
Skeletal Muscle Metabolism: Origin or Prognostic Factor for Amyotrophic Lateral Sclerosis (ALS) Development?

 ,
,  and
and
Abstract
1. Introduction
1.1. Amyotrophic Lateral Sclerosis
1.2. Neuromuscular Junction
2. Mitochondrial Failure and Oxidative Stress in ALS
2.1. Muscle Mitochondria and Respiratory Complexes
2.2. ROS and Oxidative Stress
3. Metabolic Alterations in Amyotrophic Lateral Sclerosis
3.1. Discovery of Hypermetabolism
3.2. Impairment of Skeletal Muscle Metabolism by Physical Activity
3.3. The Metabolic Switch of Muscle Fiber Types in ALS
3.4. Main Actors of the Randle Cycle
4. Pharmacological Strategies for Targeting Energetic Imbalance in ALS
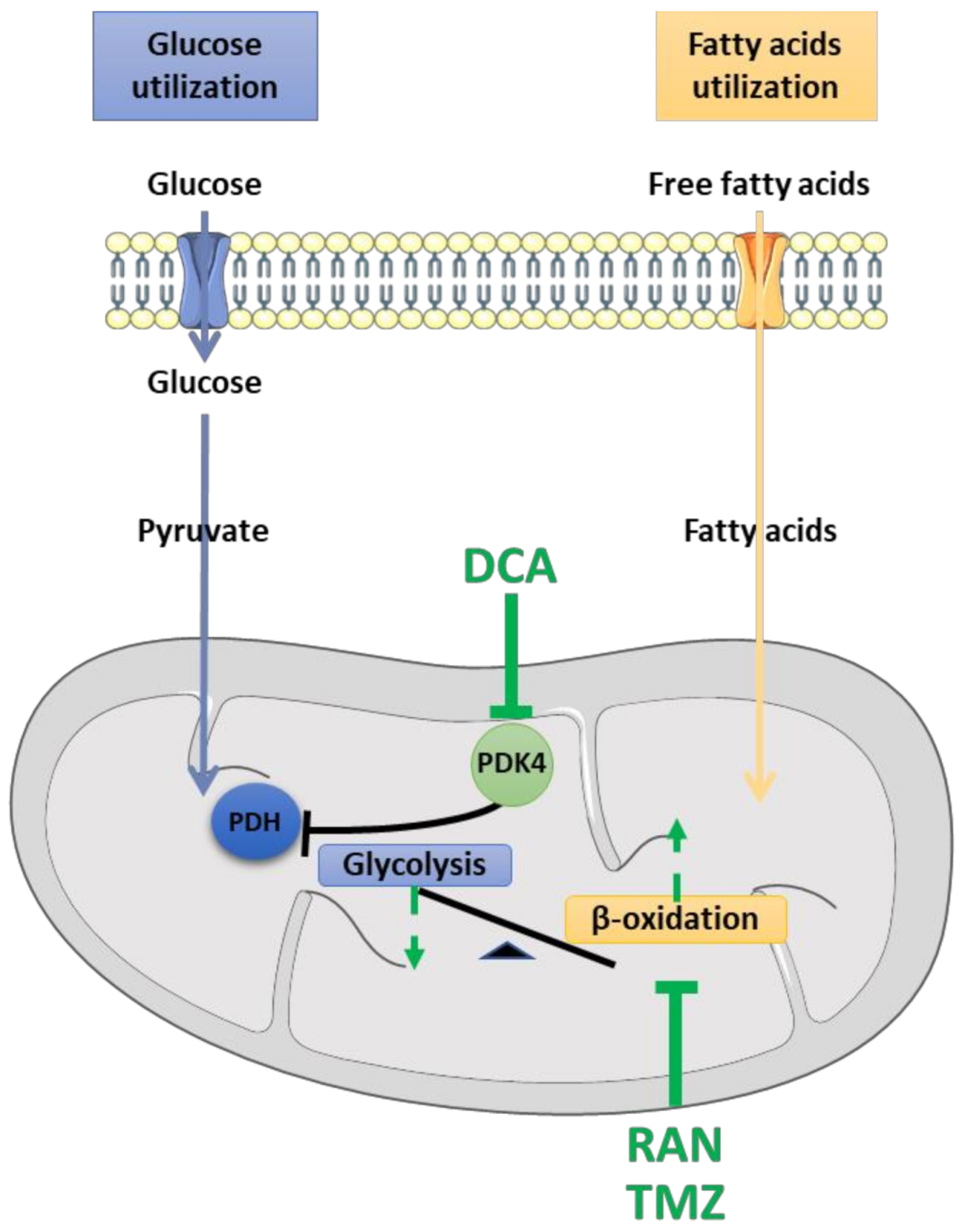
4.1. Dichloroacetate (DCA)
4.2. Ranolazine (RAN)
4.3. Trimetazidine (TMZ)
5. Open Questions and Future Directions
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 13. [Google Scholar] [CrossRef] [PubMed]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic Lateral Sclerosis: A Clinical Review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Couratier, P.; Corcia, P.; Lautrette, G.; Nicol, M.; Preux, P.-M.; Marin, B. Epidemiology of Amyotrophic Lateral Sclerosis: A Review of Literature. Rev. Neurol. 2016, 172, 37–45. [Google Scholar] [CrossRef]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of Play in Amyotrophic Lateral Sclerosis Genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Millecamps, S.; Salachas, F.; Cazeneuve, C.; Gordon, P.; Bricka, B.; Camuzat, A.; Guillot-Noel, L.; Russaouen, O.; Bruneteau, G.; Pradat, P.-F.; et al. SOD1, ANG, VAPB, TARDBP, and FUS Mutations in Familial Amyotrophic Lateral Sclerosis: Genotype-Phenotype Correlations. J. Med. Genet. 2010, 47, 554–560. [Google Scholar] [CrossRef]
- Millecamps, S.; Boillée, S.; Le Ber, I.; Seilhean, D.; Teyssou, E.; Giraudeau, M.; Moigneu, C.; Vandenberghe, N.; Danel-Brunaud, V.; Corcia, P.; et al. Phenotype Difference between ALS Patients with Expanded Repeats in C9ORF72 and Patients with Mutations in Other ALS-Related Genes. J. Med. Genet. 2012, 49, 258–263. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J.; Bosco, D.A.; LeClerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Abe, K. Safety and Efficacy of Edaravone in Well Defined Patients with Amyotrophic Lateral Sclerosis: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol. 2017, 16, 8. [Google Scholar] [CrossRef]
- Dorst, J.; Ludolph, A.C.; Huebers, A. Disease-Modifying and Symptomatic Treatment of Amyotrophic Lateral Sclerosis. Adv. Neurol. Disord. 2018, 11, 1756285617734734. [Google Scholar] [CrossRef]
- Barber, S.C.; Shaw, P.J. Oxidative Stress in ALS: Key Role in Motor Neuron Injury and Therapeutic Target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef]
- Fischer-Hayes, L.R.; Brotherton, T.; Glass, J.D. Axonal Degeneration in the Peripheral Nervous System: Implications for the Pathogenesis of Amyotrophic Lateral Sclerosis. Exp. Neurol. 2013, 246, 6–13. [Google Scholar] [CrossRef]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein Aggregation in Amyotrophic Lateral Sclerosis. Acta. Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef]
- Bogaert, E.; d’Ydewalle, C.; Van Den Bosch, L. Amyotrophic Lateral Sclerosis and Excitotoxicity: From Pathological Mechanism to Therapeutic Target. CNSNDDT 2010, 9, 297–304. [Google Scholar] [CrossRef]
- Cozzolino, M.; Carrì, M.T. Mitochondrial Dysfunction in ALS. Prog. Neurobiol. 2012, 97, 54–66. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Eisen, A.; Kim, S.; Pant, B. Amyotrophic Lateral Sclerosis (ALS): A Phylogenetic Disease of the Corticomotoneuron? Muscle Nerve 1992, 15, 219–224. [Google Scholar] [CrossRef]
- Eisen, A.; Braak, H.; Del Tredici, K.; Lemon, R.; Ludolph, A.C.; Kiernan, M.C. Cortical Influences Drive Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Stuart-Lopez, G.; Burg, T.; Scekic-Zahirovic, J.; Rouaux, C. Cortical Circuit Dysfunction as a Potential Driver of Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14, 363. [Google Scholar] [CrossRef] [PubMed]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “Dying-Back” Phenomenon of Motor Neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Moloney, E.B.; de Winter, F.; Verhaagen, J. ALS as a Distal Axonopathy: Molecular Mechanisms Affecting Neuromuscular Junction Stability in the Presymptomatic Stages of the Disease. Front. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic Lateral Sclerosis Is a Distal Axonopathy: Evidence in Mice and Man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- Gonzalez de Aguilar, J.-L.; Niederhauser-Wiederkehr, C.; Halter, B.; De Tapia, M.; Di Scala, F.; Demougin, P.; Dupuis, L.; Primig, M.; Meininger, V.; Loeffler, J.-P. Gene Profiling of Skeletal Muscle in an Amyotrophic Lateral Sclerosis Mouse Model. Physiol. Genom. 2008, 32, 207–218. [Google Scholar] [CrossRef]
- Tsujihata, M.; Hazama, R.; Yoshimura, T.; Satoh, A.; Mori, M.; Nagataki, S. The Motor End-Plate Fine Structure and Ultrastructural Localization of Acetylcholine Receptors in Amyotrophic Lateral Sclerosis. Muscle Nerve. 1984, 7, 243–249. [Google Scholar] [CrossRef]
- Siklós, L.; Engelhardt, J.; Harati, Y.; Smith, R.G.; Joó, F.; Appel, S.H. Ultrastructural Evidence for Altered Calcium in Motor Nerve Terminals in Amyotrophc Lateral Sclerosis: Calcium in ALS Motor Nerve Terminals. Ann. Neurol. 1996, 39, 203–216. [Google Scholar] [CrossRef]
- Aggarwal, A. Detection of Preclinical Motor Neurone Loss in SOD1 Mutation Carriers Using Motor Unit Number Estimation. J. Neurol. Neurosurg. Psychiatry 2002, 73, 199–201. [Google Scholar] [CrossRef]
- Frey, D.; Schneider, C.; Xu, L.; Borg, J.; Spooren, W.; Caroni, P. Early and Selective Loss of Neuromuscular Synapse Subtypes with Low Sprouting Competence in Motoneuron Diseases. J. Neurosci. 2000, 20, 2534–2542. [Google Scholar] [CrossRef]
- Pun, S.; Santos, A.F.; Saxena, S.; Xu, L.; Caroni, P. Selective Vulnerability and Pruning of Phasic Motoneuron Axons in Motoneuron Disease Alleviated by CNTF. Nat. Neurosci. 2006, 9, 408–419. [Google Scholar] [CrossRef]
- Vinsant, S.; Mansfield, C.; Jimenez-Moreno, R.; Moore, V.D.G.; Yoshikawa, M.; Hampton, T.G.; Prevette, D.; Caress, J.; Oppenheim, R.W.; Milligan, C. Characterization of Early Pathogenesis in the SOD1 G93A Mouse Model of ALS: Part II, Results and Discussion. Brain Behav. 2013, 3, 431–457. [Google Scholar] [CrossRef]
- Ngo, S.T.; Baumann, F.; Ridall, P.G.; Pettitt, A.N.; Henderson, R.D.; Bellingham, M.C.; McCombe, P.A. The Relationship between Bayesian Motor Unit Number Estimation and Histological Measurements of Motor Neurons in Wild-Type and SOD1G93A Mice. Clin. Neurophysiol. 2012, 123, 2080–2091. [Google Scholar] [CrossRef]
- Rocha, M.C.; Pousinha, P.A.; Correia, A.M.; Sebastião, A.M.; Ribeiro, J.A. Early Changes of Neuromuscular Transmission in the SOD1(G93A) Mice Model of ALS Start Long before Motor Symptoms Onset. PLoS ONE 2013, 8, e73846. [Google Scholar] [CrossRef]
- Gould, T.W.; Buss, R.R.; Vinsant, S.; Prevette, D.; Sun, W.; Knudson, C.M.; Milligan, C.E.; Oppenheim, R.W. Complete Dissociation of Motor Neuron Death from Motor Dysfunction by Bax Deletion in a Mouse Model of ALS. J. Neurosci. 2006, 26, 8774–8786. [Google Scholar] [CrossRef]
- Rouaux, C.; Panteleeva, I.; Rene, F.; Gonzalez de Aguilar, J.-L.; Echaniz-Laguna, A.; Dupuis, L.; Menger, Y.; Boutillier, A.-L.; Loeffler, J.-P. Sodium Valproate Exerts Neuroprotective Effects In Vivo through CREB-Binding Protein-Dependent Mechanisms But Does Not Improve Survival in an Amyotrophic Lateral Sclerosis Mouse Model. J. Neurosci. 2007, 27, 5535–5545. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Lepore, E.; Martini, M.; Barberi, L.; Nunn, A.; Scicchitano, B.M.; Musarò, A. Metabolic Changes Associated With Muscle Expression of SOD1G93A. Front. Physiol. 2018, 9, 831. [Google Scholar] [CrossRef]
- Wong, M.; Martin, L.J. Skeletal Muscle-Restricted Expression of Human SOD1 Causes Motor Neuron Degeneration in Transgenic Mice. Hum. Mol. Genet. 2010, 19, 2284–2302. [Google Scholar] [CrossRef]
- Martin, L.J.; Wong, M. Skeletal Muscle-Restricted Expression of Human SOD1 in Transgenic Mice Causes a Fatal ALS-Like Syndrome. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef]
- Pradat, P.-F.; Dubourg, O.; de Tapia, M.; di Scala, F.; Dupuis, L.; Lenglet, T.; Bruneteau, G.; Salachas, F.; Lacomblez, L.; Corvol, J.-C.; et al. Muscle Gene Expression Is a Marker of Amyotrophic Lateral Sclerosis Severity. Neurodegener. Dis. 2012, 9, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Pradat, P.-F.; Bruneteau, G.; Gonzalez de Aguilar, J.-L.; Dupuis, L.; Jokic, N.; Salachas, F.; Le Forestier, N.; Echaniz-Laguna, A.; Dubourg, O.; Hauw, J.-J.; et al. Muscle Nogo-a Expression Is a Prognostic Marker in Lower Motor Neuron Syndromes. Ann. Neurol. 2007, 62, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Jokic, N.; Gonzalez de Aguilar, J.-L.; Pradat, P.-F.; Dupuis, L.; Echaniz-Laguna, A.; Muller, A.; Dubourg, O.; Seilhean, D.; Hauw, J.-J.; Loeffler, J.-P.; et al. Nogo Expression in Muscle Correlates with Amyotrophic Lateral Sclerosis Severity. Ann. Neurol. 2005, 57, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Gonzalez de Aguilar, J.-L.; di Scala, F.; Rene, F.; de Tapia, M.; Pradat, P.-F.; Lacomblez, L.; Seihlan, D.; Prinjha, R.; Walsh, F.S.; et al. Nogo Provides a Molecular Marker for Diagnosis of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2002, 10, 358–365. [Google Scholar] [CrossRef]
- Jokic, N.; Gonzalez de Aguilar, J.; Dimou, L.; Lin, S.; Fergani, A.; Ruegg, M.A.; Schwab, M.E.; Dupuis, L.; Loeffler, J. The Neurite Outgrowth Inhibitor Nogo-A Promotes Denervation in an Amyotrophic Lateral Sclerosis Model. Embo. Rep. 2006, 7, 1162–1167. [Google Scholar] [CrossRef]
- Bowling, A.C.; Schulz, J.B.; Brown, R.H.; Beal, M.F. Superoxide Dismutase Activity, Oxidative Damage, and Mitochondrial Energy Metabolism in Familial and Sporadic Amyotrophic Lateral Sclerosis. J. Neurochem. 1993, 61, 2322–2325. [Google Scholar] [CrossRef]
- Dupuis, L.; Gonzalez de Aguilar, J.-L.; Oudart, H.; de Tapia, M.; Barbeito, L.; Loeffler, J.-P. Mitochondria in Amyotrophic Lateral Sclerosis: A Trigger and a Target. Neurodegener. Dis. 2004, 1, 245–254. [Google Scholar] [CrossRef]
- Sasaki, S.; Horie, Y.; Iwata, M. Mitochondrial Alterations in Dorsal Root Ganglion Cells in Sporadic Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2007, 114, 633–639. [Google Scholar] [CrossRef]
- Vandoorne, T.; De Bock, K.; Van Den Bosch, L. Energy Metabolism in ALS: An Underappreciated Opportunity? Acta. Neuropathol. 2018, 135, 489–509. [Google Scholar] [CrossRef]
- Carrì, M.T.; D’Ambrosi, N.; Cozzolino, M. Pathways to Mitochondrial Dysfunction in ALS Pathogenesis. Biochem. Biophys. Res. Commun. 2017, 483, 1187–1193. [Google Scholar] [CrossRef]
- Vielhaber, S.; Winkler, K.; Kirches, E.; Kunz, D.; Büchner, M.; Feistner, H.; Elger, C.E.; Ludolph, A.C.; Riepe, M.W.; Kunz, W.S. Visualization of Defective Mitochondrial Function in Skeletal Muscle Fibers of Patients with Sporadic Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 1999, 169, 133–139. [Google Scholar] [CrossRef]
- Chung, M.J.; Suh, Y.-L. Ultrastructural Changes of Mitochondria in the Skeletal Muscle of Patients with Amyotrophic Lateral Sclerosis. Ultrastruct. Pathol. 2002, 26, 3–7. [Google Scholar] [CrossRef]
- Menzies, F.M. Mitochondrial Dysfunction in a Cell Culture Model of Familial Amyotrophic Lateral Sclerosis. Brain 2002, 125, 1522–1533. [Google Scholar] [CrossRef]
- Crugnola, V.; Lamperti, C.; Lucchini, V.; Ronchi, D.; Peverelli, L.; Prelle, A.; Sciacco, M.; Bordoni, A.; Fassone, E.; Fortunato, F.; et al. Mitochondrial Respiratory Chain Dysfunction in Muscle from Patients with Amyotrophic Lateral Sclerosis. Arch. Neurol. 2010, 67, 6. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Zoll, J.; Ponsot, E.; N’Guessan, B.; Tranchant, C.; Loeffler, J.-P.; Lampert, E. Muscular Mitochondrial Function in Amyotrophic Lateral Sclerosis Is Progressively Altered as the Disease Develops: A Temporal Study in Man. Exp. Neurol. 2006, 198, 25–30. [Google Scholar] [CrossRef]
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective Mitochondrial Dynamics Is an Early Event in Skeletal Muscle of an Amyotrophic Lateral Sclerosis Mouse Model. PLoS ONE 2013, 8, e82112. [Google Scholar] [CrossRef]
- Zhou, J.; Yi, J.; Fu, R.; Liu, E.; Siddique, T.; Ríos, E.; Deng, H.-X. Hyperactive Intracellular Calcium Signaling Associated with Localized Mitochondrial Defects in Skeletal Muscle of an Animal Model of Amyotrophic Lateral Sclerosis*. J. Biol. Chem. 2010, 285, 705–712. [Google Scholar] [CrossRef]
- Wiedemann, F.R.; Winkler, K.; Kuznetsov, A.V.; Bartels, C.; Vielhaber, S.; Feistner, H.; Kunz, W.S. Impairment of Mitochondrial Function in Skeletal Muscle of Patients with Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 1998, 156, 65–72. [Google Scholar] [CrossRef]
- Vielhaber, S. Mitochondrial DNA Abnormalities in Skeletal Muscle of Patients with Sporadic Amyotrophic Lateral Sclerosis. Brain 2000, 123, 1339–1348. [Google Scholar] [CrossRef]
- Scaricamazza, S.; Salvatori, I.; Giacovazzo, G.; Loeffler, J.P.; Renè, F.; Rosina, M.; Quessada, C.; Proietti, D.; Heil, C.; Rossi, S.; et al. Skeletal-Muscle Metabolic Reprogramming in ALS-SOD1G93A Mice Predates Disease Onset and Is A Promising Therapeutic Target. iScience 2020, 23, 101087. [Google Scholar] [CrossRef]
- Magrané, J.; Manfredi, G. Mitochondrial Function, Morphology, and Axonal Transport in Amyotrophic Lateral Sclerosis. Antioxid. Redox Signal. 2009, 11, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.; Zech, W.-D.; Goos, M.; Leutbecher, C.; Ferri, A.; Zippelius, A.; Carrì, M.; Nau, R.; Keller, B.U. Impairment of Mitochondrial Calcium Handling in a MtSOD1 Cell Culture Model of Motoneuron Disease. BMC Neurosci. 2009, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Carrì, M.T. Expression of a Cu, Zn Superoxide Dismutase Typical of Familial Amyotrophic Lateral Sclerosis Induces Mitochondrial Alteration and Increase of Cytosolic Ca 2+ Concentration in Transfected Neuroblastoma SH-SY5Y Cells. FEBS Lett. 1997, 414, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Damiano, M.; Starkov, A.A.; Petri, S.; Kipiani, K.; Kiaei, M.; Mattiazzi, M.; Flint Beal, M.; Manfredi, G. Neural Mitochondrial Ca 2+ Capacity Impairment Precedes the Onset of Motor Symptoms in G93A Cu/Zn-Superoxide Dismutase Mutant Mice: Mitochondrial Ca 2+ Capacity in Mutant SOD1 Mice. J. Neurochem. 2006, 96, 1349–1361. [Google Scholar] [CrossRef]
- Rossi, A.E.; Boncompagni, S.; Dirksen, R.T. Sarcoplasmic Reticulum-Mitochondrial Symbiosis: Bidirectional Signaling in Skeletal Muscle. Exerc. Sport Sci. Rev. 2009, 37, 29–35. [Google Scholar] [CrossRef]
- Al-Sarraj, S.; King, A.; Cleveland, M.; Pradat, P.-F.; Corse, A.; Rothstein, J.D.; Leigh, P.N.; Abila, B.; Bates, S.; Wurthner, J.; et al. Mitochondrial Abnormalities and Low Grade Inflammation Are Present in the Skeletal Muscle of a Minority of Patients with Amyotrophic Lateral Sclerosis; an Observational Myopathology Study. Acta Neuropathol. Commun. 2014, 2, 165. [Google Scholar] [CrossRef]
- Capitanio, D.; Vasso, M.; Ratti, A.; Grignaschi, G.; Volta, M.; Moriggi, M.; Daleno, C.; Bendotti, C.; Silani, V.; Gelfi, C. Molecular Signatures of Amyotrophic Lateral Sclerosis Disease Progression in Hind and Forelimb Muscles of an SOD1 G93A Mouse Model. Antioxid. Redox Signal. 2012, 17, 1333–1350. [Google Scholar] [CrossRef]
- Dupuis, L.; Scala, F.; Rene, F.; Tapia, M.; Oudart, H.; Pradat, P.-F.; Meininger, V.; Loeffler, J.-P. Up-regulation of Mitochondrial Uncoupling Protein 3 Reveals an Early Muscular Metabolic Defect in Amyotrophic Lateral Sclerosis. FASEB J. 2003, 17, 1–19. [Google Scholar] [CrossRef]
- Nabben, M.; Hoeks, J. Mitochondrial Uncoupling Protein 3 and Its Role in Cardiac- and Skeletal Muscle Metabolism. Physiol. Behav. 2008, 94, 259–269. [Google Scholar] [CrossRef]
- Aguer, C.; Fiehn, O.; Seifert, E.L.; Bézaire, V.; Meissen, J.K.; Daniels, A.; Scott, K.; Renaud, J.; Padilla, M.; Bickel, D.R.; et al. Muscle Uncoupling Protein 3 Overexpression Mimics Endurance Training and Reduces Circulating Biomarkers of Incomplete Β-oxidation. FASEB J. 2013, 27, 4213–4225. [Google Scholar] [CrossRef]
- Dupuis, L.; Gonzalez de Aguilar, J.-L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle Mitochondrial Uncoupling Dismantles Neuromuscular Junction and Triggers Distal Degeneration of Motor Neurons. PLoS ONE 2009, 4, e5390. [Google Scholar] [CrossRef]
- Loeffler, J.; Picchiarelli, G.; Dupuis, L.; Gonzalez De Aguilar, J. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 227–236. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Bonconpagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal Muscle Is a Primary Target of SOD1G93A-Mediated Toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef]
- Ioannides, Z.A.; Ngo, S.T.; Henderson, R.D.; McCombe, P.A.; Steyn, F.J. Altered Metabolic Homeostasis in Amyotrophic Lateral Sclerosis: Mechanisms of Energy Imbalance and Contribution to Disease Progression. Neurodegener. Dis. 2016, 16, 382–397. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The Role of Mitochondria in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Gandhi, S.; Abramov, A.Y. Mechanism of Oxidative Stress in Neurodegeneration. Oxidative Med. Cell. Longev. 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- Zhang, L.; Keung, W.; Samokhvalov, V.; Wang, W.; Lopaschuk, G.D. Role of Fatty Acid Uptake and Fatty Acid β-Oxidation in Mediating Insulin Resistance in Heart and Skeletal Muscle. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2010, 1801, 1–22. [Google Scholar] [CrossRef]
- Aon, M.A.; Bhatt, N.; Cortassa, S.C. Mitochondrial and Cellular Mechanisms for Managing Lipid Excess. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative Stress in ALS: A Mechanism of Neurodegeneration and a Therapeutic Target. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2006, 1762, 1051–1067. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Kaczor, J.J.; Bourgeois, J.; Yasuda, N.; Tarnopolsky, M.A. Oxidative Stress and Antioxidant Enzyme Upregulation in SOD1-G93A Mouse Skeletal Muscle. Muscle Nerve. 2006, 33, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.G.; Henry, Y.K.; Mattson, M.P.; Appel, S.H. Presence of 4-Hydroxynonenal in Cerebrospinal Fluid of Patients with Sporadic Amyotrophic Lateral Sclerosis. Ann. Neurol. 1998, 44, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.-C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative Stress Biomarkers in Sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Ince, P.G.; Falkous, G.; Mantle, D. Oxidative Damage to Protein in Sporadic Motor Neuron Disease Spinal Cord. Ann. Neurol. 1995, 38, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Nagai, R.; Uchida, K.; Horiuchi, S.; Yamada, S.; Hirano, A.; Kawaguchi, M.; Yamamoto, T.; Sasaki, S.; Kobayashi, M. Morphological Evidence for Lipid Peroxidation and Protein Glycoxidation in Spinal Cords from Sporadic Amyotrophic Lateral Sclerosis Patients. Brain Res. 2001, 917, 97–104. [Google Scholar] [CrossRef]
- Chang, Y.; Kong, Q.; Shan, X.; Tian, G.; Ilieva, H.; Cleveland, D.W.; Rothstein, J.D.; Borchelt, D.R.; Wong, P.C.; Lin, C.G. Messenger RNA Oxidation Occurs Early in Disease Pathogenesis and Promotes Motor Neuron Degeneration in ALS. PLoS ONE 2008, 3, e2849. [Google Scholar] [CrossRef] [PubMed]
- Halter, B.; Gonzalez de Aguilar, J.-L.; Rene, F.; Petri, S.; Fricker, B.; Echaniz-Laguna, A.; Dupuis, L.; Larmet, Y.; Loeffler, J.-P. Oxidative Stress in Skeletal Muscle Stimulates Early Expression of Rad in a Mouse Model of Amyotrophic Lateral Sclerosis. Free Radic. Biol. Med. 2010, 48, 915–923. [Google Scholar] [CrossRef]
- Leclerc, N.; Ribera, F.; Zoll, J.; Warter, J.-M.; Poindron, P.; Lampert, E.; Borg, J. Selective Changes in Mitochondria Respiratory Properties in Oxidative or Glycolytic Muscle Fibers Isolated from G93AhumanSOD1 Transgenic Mice. Neuromuscul. Disord. 2001, 11, 722–727. [Google Scholar] [CrossRef]
- Xiao, Y.; Karam, C.; Yi, J.; Zhang, L.; Li, X.; Yoon, D.; Wang, H.; Dhakal, K.; Ramlow, P.; Yu, T.; et al. ROS-Related Mitochondrial Dysfunction in Skeletal Muscle of an ALS Mouse Model during the Disease Progression. Pharmacol. Res. 2018, 138, 25–36. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Zoll, J.; Ribera, F.; Tranchant, C.; Warter, J.-M.; Lonsdorfer, J.; Lampert, E. Mitochondrial Respiratory Chain Function in Skeletal Muscle of ALS Patients. Ann. Neurol. 2002, 52, 623–627. [Google Scholar] [CrossRef]
- Krasnianski, A.; Deschauer, M.; Neudecker, S.; Gellerich, F.N.; Müller, T.; Schoser, B.G.; Krasnianski, M.; Zierz, S. Mitochondrial Changes in Skeletal Muscle in Amyotrophic Lateral Sclerosis and Other Neurogenic Atrophies. Brain 2005, 128, 1870–1876. [Google Scholar] [CrossRef]
- Ryan, T.E.; Erickson, M.L.; Verma, A.; Chavez, J.; Rivner, M.H.; Mccully, K.K. Skeletal Muscle Oxidative Capacity in Amyotrophic Lateral Sclerosis: Muscle Metabolism in ALS. Muscle Nerve. 2014, 50, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Kasarskis, E.J.; Berryman, S.; Vanderleest, J.G.; Schneider, A.R.; McClain, C.J. Nutritional Status of Patients with Amyotrophic Lateral Sclerosis: Relation to the Proximity of Death. Am. J. Clin. Nutr. 1996, 63, 130–137. [Google Scholar] [CrossRef]
- Reyes, E.T.; Perurena, O.H.; Festoff, B.W.; Jorgensen, R.; Moore, W.V. Insulin Resistance in Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 1984, 63, 317–324. [Google Scholar] [CrossRef]
- Pradat, P.-F.; Bruneteau, G.; Gordon, P.H.; Dupuis, L.; Bonnefont-Rousselot, D.; Simon, D.; Salachas, F.; Corcia, P.; Frochot, V.; Lacorte, J.-M.; et al. Impaired Glucose Tolerance in Patients with Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. 2010, 11, 166–171. [Google Scholar] [CrossRef]
- Desport, J.C.; Preux, P.M.; Truong, T.C.; Vallat, J.M.; Sautereau, D.; Couratier, P. Nutritional Status Is a Prognostic Factor for Survival in ALS Patients. Neurology 1999, 53, 1059. [Google Scholar] [CrossRef]
- Marin, B.; Desport, J.C.; Kajeu, P.; Jesus, P.; Nicolaud, B.; Nicol, M.; Preux, P.M.; Couratier, P. Alteration of Nutritional Status at Diagnosis Is a Prognostic Factor for Survival of Amyotrophic Lateral Sclerosis Patients. J. Neurol. Neurosurg. Psychiatry 2011, 82, 628–634. [Google Scholar] [CrossRef]
- Nakken, O.; Meyer, H.E.; Stigum, H.; Holmøy, T. High BMI Is Associated with Low ALS Risk: A Population-Based Study. Neurology 2019, 93, e424–e432. [Google Scholar] [CrossRef]
- Pape, J.A.; Grose, J.H. The Effects of Diet and Sex in Amyotrophic Lateral Sclerosis. Rev. Neurol. 2020, 176, 301–315. [Google Scholar] [CrossRef]
- Jawaid, A.; Murthy, S.B.; Wilson, A.M.; Qureshi, S.U.; Amro, M.J.; Wheaton, M.; Simpson, E.; Harati, Y.; Strutt, A.M.; York, M.K.; et al. A Decrease in Body Mass Index Is Associated with Faster Progression of Motor Symptoms and Shorter Survival in ALS. Amyotroph. Lateral Scler. 2010, 11, 542–548. [Google Scholar] [CrossRef]
- Paganoni, S.; Deng, J.; Jaffa, M.; Cudkowicz, M.E.; Wills, A.-M. Body Mass Index, Not Dyslipidemia, Is an Independent Predictor of Survival in Amyotrophic Lateral Sclerosis. Muscle Nerve. 2011, 44, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Dardiotis, E.; Siokas, V.; Sokratous, M.; Tsouris, Z.; Aloizou, A.-M.; Florou, D.; Dastamani, M.; Mentis, A.-F.A.; Brotis, A.G. Body Mass Index and Survival from Amyotrophic Lateral Sclerosis: A Meta-Analysis. Neurol. Clin. Pract. 2018, 8, 437–444. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, É.J.; Wang, H.; Weisskopf, M.G.; Fitzgerald, K.C.; Falcone, G.; McCullough, M.L.; Thun, M.; Park, Y.; Kolonel, L.N.; Ascherio, A. Premorbid Body Mass Index and Risk of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Desport, J.C.; Preux, P.M.; Magy, L.; Boirie, Y.; Vallat, J.M.; Beaufrère, B.; Couratier, P. Factors Correlated with Hypermetabolism in Patients with Amyotrophic Lateral Sclerosis. Am. J. Clin. Nutr. 2001, 74, 328–334. [Google Scholar] [CrossRef]
- Desport, J.-C.; Torny, F.; Lacoste, M.; Preux, P.-M.; Couratier, P. Hypermetabolism in ALS: Correlations with Clinical and Paraclinical Parameters. Neurodegener. Dis. 2005, 2, 202–207. [Google Scholar] [CrossRef]
- Fayemendy, P.; Marin, B.; Labrunie, A.; Boirie, Y.; Walrand, S.; Achamrah, N.; Coëffier, M.; Preux, P.-M.; Lautrette, G.; Desport, J.-C.; et al. Hypermetabolism Is a Reality in Amyotrophic Lateral Sclerosis Compared to Healthy Subjects. J. Neurol. Sci. 2021, 420, 117257. [Google Scholar] [CrossRef]
- Jésus, P.; Fayemendy, P.; Nicol, M.; Lautrette, G.; Sourisseau, H.; Preux, P.-M.; Desport, J.-C.; Marin, B.; Couratier, P. Hypermetabolism Is a Deleterious Prognostic Factor in Patients with Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2018, 25, 97–104. [Google Scholar] [CrossRef]
- Bouteloup, C.; Desport, J.-C.; Clavelou, P.; Guy, N.; Derumeaux-Burel, H.; Ferrier, A.; Couratier, P. Hypermetabolism in ALS Patients: An Early and Persistent Phenomenon. J. Neurol. 2009, 256, 1236–1242. [Google Scholar] [CrossRef]
- Steyn, F.J.; Ioannides, Z.A.; van Eijk, R.P.A.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; van den Berg, L.H.; et al. Hypermetabolism in ALS Is Associated with Greater Functional Decline and Shorter Survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef]
- Funalot, B.; Desport, J.-C.; Sturtz, F.; Camu, W.; Couratier, P. High Metabolic Level in Patients with Familial Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. 2009, 10, 113–117. [Google Scholar] [CrossRef]
- Peter, R.S.; Rosenbohm, A.; Dupuis, L.; Brehme, T.; Kassubek, J.; Rothenbacher, D.; Nagel, G.; Ludolph, A.C. Life Course Body Mass Index and Risk and Prognosis of Amyotrophic Lateral Sclerosis: Results from the ALS Registry Swabia. Eur. J. Epidemiol. 2017, 32, 901–908. [Google Scholar] [CrossRef]
- Moglia, C.; Calvo, A.; Grassano, M.; Canosa, A.; Manera, U.; D’Ovidio, F.; Bombaci, A.; Bersano, E.; Mazzini, L.; Mora, G.; et al. Early Weight Loss in Amyotrophic Lateral Sclerosis: Outcome Relevance and Clinical Correlates in a Population-Based Cohort. J. Neurol. Neurosurg. Psychiatry 2019, 90, 666–673. [Google Scholar] [CrossRef]
- Dupuis, L.; Corcia, P.; Fergani, A. Dyslipidemia Is a Protective Factor in Amyotrophic Lateral Sclerosis. Neurology 2008, 7, 1004–1009. [Google Scholar] [CrossRef]
- Ngo, S.T.; Steyn, F.J.; Huang, L.; Mantovani, S.; Pfluger, C.M.M.; Woodruff, T.M.; O’Sullivan, J.D.; Henderson, R.D.; McCombe, P.A. Altered Expression of Metabolic Proteins and Adipokines in Patients with Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 2015, 357, 22–27. [Google Scholar] [CrossRef]
- Dupuis, L.; Oudart, H.; Rene, F.; de Aguilar, J.-L.G.; Loeffler, J.-P. Evidence for Defective Energy Homeostasis in Amyotrophic Lateral Sclerosis: Benefit of a High-Energy Diet in a Transgenic Mouse Model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef]
- Palamiuc, L.; Schlagowski, A.; Ngo, S.T.; Vernay, A.; Dirrig-Grosch, S.; Henriques, A.; Boutillier, A.; Zoll, J.; Echaniz-Laguna, A.; Loeffler, J.; et al. A Metabolic Switch toward Lipid Use in Glycolytic Muscle Is an Early Pathologic Event in a Mouse Model of Amyotrophic Lateral Sclerosis. Embo. Mol. Med. 2015, 7, 526–546. [Google Scholar] [CrossRef]
- Joardar, A.; Manzo, E.; Zarnescu, D.C. Metabolic Dysregulation in Amyotrophic Lateral Sclerosis: Challenges and Opportunities. Curr. Genet. Med. Rep. 2017, 5, 108–114. [Google Scholar] [CrossRef]
- Jeoung, N.H.; Wu, P.; Joshi, M.A.; Jaskiewicz, J.; Bock, C.B.; Depaoli-Roach, A.A.; Harris, R.A. Role of Pyruvate Dehydrogenase Kinase Isoenzyme 4 (PDHK4) in Glucose Homoeostasis during Starvation. Biochem. J. 2006, 397, 417–425. [Google Scholar] [CrossRef]
- Zhang, S.; Hulver, M.W.; McMillan, R.P.; Cline, M.A.; Gilbert, E.R. The Pivotal Role of Pyruvate Dehydrogenase Kinases in Metabolic Flexibility. Nutr. Metab. (Lond.) 2014, 11, 10. [Google Scholar] [CrossRef]
- Desseille, C.; Deforges, S.; Biondi, O.; Houdebine, L.; D’amico, D.; Lamazière, A.; Caradeuc, C.; Bertho, G.; Bruneteau, G.; Weill, L.; et al. Specific Physical Exercise Improves Energetic Metabolism in the Skeletal Muscle of Amyotrophic-Lateral- Sclerosis Mice. Front. Mol. Neurosci. 2017, 10, 332. [Google Scholar] [CrossRef]
- Telerman-Toppet, N.; Coërs, C. Motor Innervation and Fiber Type Pattern in Amyotrophic Lateral Sclerosis and in Charcot-Marie-Tooth Disease: Motor Units in ALS and CMT. Muscle Nerve. 1978, 1, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, J.; Putman, C.T.; Gordon, T. Time Course of Preferential Motor Unit Loss in the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2007, 28, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, J.; Putman, C.T.; Tyreman, N.; Gordon, T. Preferential Motor Unit Loss in the SOD1 G93A Transgenic Mouse Model of Amyotrophic Lateral Sclerosis: Motor Units in Mouse ALS. J. Physiol. 2008, 586, 3337–3351. [Google Scholar] [CrossRef] [PubMed]
- Atkin, J.D.; Scott, R.L.; West, J.M.; Lopes, E.; Quah, A.K.J.; Cheema, S.S. Properties of Slow- and Fast-Twitch Muscle Fibres in a Mouse Model of Amyotrophic Lateral Sclerosis. Neuromuscul. Disord. 2005, 15, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Deforges, S.; Branchu, J.; Biondi, O.; Grondard, C.; Pariset, C.; Lécolle, S.; Lopes, P.; Vidal, P.-P.; Chanoine, C.; Charbonnier, F. Motoneuron Survival Is Promoted by Specific Exercise in a Mouse Model of Amyotrophic Lateral Sclerosis: Motoneuron Activation and Neuroprotection in ALS Mice. J. Physiol. 2009, 587, 3561–3572. [Google Scholar] [CrossRef] [PubMed]
- Flis, D.J.; Dzik, K.; Kaczor, J.J.; Cieminski, K.; Halon-Golabek, M.; Antosiewicz, J.; Wieckowski, M.R.; Ziolkowski, W. Swim Training Modulates Mouse Skeletal Muscle Energy Metabolism and Ameliorates Reduction in Grip Strength in a Mouse Model of Amyotrophic Lateral Sclerosis. IJMS 2019, 20, 233. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The Glucose Fatty-Acid Cycle. Its Role in Insulin Sensitivity and the Metabolic Disturbances of Diabetes Mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Bassel-Duby, R.; Olson, E.N. Signaling Pathways in Skeletal Muscle Remodeling. Annu. Rev. Biochem. 2006, 75, 19–37. [Google Scholar] [CrossRef]
- Talanian, J.L.; Holloway, G.P.; Snook, L.A.; Heigenhauser, G.J.F.; Bonen, A.; Spriet, L.L. Exercise Training Increases Sarcolemmal and Mitochondrial Fatty Acid Transport Proteins in Human Skeletal Muscle. Endocrinol. Metab. 2010, 299, 9. [Google Scholar] [CrossRef]
- Pette, D.; Staron, R.S. Transitions of Muscle Fiber Phenotypic Profiles. Histochem Cell Biol. 2001, 115, 359–372. [Google Scholar] [CrossRef]
- Lacorte, E.; Ferrigno, L.; Leoncini, E.; Corbo, M.; Boccia, S.; Vanacore, N. Physical Activity, and Physical Activity Related to Sports, Leisure and Occupational Activity as Risk Factors for ALS: A Systematic Review. Neurosci. Biobehav. Rev. 2016, 66, 61–79. [Google Scholar] [CrossRef]
- Bozzoni, V. Amyotrophic Lateral Sclerosis and Environmental Factors. Funct. Neurol. 2016, 31. [Google Scholar] [CrossRef]
- Chio, A. Severely Increased Risk of Amyotrophic Lateral Sclerosis among Italian Professional Football Players. Brain 2005, 128, 472–476. [Google Scholar] [CrossRef]
- Chio, A.; Mora, G.; Calvo, A.; Mazzini, L.; Bottacchi, E.; Mutani, R. On behalf of the PARALS Epidemiology of ALS in Italy: A 10-Year Prospective Population-Based Study. Neurology 2009, 72, 725–731. [Google Scholar] [CrossRef]
- Beghi, E. Are Professional Soccer Players at Higher Risk for ALS? Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 501–506. [Google Scholar] [CrossRef]
- Piazza, O.; Sirén, A.-L.; Ehrenreich, H. Soccer, Neurotrauma and Amyotrophic Lateral Sclerosis: Is There a Connection? Curr. Med. Res. Opin. 2004, 20, 505–508. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O. The Epidemiology of ALS: A Conspiracy of Genes, Environment and Time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef]
- Scarmeas, N.; Shih, T.; Stern, Y.; Ottman, R.; Rowland, L.P. Premorbid Weight, Body Mass, and Varsity Athletics in ALS. Neurology 2002, 59, 773–775. [Google Scholar] [CrossRef]
- Huisman, M.H.B.; Seelen, M.; de Jong, S.W.; Dorresteijn, K.R.I.S.; van Doormaal, P.T.C.; van der Kooi, A.J.; de Visser, M.; Schelhaas, H.J.; van den Berg, L.H.; Veldink, J.H. Lifetime Physical Activity and the Risk of Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 976–981. [Google Scholar] [CrossRef]
- Longstreth, W.T.; McGuire, V.; Koepsell, T.D.; Wang, Y.; van Belle, G. Risk of Amyotrophic Lateral Sclerosis and History of Physical Activity: A Population-Based Case-Control Study. Arch Neurol 1998, 55, 201. [Google Scholar] [CrossRef]
- Valenti, M.; Pontieri, F.E.; Conti, F.; Altobelli, E.; Manzoni, T.; Frati, L. Amyotrophic Lateral Sclerosis and Sports: A Case-Control Study. Eur. J. Neurol. 2005, 12, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Goldacre, R.; Ramagopalan, S.; Talbot, K.; Goldacre, M.J. Autoimmune Disease Preceding Amyotrophic Lateral Sclerosis: An Epidemiologic Study. Neurology 2013, 81, 1222–1225. [Google Scholar] [CrossRef] [PubMed]
- Dal Bello-Haas, V.; Florence, J.M. Therapeutic Exercise for People with Amyotrophic Lateral Sclerosis or Motor Neuron Disease. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, D.J.; Rodriguez, C.; Devries, M.; Yasuda, N.; Tarnopolsky, M.A. Effects of High-Intensity Endurance Exercise Training in the G93A Mouse Model of Amyotrophic Lateral Sclerosis. Muscle Nerve. 2004, 29, 656–662. [Google Scholar] [CrossRef]
- Carreras, I.; Yuruker, S.; Aytan, N.; Hossain, L.; Choi, J.-K.; Jenkins, B.G.; Kowall, N.W.; Dedeoglu, A. Moderate Exercise Delays the Motor Performance Decline in a Transgenic Model of ALS. Brain Res. 2010, 1313, 192–201. [Google Scholar] [CrossRef]
- Kaspar, B.K.; Frost, L.M.; Christian, L.; Umapathi, P.; Gage, F.H. Synergy of Insulin-like Growth Factor-1 and Exercise in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2005, 57, 649–655. [Google Scholar] [CrossRef]
- Grondard, C.; Biondi, O.; Pariset, C.; Lopes, P.; Deforges, S.; Lécolle, S.; Gaspera, B.D.; Gallien, C.-L.; Chanoine, C.; Charbonnier, F. Exercise-Induced Modulation of Calcineurin Activity Parallels the Time Course of Myofibre Transitions. J. Cell. Physiol. 2008, 214, 126–135. [Google Scholar] [CrossRef]
- Smittkamp, S.E.; Morris, J.K.; Bomhoff, G.L.; Chertoff, M.E.; Geiger, P.C.; Stanford, J.A. SOD1-G93A Mice Exhibit Muscle-Fiber-Type-Specific Decreases in Glucose Uptake in the Absence of Whole-Body Changes in Metabolism. Neurodegener. Dis. 2013. [Google Scholar] [CrossRef]
- Zubiri, I.; Lombardi, V.; Bremang, M.; Mitra, V.; Nardo, G.; Adiutori, R.; Lu, C.-H.; Leoni, E.; Yip, P.; Yildiz, O.; et al. Tissue-Enhanced Plasma Proteomic Analysis for Disease Stratification in Amyotrophic Lateral Sclerosis. Mol. Neurodegener. 2018, 13, 60. [Google Scholar] [CrossRef]
- Ren, J.M.; Marshall, B.A.; Mueckler, M.M.; McCaleb, M.; Amatruda, J.M.; Shulman, G.I. Overexpression of Glut4 Protein in Muscle Increases Basal and Insulin-Stimulated Whole Body Glucose Disposal in Conscious Mice. J. Clin. Investig. 1995, 95, 429–432. [Google Scholar] [CrossRef]
- Leturque, A.; Loizeau, M.; Vaulont, S.; Salminen, M.; Girard, J. Improvement of Insulin Action in Diabetic Transgenic Mice Selectively Overexpressing GLUT4 in Skeletal Muscle. Diabetes 1996, 45, 23–27. [Google Scholar] [CrossRef]
- Tsao, T.-S.; Burcelin, R.; Katz, E.B.; Huang, L.; Charron, M.J. Enhanced Insulin Action Due to Targeted GLUT4 Overexpression Exclusively in Muscle. Diabetes 1996, 45, 28–36. [Google Scholar] [CrossRef]
- Dupuis, L.; Pradat, P.-F.; Ludolph, A.C.; Loeffler, J.-P. Energy Metabolism in Amyotrophic Lateral Sclerosis. Lancet. Neurol. 2011, 10, 75–82. [Google Scholar] [CrossRef]
- Saccà, F.; Quarantelli, M.; Rinaldi, C.; Tucci, T.; Piro, R.; Perrotta, G.; Carotenuto, B.; Marsili, A.; Palma, V.; Michele, G.; et al. A Randomized Controlled Clinical Trial of Growth Hormone in Amyotrophic Lateral Sclerosis: Clinical, Neuroimaging, and Hormonal Results. J. Neurol. 2012, 259, 132–138. [Google Scholar] [CrossRef]
- Massao Hirabara, S.; de Oliveira Carvalho, C.R.; Mendonça, J.R.; Piltcher Haber, E.; Fernandes, L.C.; Curi, R. Palmitate Acutely Raises Glycogen Synthesis in Rat Soleus Muscle by a Mechanism That Requires Its Metabolization (Randle Cycle). FEBS Lett. 2003, 541, 109–114. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α (PGC-1α): Transcriptional Coactivator and Metabolic Regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef]
- Thau, N.; Knippenberg, S.; Korner, S.; Rath, K.J.; Dengler, R.; Petri, S. Decreased MRNA Expression of PGC-1> and PGC-1>YRegulated Factors in the SOD1G93A ALS Mouse Model and in Human Sporadic ALS. J. Neuropathol. Exp. Neurol. 2012, 71, 11. [Google Scholar] [CrossRef]
- Cho, Y.; Hazen, B.C.; Russell, A.P.; Kralli, A. Peroxisome Proliferator-Activated Receptor γ Coactivator 1 (PGC-1)- and Estrogen-Related Receptor (ERR)-Induced Regulator in Muscle 1 (PERM1) Is a Tissue-Specific Regulator of Oxidative Capacity in Skeletal Muscle Cells. J. Biol. Chem. 2013, 288, 25207–25218. [Google Scholar] [CrossRef]
- Cresci, S.; Wright, L.D.; Spratt, J.A.; Briggs, F.N.; Kelly, D.P. Activation of a Novel Metabolic Gene Regulatory Pathway by Chronic Stimulation of Skeletal Muscle. Am. J. Physiol. Cell Physiol. 1996, 270, C1413–C1420. [Google Scholar] [CrossRef]
- Gulick, T.; Cresci, S.; Caira, T.; Moore, D.D.; Kelly, D.P. The Peroxisome Proliferator-Activated Receptor Regulates Mitochondrial Fatty Acid Oxidative Enzyme Gene Expression. Proc. Natl. Acad. Sci. USA 1994, 91, 11012–11016. [Google Scholar] [CrossRef]
- Finck, B.N.; Bernal-Mizrachi, C.; Han, D.H.; Coleman, T.; Sambandam, N.; LaRiviere, L.L.; Holloszy, J.O.; Semenkovich, C.F.; Kelly, D.P. A Potential Link between Muscle Peroxisome Proliferator- Activated Receptor-α Signaling and Obesity-Related Diabetes. Cell Metab. 2005, 1, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, S.; Parone, P.A.; Lopes, V.S.; Lillo, C.; McAlonis-Downes, M.; Lee, S.K.; Vetto, A.P.; Petrosyan, S.; Marsala, M.; Murphy, A.N.; et al. Elevated PGC-1α Activity Sustains Mitochondrial Biogenesis and Muscle Function without Extending Survival in a Mouse Model of Inherited ALS. Cell Metab. 2012, 15, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.-Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional Co-Activator PGC-1α Drives the Formation of Slow-Twitch Muscle Fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome Proliferator-activated Receptor δ Controls Muscle Development and Oxydative Capability. FASEB J. 2003, 17, 2299–2301. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Zhang, C.-L.; Yu, R.T.; Cho, H.K.; Nelson, M.C.; Bayuga-Ocampo, C.R.; Ham, J.; Kang, H.; Evans, R.M. Regulation of Muscle Fiber Type and Running Endurance by PPARδ. PLoS Biol. 2004, 2, e294. [Google Scholar] [CrossRef]
- McVeigh, J.J.; Lopaschuk, G.D. Dichloroacetate Stimulation of Glucose Oxidation Improves Recovery of Ischemic Rat Hearts. Am. J. Physiol.-Heart Circ. Physiol. 1990, 259, H1079–H1085. [Google Scholar] [CrossRef]
- Whitehouse, S.; Randle, P.J. Activation of Pyruvate Dehydrogenase in Perfused Rat Heart by Dichloroacetate (Short Communication). Biochem. J. 1973, 134, 651–653. [Google Scholar] [CrossRef]
- Miquel, E.; Cassina, A.; Martínez-Palma, L.; Bolatto, C.; Trías, E.; Gandelman, M.; Radi, R.; Barbeito, L.; Cassina, P. Modulation of Astrocytic Mitochondrial Function by Dichloroacetate Improves Survival and Motor Performance in Inherited Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e34776. [Google Scholar] [CrossRef]
- DeAngelo, A.B.; George, M.H.; House, D.E. Hepatocarcinogenicity In The Male B6c3f1 Mouse Following A Lifetime Exposure To Dichloroacetic Acid In The Drinking Water: Dose-Response Determination And Modes Of Action. J. Toxicol. Environ. Health 1999, 58, 485–507. [Google Scholar] [CrossRef]
- Lionetti, V.; Stanley, W.C.; Recchia, F.A. Modulating Fatty Acid Oxidation in Heart Failure. Cardiovasc. Res. 2011, 90, 202–209. [Google Scholar] [CrossRef]
- McCormack, J.G.; Stanley, W.C.; Wolff, A.A. Ranolazine: A Novel Metabolic Modulator for the Treatment of Angina. Gen. Pharmacol. Vasc. Syst. 1998, 30, 639–645. [Google Scholar] [CrossRef]
- Tafreshi, M.J.; Fisher, E. Ranolazine: A New Approach to Management of Patients with Angina. Ann. Pharm. 2006, 40, 689–693. [Google Scholar] [CrossRef]
- Hill, J.A.; Schofield, R.S. The Use of Ranolazine in Cardiovascular Disease. Expert Opin. Investig. Drugs 2002, 11, 117–123. [Google Scholar] [CrossRef]
- Clarke, B. Ranolazine Increases Active Pyruvate Dehydrogenase in Perfused Normoxic Rat Hearts: Evidence for an Indirect Mechanism. J. Mol. Cell. Cardiol. 1996, 28, 341–350. [Google Scholar] [CrossRef]
- Stanley, W.C. Partial Fatty Acid Oxidation Inhibitors for Stable Angina. Expert Opin. Investig. Drugs 2002, 11, 615–629. [Google Scholar] [CrossRef]
- Mourouzis, I.; Mantzouratou, P.; Galanopoulos, G.; Kostakou, E.; Dhalla, A.K.; Belardinelli, L.; Pantos, C. The Beneficial Effects of Ranolazine on Cardiac Function After Myocardial Infarction Are Greater in Diabetic Than in Nondiabetic Rats. J. Cardiovasc Pharm. 2014, 19, 457–469. [Google Scholar] [CrossRef]
- Chaitman, B.R.; Pepine, C.J.; Parker, J.O.; Skopal, J.; Chumakova, G.; Kuch, J.; Wang, W.; Skettino, S.L.; Wolff, A.A. Combination Assessment of Ranolazine In Stable Angina (CARISA) Investigators Effects of Ranolazine with Atenolol, Amlodipine, or Diltiazem on Exercise Tolerance and Angina Frequency in Patients with Severe Chronic Angina: A Randomized Controlled Trial. JAMA 2004, 291, 309–316. [Google Scholar] [CrossRef]
- Novak, K.R.; Norman, J.; Mitchell, J.R.; Pinter, M.J.; Rich, M.M. Sodium Channel Slow Inactivation as a Therapeutic Target for Myotonia Congenita. Ann. Neurol. 2015, 77, 320–332. [Google Scholar] [CrossRef]
- Fu, Z.; Zhao, L.; Chai, W.; Dong, Z.; Cao, W.; Liu, Z. Ranolazine Recruits Muscle Microvasculature and Enhances Insulin Action in Rats: Ranolazine, Microvasculature and Insulin Action. J. Physiol. 2013, 591, 5235–5249. [Google Scholar] [CrossRef]
- Eckel, R.H.; Henry, R.R.; Yue, P.; Dhalla, A.; Wong, P.; Jochelson, P.; Belardinelli, L.; Skyler, J.S. Effect of Ranolazine Monotherapy on Glycemic Control in Subjects With Type 2 Diabetes. Diabetes Care 2015, 38, 1189–1196. [Google Scholar] [CrossRef]
- Caminiti, G.; Fossati, C.; Battaglia, D.; Massaro, R.; Rosano, G.; Volterrani, M. Ranolazine Improves Insulin Resistance in Non-Diabetic Patients with Coronary Heart Disease. A Pilot Study. Int. J. Cardiol. 2016, 219, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.A.; Rotmensch, H.H.; Stanley, W.C.; Ferrari, R. Metabolic Approaches to the Treatment of Ischemic Heart Disease: The Clinicians’ Perspective. Heart Fail. Rev. 2020, 7, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial Fatty Acid Metabolism in Health and Disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef] [PubMed]
- Kantor, P.F.; Lucien, A.; Kozak, R.; Lopaschuk, G.D. The Antianginal Drug Trimetazidine Shifts Cardiac Energy Metabolism From Fatty Acid Oxidation to Glucose Oxidation by Inhibiting Mitochondrial Long-Chain 3-Ketoacyl Coenzyme A Thiolase. Circ. Res. 2000, 86, 580–588. [Google Scholar] [CrossRef]
- Fang, Y.-H.; Piao, L.; Hong, Z.; Toth, P.T.; Marsboom, G.; Bache-Wiig, P.; Rehman, J.; Archer, S.L. Therapeutic Inhibition of Fatty Acid Oxidation in Right Ventricular Hypertrophy: Exploiting Randle’s Cycle. J. Mol. Med. 2012, 90, 31–43. [Google Scholar] [CrossRef]
- Ferraro, E.; Pin, F.; Gorini, S.; Pontecorvo, L.; Ferri, A.; Mollace, V.; Costelli, P.; Rosano, G. Improvement of Skeletal Muscle Performance in Ageing by the Metabolic Modulator Trimetazidine: Metabolism Remodeling in Skeletal Muscle. J. CachexiaSarcopenia Muscle 2016, 7, 449–457. [Google Scholar] [CrossRef]
- Gatta, L.; Vitiello, L.; Gorini, S.; Chiandotto, S.; Costelli, P.; Giammarioli, A.M.; Malorni, W.; Rosano, G.; Ferraro, E. Modulating the Metabolism by Trimetazidine Enhances Myoblast Differentiation and Promotes Myogenesis in Cachectic Tumor-Bearing C26 Mice. Oncotarget 2017, 8, 113938–113956. [Google Scholar] [CrossRef]
- Karahan, G.; Kaya, H.; Erdogan, M.A.; Yigitturk, G.; Gokyayla, E.; Erbas, O. Effects of Trimetazidine on Nerve Regeneration in a Rat Sciatic Nerve Injury Model. Bratisl. Lek. Listy 2019, 120, 777–782. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Change Relative to Onset of Symptoms | Results | Ref. | ||
|---|---|---|---|---|
| NMJ alterations | Patients | Before |
| [30] |
| After |
| [28] | ||
| SOD1G93A mice | Before |
| [26] | |
| [31] | |||
| [32] | |||
| [33] | |||
| [34] |
| Change Relative to Onset of Symptoms | Results | Ref. | ||
|---|---|---|---|---|
| Mitochondrial alterations in skeletal muscle | Patients | After |
| [51] |
| [52] | |||
| [54] | |||
| [55] | |||
| [58] | |||
| [59] | |||
| SOD1G93A mice | Before |
| [57] | |
| [60] |
| Change Relative to Onset of Symptoms | Results | Ref. | ||
|---|---|---|---|---|
| Alteration of skeletal muscle metabolism and contractile properties | Patients | After |
| [116] |
| [120] | |||
| [121] | |||
| SOD1G93A mice | Before |
| [60] | |
| [120] | |||
| [122] | |||
→ Preferential denervation of fast motor neurons | [123] | |||
| [124] | |||
| After |
| [125] | ||
→ Physical activity improves metabolism | [120] | |||
→ Swimming exercise modulates skeletal muscle energy metabolism | [126] | |||
| Sod1G86R mice | Before |
| [116] | |
| After |
| [116] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quessada, C.; Bouscary, A.; René, F.; Valle, C.; Ferri, A.; Ngo, S.T.; Loeffler, J.-P. Skeletal Muscle Metabolism: Origin or Prognostic Factor for Amyotrophic Lateral Sclerosis (ALS) Development? Cells 2021, 10, 1449. https://doi.org/10.3390/cells10061449
Quessada C, Bouscary A, René F, Valle C, Ferri A, Ngo ST, Loeffler J-P. Skeletal Muscle Metabolism: Origin or Prognostic Factor for Amyotrophic Lateral Sclerosis (ALS) Development? Cells. 2021; 10(6):1449. https://doi.org/10.3390/cells10061449
Chicago/Turabian StyleQuessada, Cyril, Alexandra Bouscary, Frédérique René, Cristiana Valle, Alberto Ferri, Shyuan T. Ngo, and Jean-Philippe Loeffler. 2021. "Skeletal Muscle Metabolism: Origin or Prognostic Factor for Amyotrophic Lateral Sclerosis (ALS) Development?" Cells 10, no. 6: 1449. https://doi.org/10.3390/cells10061449
APA StyleQuessada, C., Bouscary, A., René, F., Valle, C., Ferri, A., Ngo, S. T., & Loeffler, J.-P. (2021). Skeletal Muscle Metabolism: Origin or Prognostic Factor for Amyotrophic Lateral Sclerosis (ALS) Development? Cells, 10(6), 1449. https://doi.org/10.3390/cells10061449