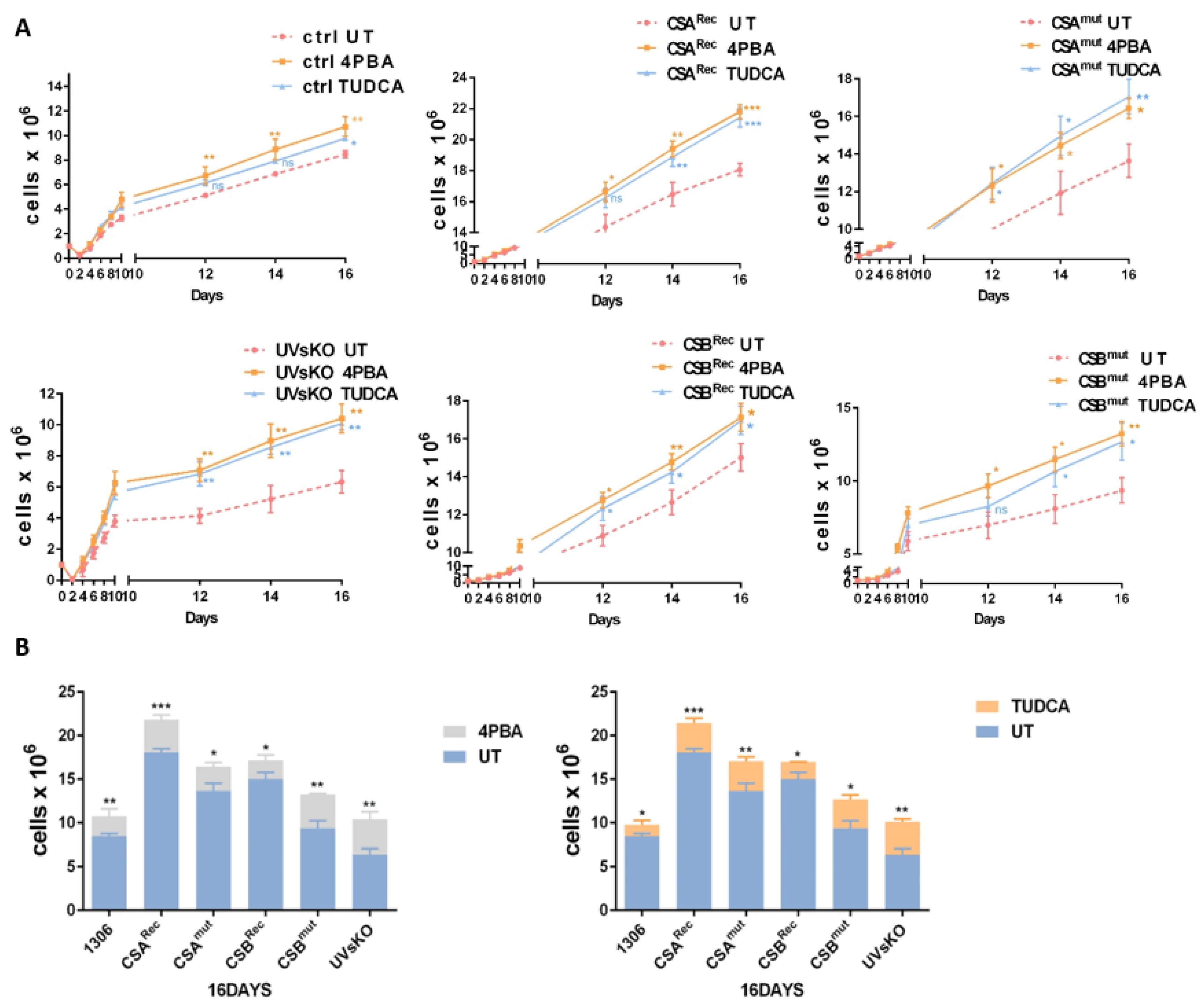
3.4. Pharmaceutical Chaperones Enhance Cellular Proliferation
4. Discussion
Simple Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spitz, M.A.; Severac, F.; Obringer, C.; Baer, S.; Le May, N.; Calmels, N.; Laugel, V. Diagnostic and severity scores for Cockayne syndrome. Orphanet J. Rare Dis. 2021, 16, 63. [Google Scholar] [CrossRef] [PubMed]
- Laugel, V. Cockayne syndrome: The expanding clinical and mutational spectrum. Mech. Ageing Dev. 2013, 134, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, G.S.; Nascimento, L.L.S.; Neves, M.R.C.; Leandro, G.D.S.; Garcia, C.C.M.; Menck, C.F.M. Transcription blockage by DNA damage in nucleotide excision repair-related neurological dysfunctions. Semin. Cell Dev. Biol. 2021, 114, 20–35. [Google Scholar] [CrossRef]
- Horibata, K.; Iwamoto, Y.; Kuraoka, I.; Jaspers, N.G.; Kurimasa, A.; Oshimura, M.; Ichihashi, M.; Tanaka, K. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 15410–15415. [Google Scholar] [CrossRef] [Green Version]
- Brooks, P.J. Blinded by the UV light: How the focus on transcription-coupled NER has distracted from understanding the mechanisms of Cockayne syndrome neurologic disease. DNA Repair (Amst) 2013, 12, 656–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iben, S.; Tschochner, H.; Bier, M.; Hoogstraten, D.; Hozak, P.; Egly, J.M.; Grummt, I. TFIIH plays an essential role in RNA polymerase I transcription. Cell 2002, 109, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Bradsher, J.; Auriol, J.; Proietti de Santis, L.; Iben, S.; Vonesch, J.L.; Grummt, I.; Egly, J.M. CSB is a component of RNA pol I transcription. Mol. Cell 2002, 10, 819–829. [Google Scholar] [CrossRef]
- Koch, S.; Garcia Gonzalez, O.; Assfalg, R.; Schelling, A.; Schafer, P.; Scharffetter-Kochanek, K.; Iben, S. Cockayne syndrome protein A is a transcription factor of RNA polymerase I and stimulates ribosomal biogenesis and growth. Cell Cycle 2014, 13, 2029–2037. [Google Scholar] [CrossRef] [Green Version]
- Kressler, D.; Hurt, E.; Bassler, J. A Puzzle of Life: Crafting Ribosomal Subunits. Trends Biochem. Sci. 2017, 42, 640–654. [Google Scholar] [CrossRef] [Green Version]
- Alupei, M.C.; Maity, P.; Esser, P.R.; Krikki, I.; Tuorto, F.; Parlato, R.; Penzo, M.; Schelling, A.; Laugel, V.; Montanaro, L.; et al. Loss of Proteostasis Is a Pathomechanism in Cockayne Syndrome. Cell Rep. 2018, 23, 1612–1619. [Google Scholar] [CrossRef]
- Troelstra, C.; van Gool, A.; de Wit, J.; Vermeulen, W.; Bootsma, D.; Hoeijmakers, J.H. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell 1992, 71, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Henning, K.A.; Li, L.; Iyer, N.; McDaniel, L.D.; Reagan, M.S.; Legerski, R.; Schultz, R.A.; Stefanini, M.; Lehmann, A.R.; Mayne, L.V.; et al. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell 1995, 82, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Penzo, M.; Carnicelli, D.; Montanaro, L.; Brigotti, M. A reconstituted cell-free assay for the evaluation of the intrinsic activity of purified human ribosomes. Nat. Protoc. 2016, 11, 1309–1325. [Google Scholar] [CrossRef]
- Treaster, S.B.; Ridgway, I.D.; Richardson, C.A.; Gaspar, M.B.; Chaudhuri, A.R.; Austad, S.N. Superior proteome stability in the longest lived animal. Age (Dordr) 2014, 36, 9597. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Liu, H.; Sun, Q.; Yuan, G.; Zhang, L.; Chen, Z. Establishment and characterization of a tamoxifen-mediated reversible immortalized mouse dental papilla cell line. In Vitr. Cell Dev. Biol. Anim. 2013, 49, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Pestov, D.G. Quantitative Northern Blot Analysis of Mammalian rRNA Processing. Methods Mol. Biol. 2016, 1455, 147–157. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Ridley, A.J.; Colley, J.; Wynford-Thomas, D.; Jones, C.J. Characterisation of novel mutations in Cockayne syndrome type A and xeroderma pigmentosum group C subjects. J. Hum. Genet. 2005, 50, 151–154. [Google Scholar] [CrossRef] [Green Version]
- Hung, S.S.; Lesmana, A.; Peck, A.; Lee, R.; Tchoubrieva, E.; Hannan, K.M.; Lin, J.; Sheppard, K.E.; Jastrzebski, K.; Quinn, L.M.; et al. Cell cycle and growth stimuli regulate different steps of RNA polymerase I transcription. Gene 2017, 612, 36–48. [Google Scholar] [CrossRef] [Green Version]
- Lebedev, A.; Scharffetter-Kochanek, K.; Iben, S. Truncated Cockayne syndrome B protein represses elongation by RNA polymerase I. J. Mol. Biol. 2008, 382, 266–274. [Google Scholar] [CrossRef]
- Okur, M.N.; Lee, J.-H.; Osmani, W.; Kimura, R.; Demarest, T.G.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome group A and B proteins function in rRNA transcription through nucleolin regulation. Nucleic Acids Res. 2020, 48, 2473–2485. [Google Scholar] [CrossRef]
- Scull, C.E.; Schneider, D.A. Coordinated Control of rRNA Processing by RNA Polymerase I. Trends Genet. 2019, 35, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Nonnekens, J.; Perez-Fernandez, J.; Theil, A.F.; Gadal, O.; Bonnart, C.; Giglia-Mari, G. Mutations in TFIIH causing trichothiodystrophy are responsible for defects in ribosomal RNA production and processing. Hum. Mol. Genet. 2013, 22, 2881–2893. [Google Scholar] [CrossRef] [Green Version]
- Mullineux, S.T.; Lafontaine, D.L. Mapping the cleavage sites on mammalian pre-rRNAs: Where do we stand? Biochimie 2012, 94, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Baßler, J.; Hurt, E. Eukaryotic Ribosome Assembly. Annu Rev. Biochem. 2019, 88, 281–306. [Google Scholar] [CrossRef] [PubMed]
- Hoogstraten, D.; Nigg, A.L.; Heath, H.; Mullenders, L.H.; van Driel, R.; Hoeijmakers, J.H.; Vermeulen, W.; Houtsmuller, A.B. Rapid switching of TFIIH between RNA polymerase I and II transcription and DNA repair in vivo. Mol. Cell 2002, 10, 1163–1174. [Google Scholar] [CrossRef]
- Assfalg, R.; Lebedev, A.; Gonzalez, O.G.; Schelling, A.; Koch, S.; Iben, S. TFIIH is an elongation factor of RNA polymerase I. Nucleic Acids Res. 2012, 40, 650–659. [Google Scholar] [CrossRef]
- Badertscher, L.; Wild, T.; Montellese, C.; Alexander, L.T.; Bammert, L.; Sarazova, M.; Stebler, M.; Csucs, G.; Mayer, T.U.; Zamboni, N.; et al. Genome-wide RNAi Screening Identifies Protein Modules Required for 40S Subunit Synthesis in Human Cells. Cell Rep. 2015, 13, 2879–2891. [Google Scholar] [CrossRef] [Green Version]
- Bohnsack, K.E.; Bohnsack, M.T. Uncovering the assembly pathway of human ribosomes and its emerging links to disease. Embo J. 2019, 38, e100278. [Google Scholar] [CrossRef]
- Venturi, G.; Montanaro, L. How Altered Ribosome Production Can Cause or Contribute to Human Disease: The Spectrum of Ribosomopathies. Cells 2020, 9, 2300. [Google Scholar] [CrossRef]
- Lezzerini, M.; Penzo, M.; O’Donohue, M.F.; Marques Dos Santos Vieira, C.; Saby, M.; Elfrink, H.L.; Diets, I.J.; Hesse, A.M.; Couté, Y.; Gastou, M.; et al. Ribosomal protein gene RPL9 variants can differentially impair ribosome function and cellular metabolism. Nucleic Acids Res. 2020, 48, 770–787. [Google Scholar] [CrossRef] [Green Version]
- Paolini, N.A.; Attwood, M.; Sondalle, S.B.; Vieira, C.M.D.S.; van Adrichem, A.M.; di Summa, F.M.; O’Donohue, M.F.; Gleizes, P.E.; Rachuri, S.; Briggs, J.W.; et al. A Ribosomopathy Reveals Decoding Defective Ribosomes Driving Human Dysmorphism. Am. J. Hum. Genet. 2017, 100, 506–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetman, M.; Slomnicki, L.P. Ribosomal biogenesis as an emerging target of neurodevelopmental pathologies. J. Neurochem. 2019, 148, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Maslov, A.Y.; Ganapathi, S.; Westerhof, M.; Quispe-Tintaya, W.; White, R.R.; Van Houten, B.; Reiling, E.; Dollé, M.E.; van Steeg, H.; Hasty, P.; et al. DNA damage in normally and prematurely aged mice. Aging Cell 2013, 12, 467–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheibye-Knudsen, M.; Tseng, A.; Borch Jensen, M.; Scheibye-Alsing, K.; Fang, E.F.; Iyama, T.; Bharti, S.K.; Marosi, K.; Froetscher, L.; Kassahun, H.; et al. Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc. Natl. Acad. Sci. USA 2016, 113, 12502–12507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamenisch, Y.; Fousteri, M.; Knoch, J.; Von Thaler, A.K.; Fehrenbacher, B.; Kato, H.; Becker, T.; Dolle, M.E.; Kuiper, R.; Majora, M.; et al. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J. Exp. Med. 2010, 207, 379–390. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiang, M.; Khalid, F.; Phan, T.; Ludwig, C.; Scharffetter-Kochanek, K.; Iben, S. Cockayne Syndrome-Associated CSA and CSB Mutations Impair Ribosome Biogenesis, Ribosomal Protein Stability, and Global Protein Folding. Cells 2021, 10, 1616. https://doi.org/10.3390/cells10071616
Qiang M, Khalid F, Phan T, Ludwig C, Scharffetter-Kochanek K, Iben S. Cockayne Syndrome-Associated CSA and CSB Mutations Impair Ribosome Biogenesis, Ribosomal Protein Stability, and Global Protein Folding. Cells. 2021; 10(7):1616. https://doi.org/10.3390/cells10071616
Chicago/Turabian StyleQiang, Mingyue, Fatima Khalid, Tamara Phan, Christina Ludwig, Karin Scharffetter-Kochanek, and Sebastian Iben. 2021. "Cockayne Syndrome-Associated CSA and CSB Mutations Impair Ribosome Biogenesis, Ribosomal Protein Stability, and Global Protein Folding" Cells 10, no. 7: 1616. https://doi.org/10.3390/cells10071616
APA StyleQiang, M., Khalid, F., Phan, T., Ludwig, C., Scharffetter-Kochanek, K., & Iben, S. (2021). Cockayne Syndrome-Associated CSA and CSB Mutations Impair Ribosome Biogenesis, Ribosomal Protein Stability, and Global Protein Folding. Cells, 10(7), 1616. https://doi.org/10.3390/cells10071616