
Chemotherapy Shifts the Balance in Favor of CD8+ TNFR2+ TILs in Triple-Negative Breast Tumors

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. Immunohistochemistry
2.3. PD-L1 Expressing 4T1 Cells
2.4. Tumor Establishment and Chemotherapy Administration
2.5. Preparation of Dissociated Cells from Tumors and Spleens
2.6. Analysis of Lymphocyte Subsets
3. Results
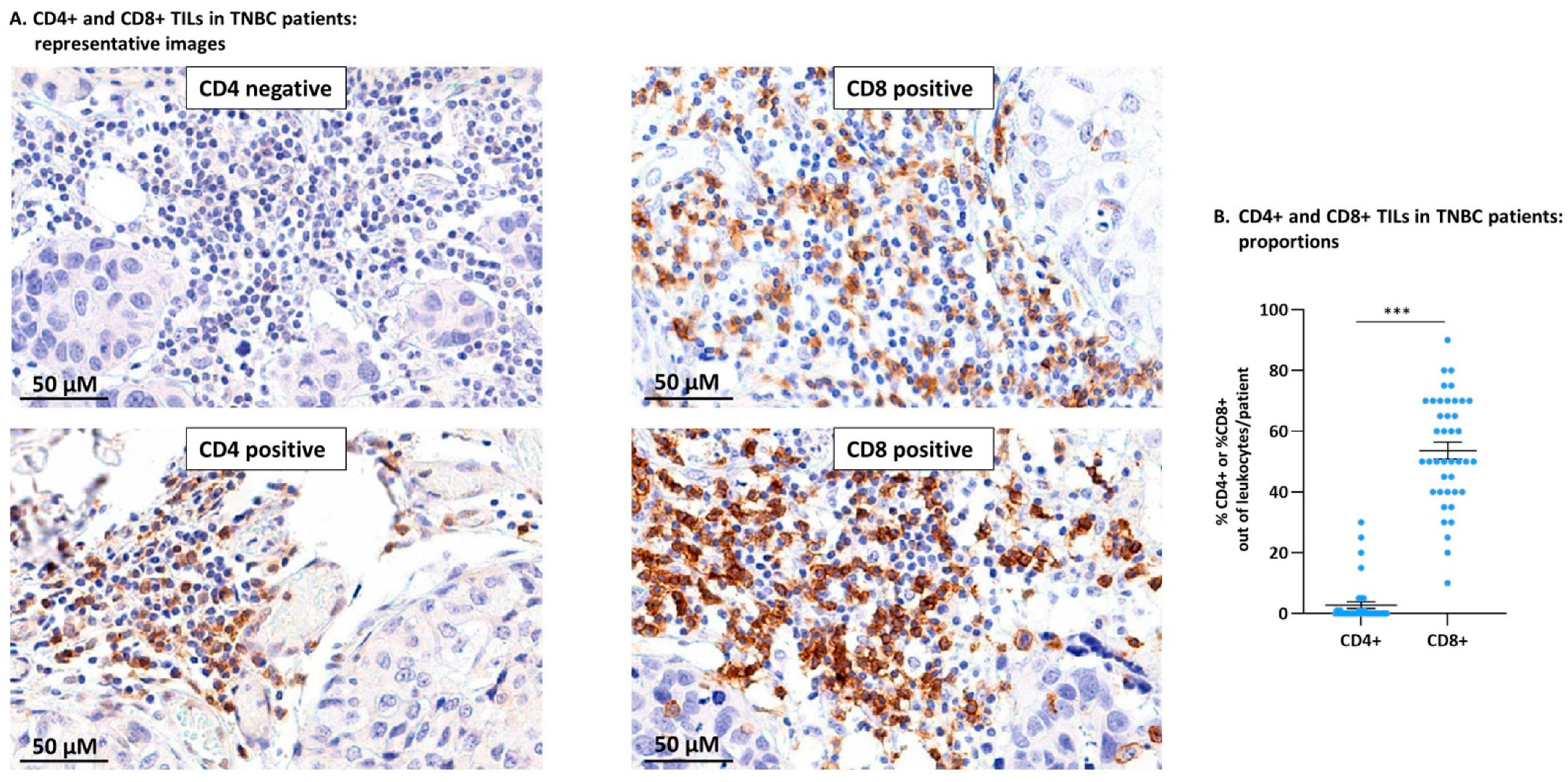
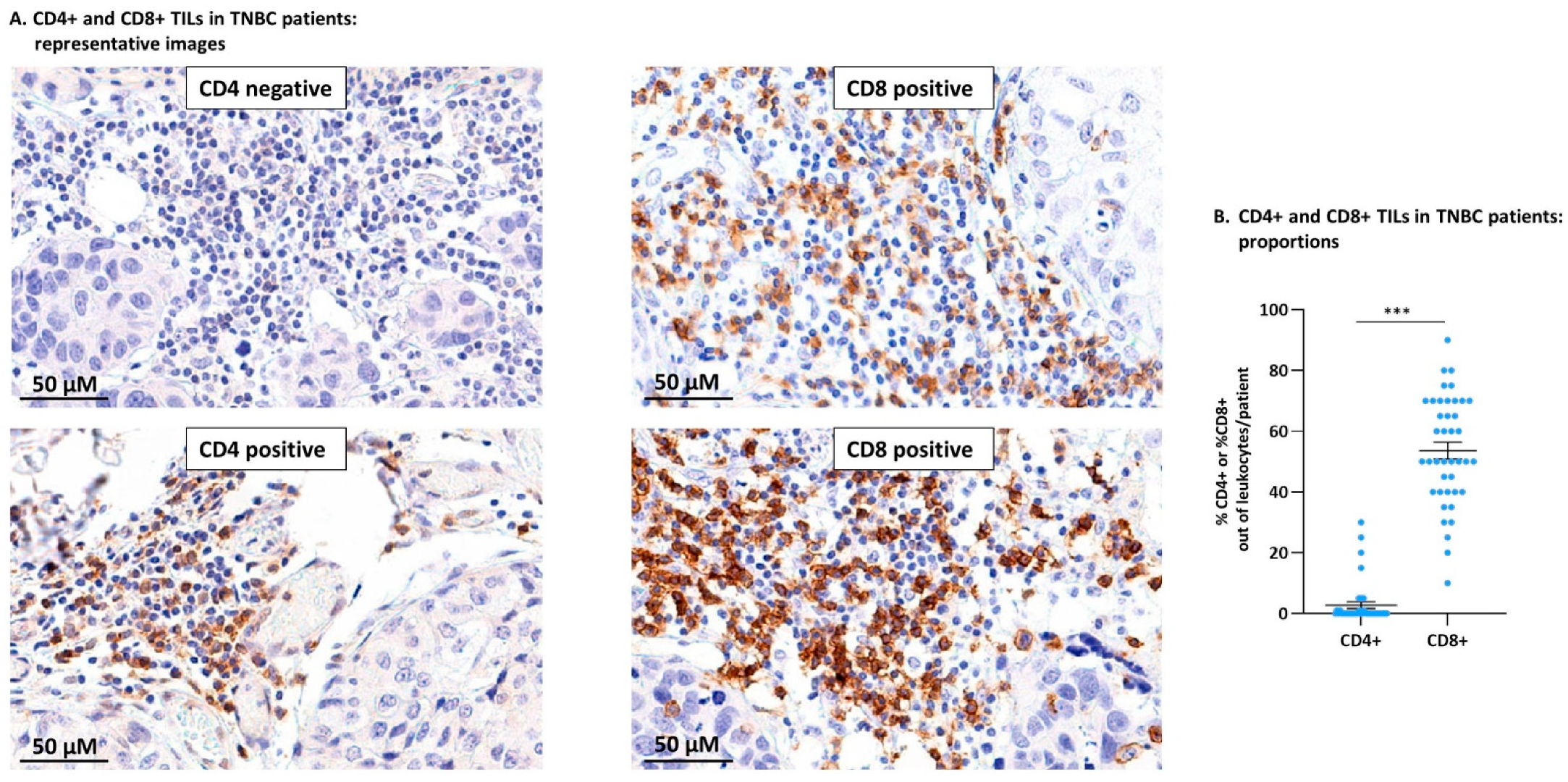
3.1. TNBC Patient Tumors Contained CD4+ TILs and Were Particularly Enriched with CD8+ TILs
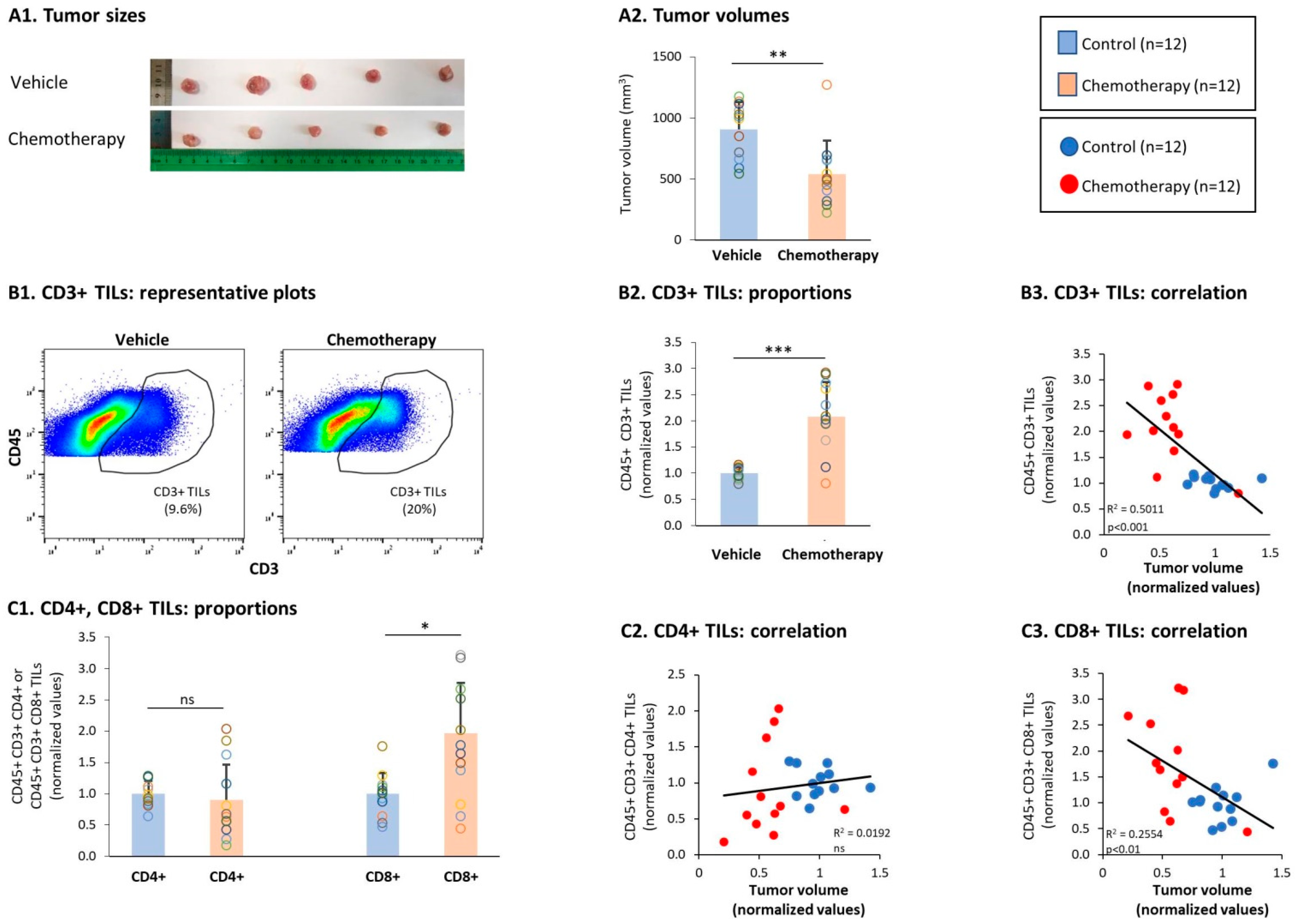
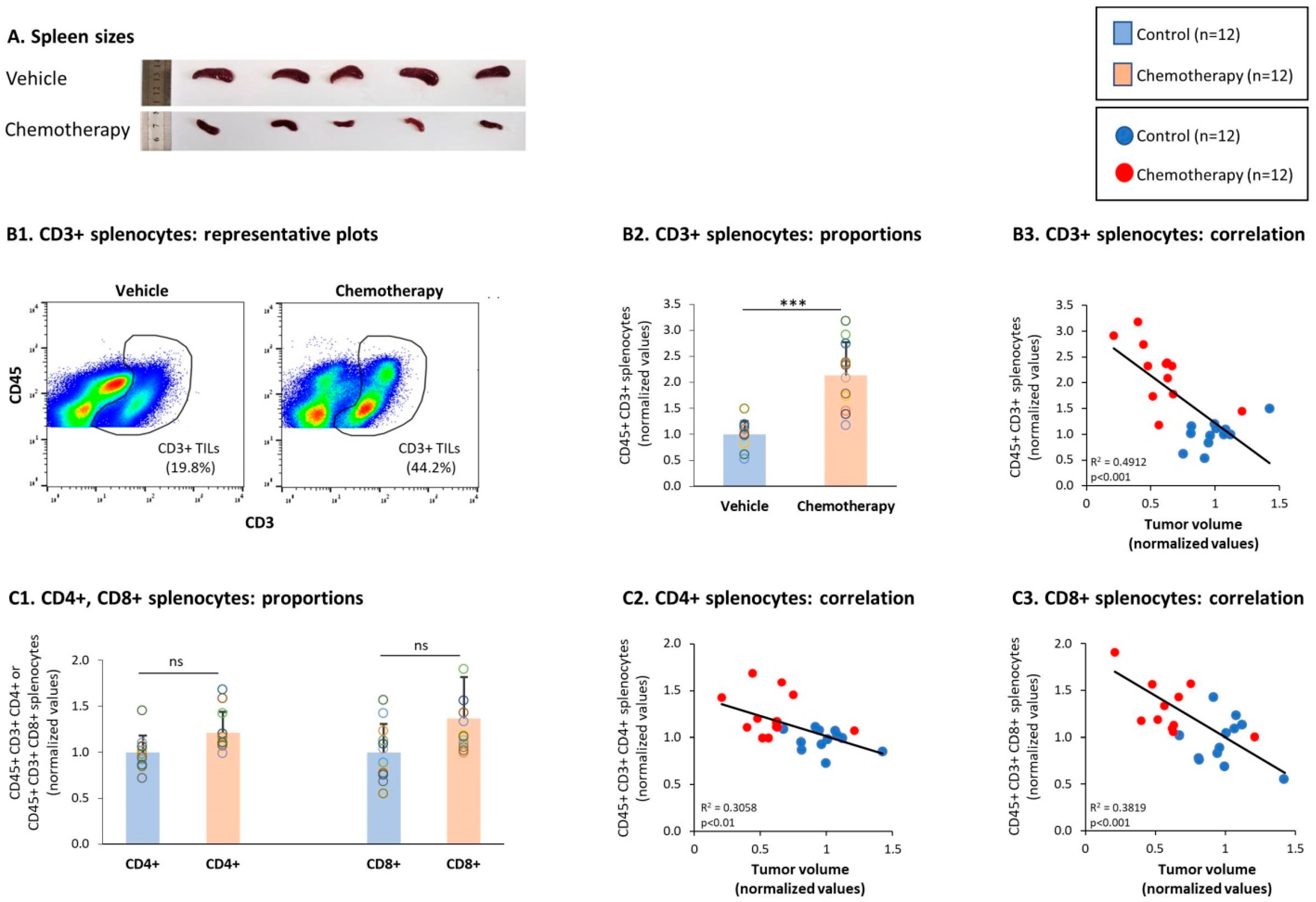
3.2. General Characteristics of T Cell Subsets in TILs and Splenocytes of TNBC-Bearing Mice
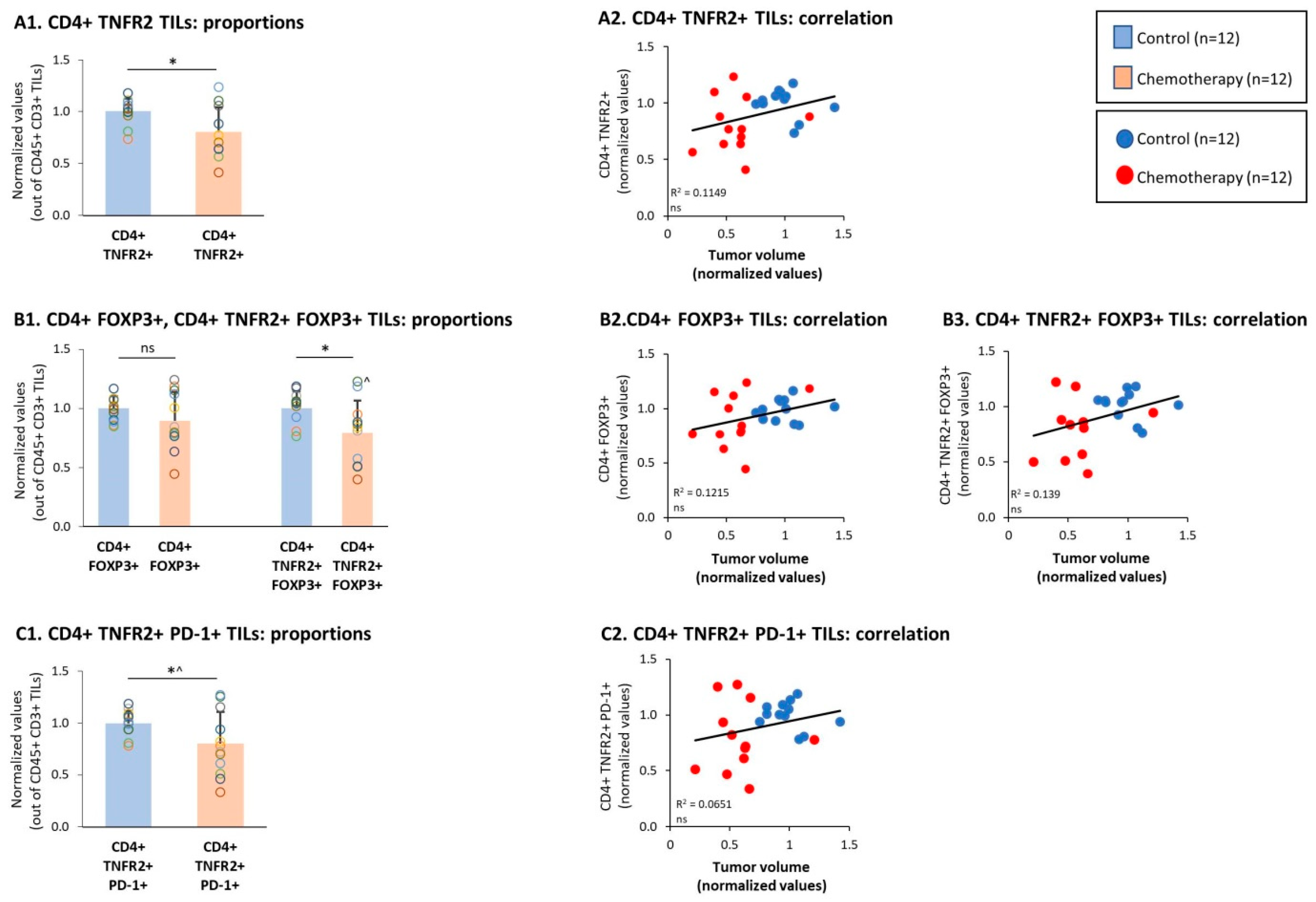
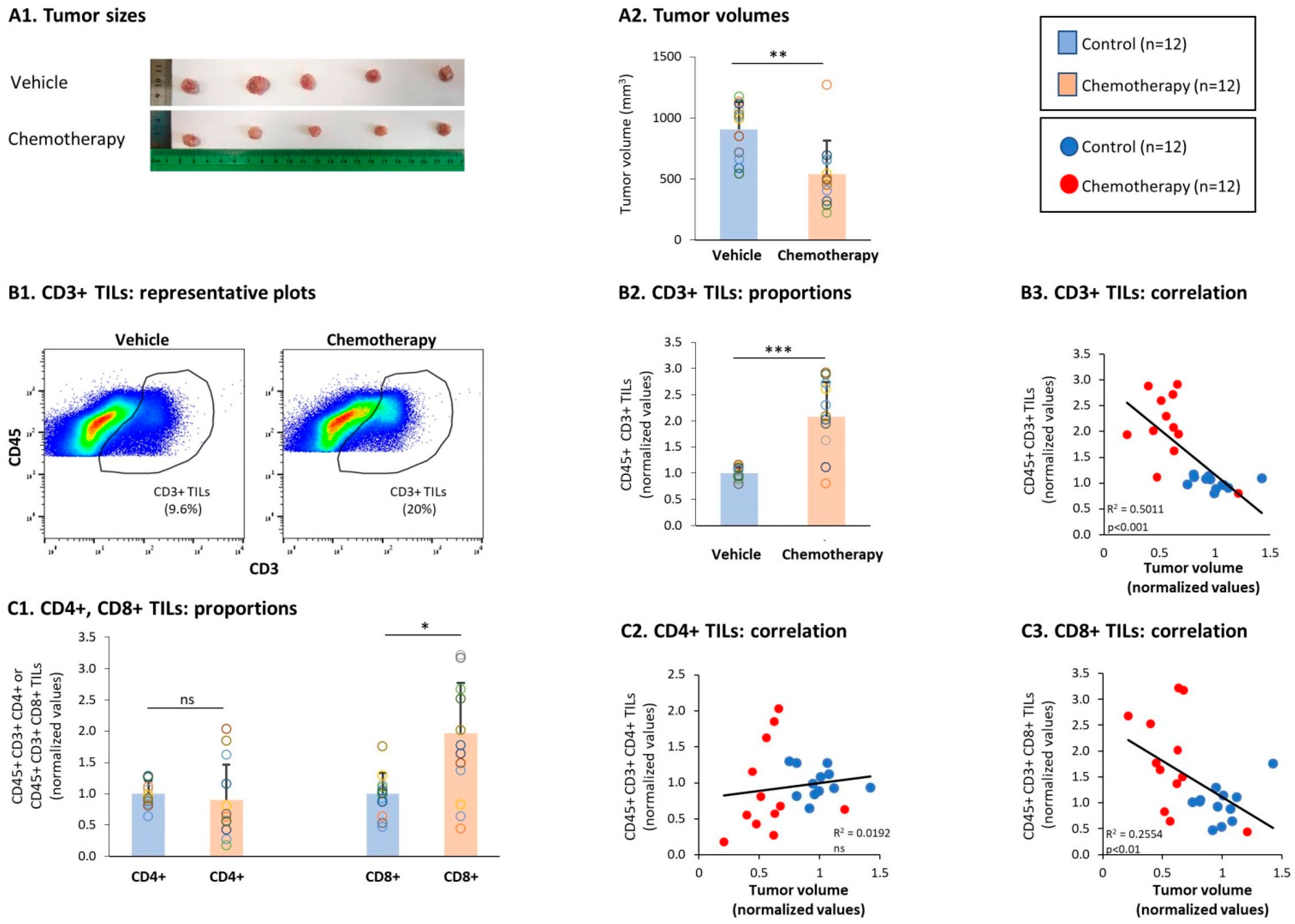
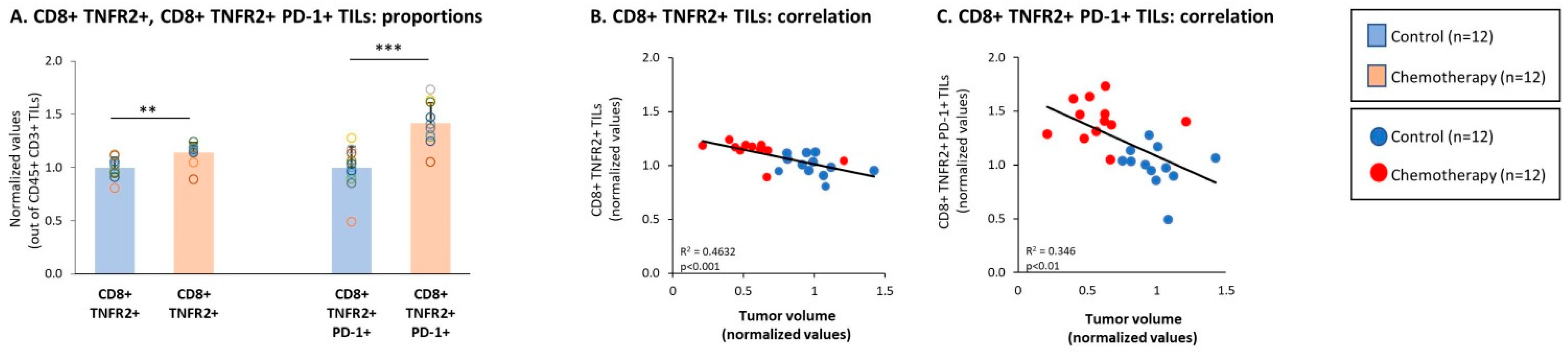
3.3. Chemotherapy Led to Down-Regulation of CD4+ TNFR2+ TIL Subsets and Up-Regulation of CD8+ TNFR2+ TIL Subsets in Tumors of TNBC-Bearing Mice
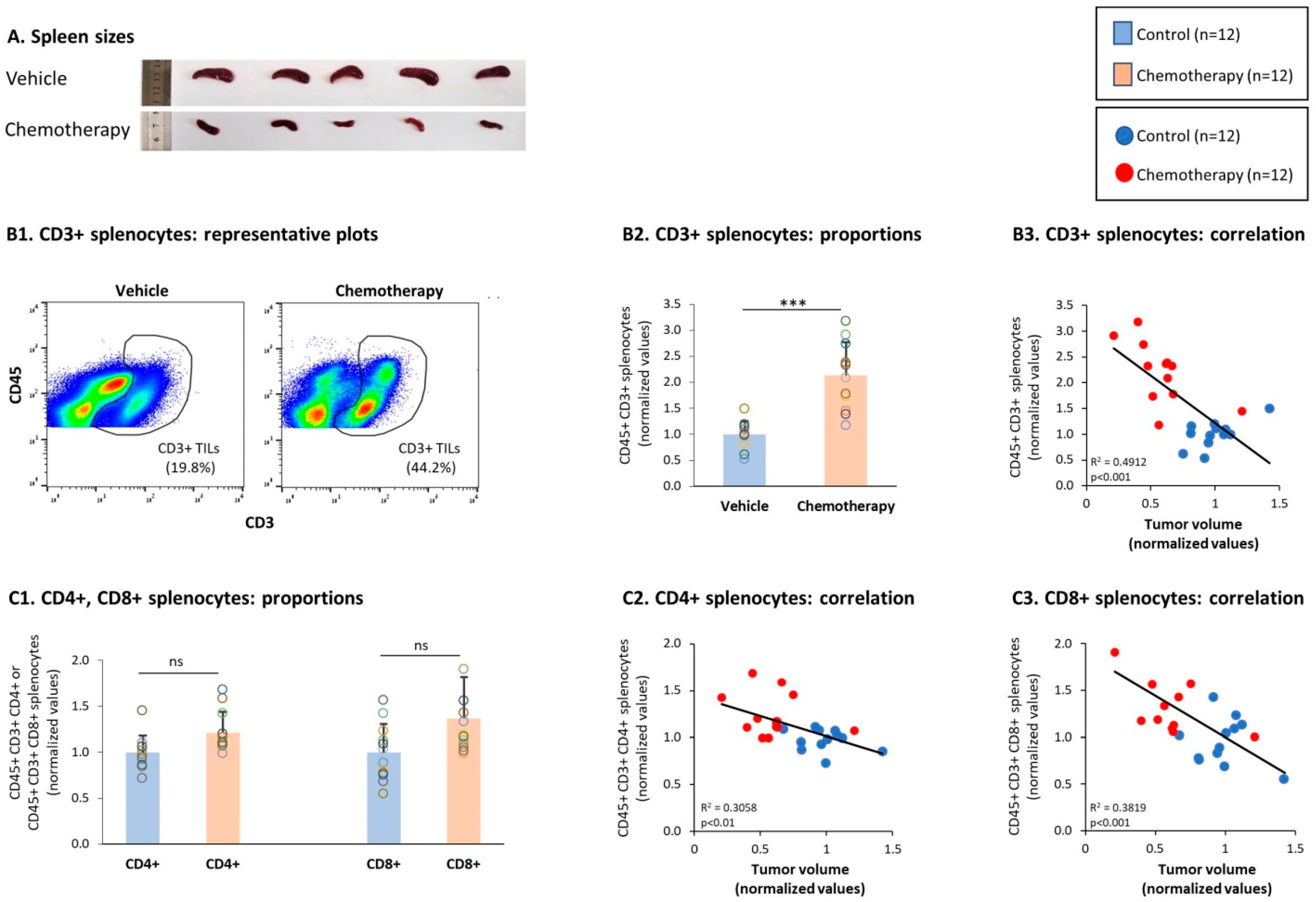
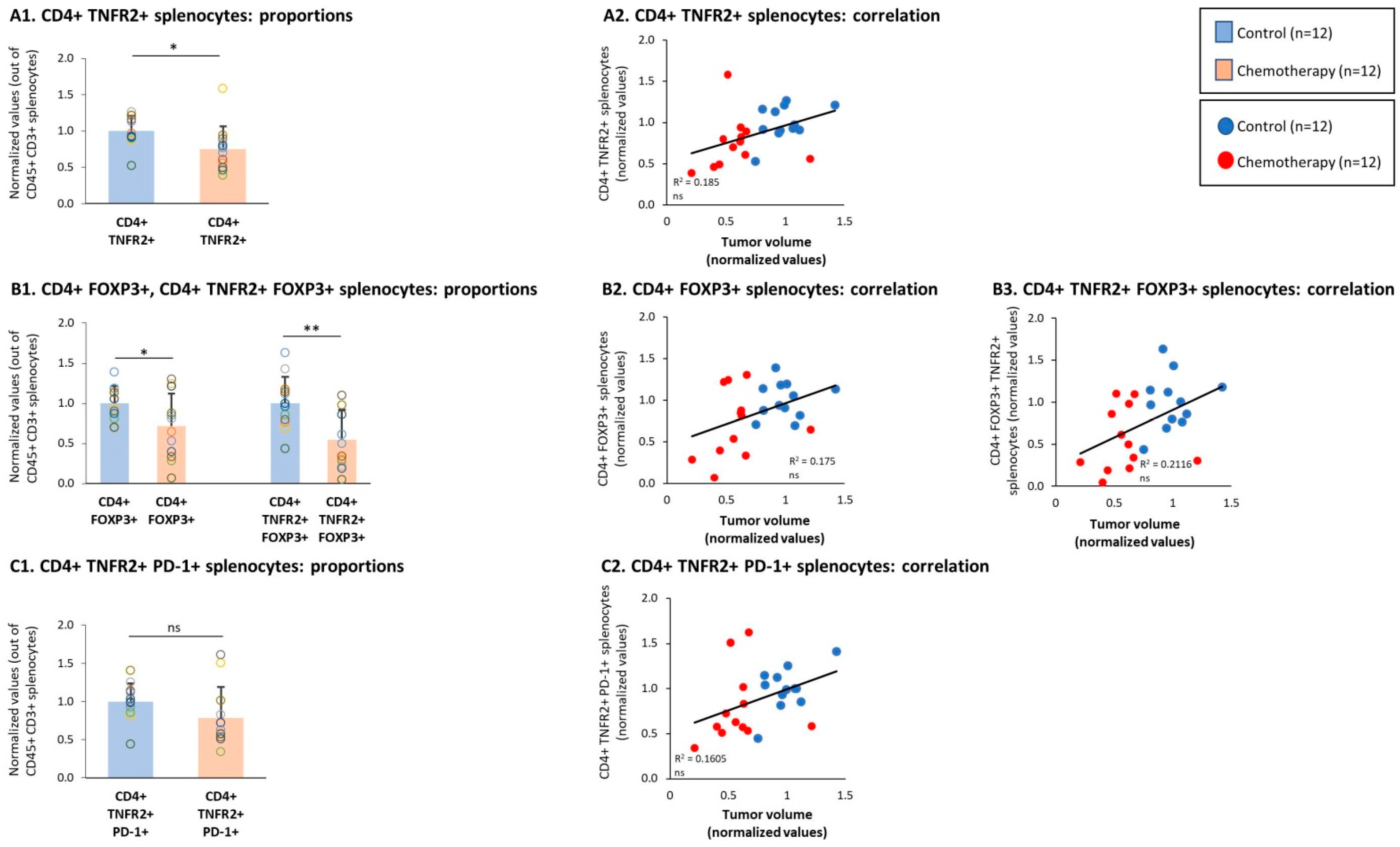
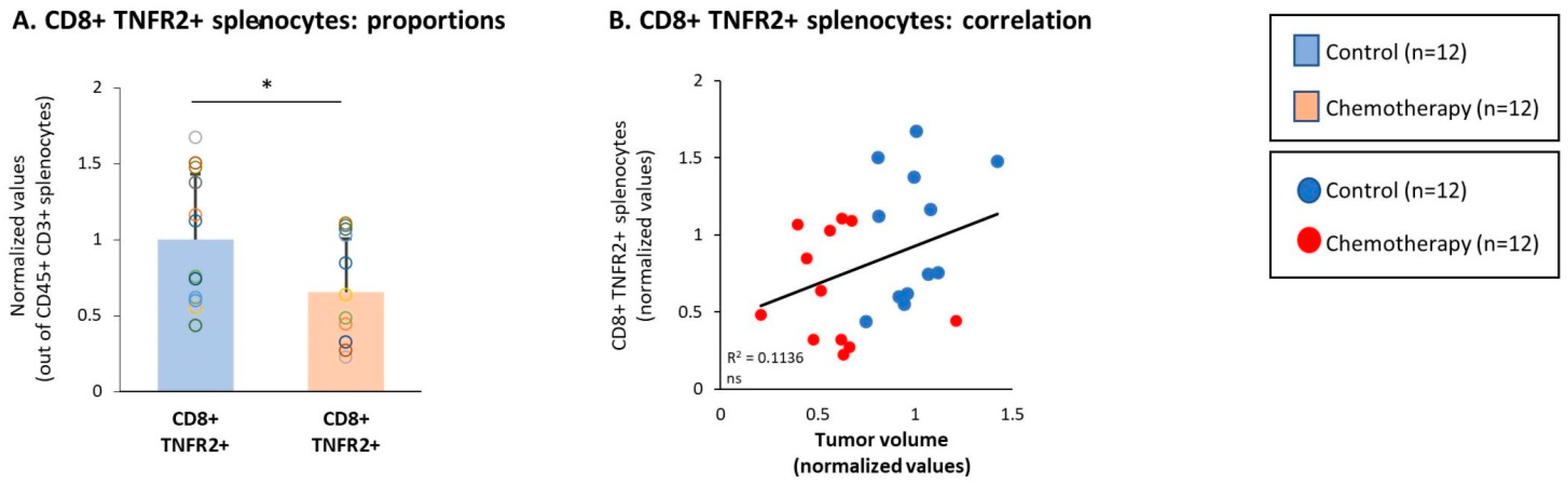
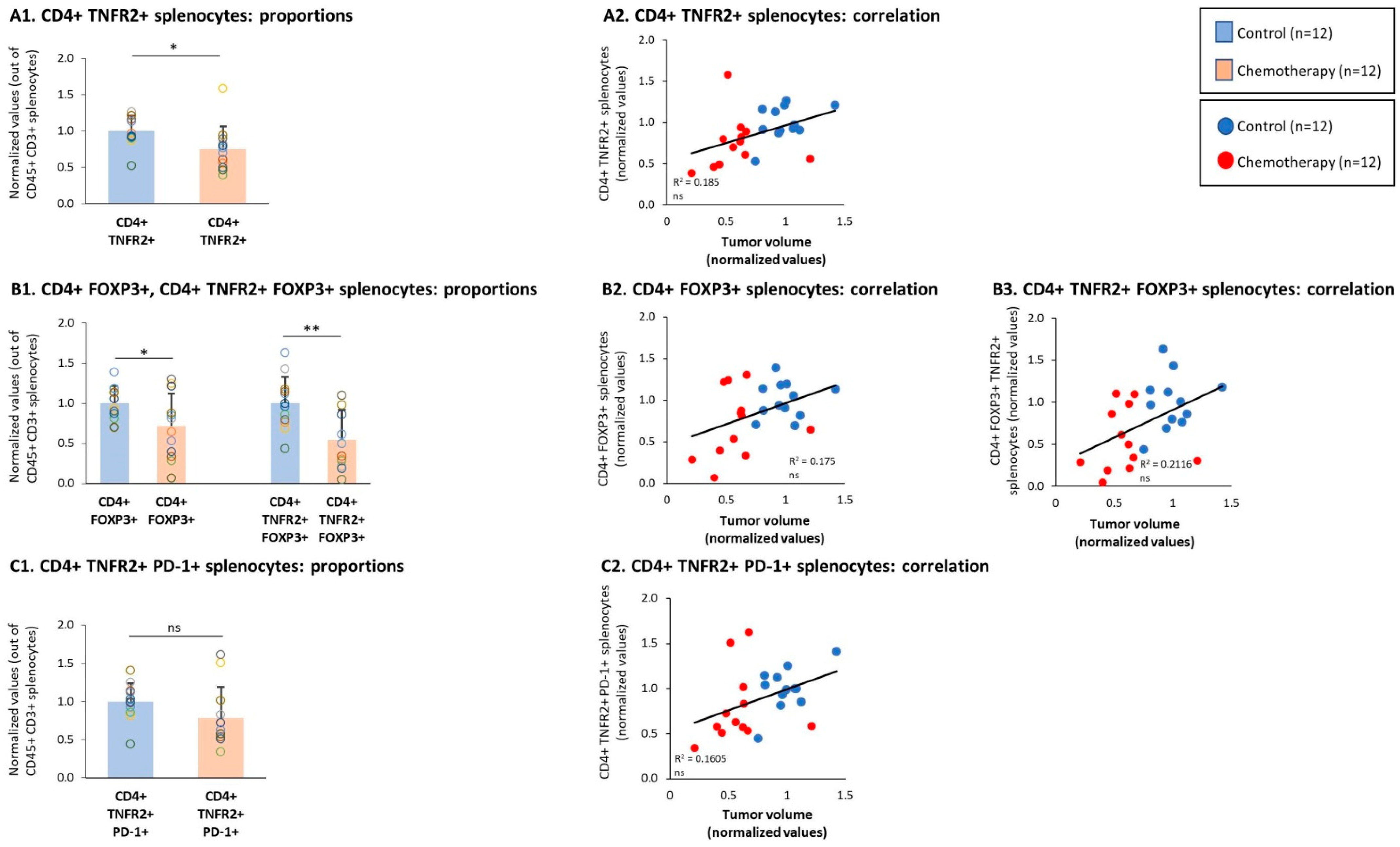
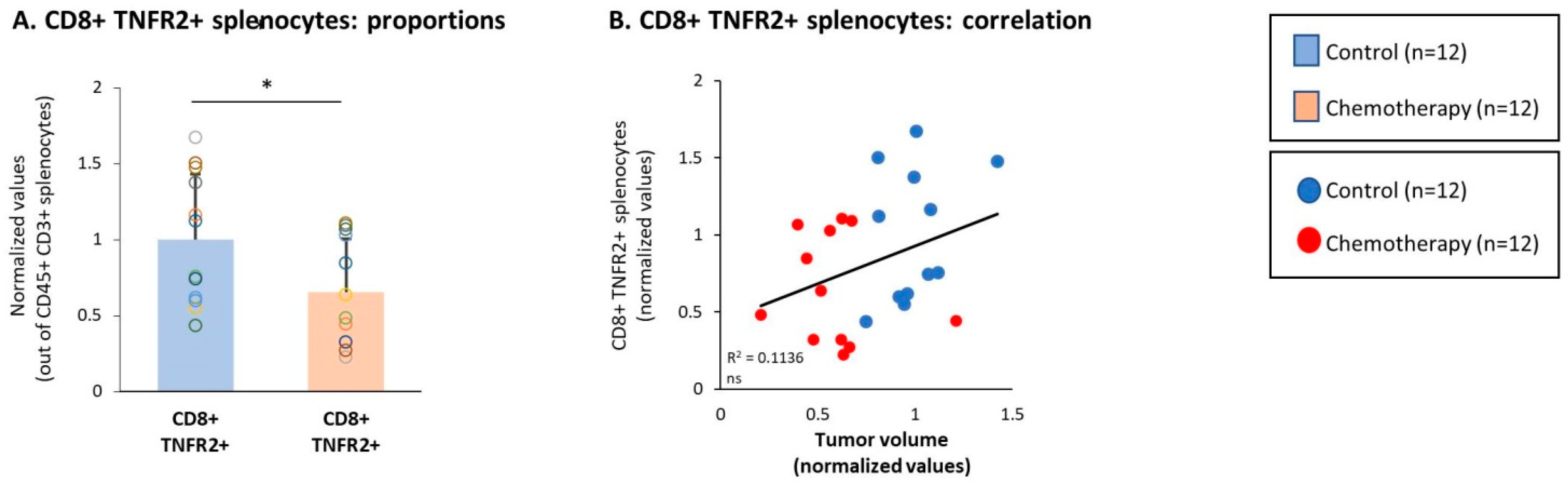
3.4. Chemotherapy Led to Down-Regulation of CD4+ TNFR2+ Subsets in Spleens of TNBC-Bearing Mice, but Did Not Give Rise to Up-Regulation of CD8+ TNFR2+ Subsets in Their Spleens
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eroles, P.; Bosch, A.; Pérez-Fidalgo, J.A.; Lluch, A. Molecular biology in breast cancer: Intrinsic subtypes and signaling pathways. Cancer Treat. Rev. 2012, 38, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Gerratana, L.; Fanotto, V.; Bonotto, M.; Bolzonello, S.; Andreetta, C.; Moroso, S.; Pascoletti, G.; Fasola, G.; Puglisi, F. Pattern of metastatic spread and prognosis of breast cancer biologic subtypes. J. Clin. Oncol. 2014, 32, e12532. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Domchek, S.M.; Clark, A.S. Immunotherapy for breast cancer: What are we missing? Clin. Cancer Res. 2017, 23, 2640–2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartkopf, A.D.; Taran, F.-A.; Wallwiener, M.; Walter, C.B.; Krämer, B.; Grischke, E.-M.; Brucker, S.Y. PD-1 and PD-L1 immune checkpoint blockade to treat breast cancer. Breast Care 2016, 11, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Mediratta, K.; El-Sahli, S.; D’Costa, V.; Wang, L. Current progresses and challenges of immunotherapy in triple-negative breast cancer. Cancers 2020, 12, 3529. [Google Scholar] [CrossRef] [PubMed]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 2018, 167, 671–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef]
- Emens, L.A.; Cruz, C.; Eder, J.P.; Braiteh, F.; Chung, C.; Tolaney, S.M.; Kuter, I.; Nanda, R.; Cassier, P.A.; Delord, J.P.; et al. Long-term clinical outcomes and biomarker analyses of atezolizumab therapy for patients with metastatic triple-negative breast cancer: A phase 1 study. JAMA Oncol. 2019, 5, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Makhoul, I.; Atiq, M.; Alwbari, A.; Kieber-Emmons, T. Breast cancer immunotherapy: An update. Breast Cancer 2018, 12, 1178223418774802. [Google Scholar] [CrossRef] [PubMed]
- Szekely, B.; Silber, A.L.; Pusztai, L. New therapeutic strategies for triple-negative breast cancer. Oncology 2017, 31, 130–137. [Google Scholar]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Krishnamurti, U.; Wetherilt, C.S.; Yang, J.; Peng, L.; Li, X. Tumor-infiltrating lymphocytes are significantly associated with better overall survival and disease-free survival in triple-negative but not estrogen receptor-positive breast cancers. Hum. Pathol. 2017, 64, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Kubo, M.; Yamaguchi, R.; Nishimura, R.; Osako, T.; Arima, N.; Okumura, Y.; Okido, M.; Yamada, M.; Kai, M.; et al. The combination of PD-L1 expression and decreased tumor-infiltrating lymphocytes is associated with a poor prognosis in triple-negative breast cancer. Oncotarget 2017, 8, 15584–15592. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.; Goldstein, L.J.; Sparano, J.A.; Demaria, S.; Badve, S.S. Tumor infiltrating lymphocytes (TILs) improve prognosis in patients with triple negative breast cancer (TNBC). Oncoimmunology 2015, 4, e985930. [Google Scholar] [CrossRef] [Green Version]
- Loi, S.; Michiels, S.; Salgado, R.; Sirtaine, N.; Jose, V.; Fumagalli, D.; Kellokumpu-Lehtinen, P.L.; Bono, P.; Kataja, V.; Desmedt, C.; et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Ann. Oncol. 2014, 25, 1544–1550. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hamano, R.; Subleski, J.J.; Hurwitz, A.A.; Howard, O.M.; Oppenheim, J.J. Expression of costimulatory TNFR2 induces resistance of CD4+FoxP3- conventional T cells to suppression by CD4+FoxP3+ regulatory T cells. J. Immunol. 2010, 185, 174–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Subleski, J.J.; Hamano, R.; Howard, O.M.; Wiltrout, R.H.; Oppenheim, J.J. Co-expression of TNFR2 and CD25 identifies more of the functional CD4+FOXP3+ regulatory T cells in human peripheral blood. Eur. J. Immunol. 2010, 40, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Plebanski, M. Editorial: The Role of TNF-TNFR2 Signal in Immunosuppressive Cells and Its Therapeutic Implications. Front. Immunol. 2019, 10, 2126. [Google Scholar] [CrossRef]
- Qu, Y.; Zhao, G.; Li, H. Forward and reverse signaling mediated by transmembrane tumor necrosis factor-alpha and tnf receptor 2: Potential roles in an immunosuppressive tumor microenvironment. Front. Immunol. 2017, 8, 1675. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.S.; Mistry, B.; Guillard, S.; Ulrichsen, J.C.; Sandercock, A.M.; Wang, J.; Gonzalez-Munoz, A.; Parmentier, J.; Black, C.; Soden, J.; et al. Phenotypic screening reveals TNFR2 as a promising target for cancer immunotherapy. Oncotarget 2016, 7, 68278–68291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubrano di Ricco, M.; Ronin, E.; Collares, D.; Divoux, J.; Gregoire, S.; Wajant, H.; Gomes, T.; Grinberg-Bleyer, Y.; Baud, V.; Marodon, G.; et al. Tumor necrosis factor receptor family costimulation increases regulatory T-cell activation and function via NF-kappaB. Eur. J. Immunol. 2020, 50, 972–985. [Google Scholar] [CrossRef] [Green Version]
- Torrey, H.; Kühtreiber, W.M.; Okubo, Y.; Tran, L.; Case, K.; Zheng, H.; Vanamee, E.; Faustman, D.L. A novel TNFR2 agonist antibody expands highly potent regulatory T cells. Sci. Signal. 2020, 13, eaba9600. [Google Scholar] [CrossRef]
- MacEwan, D.J. TNF ligands and receptors–A matter of life and death. Br. J. Pharmacol. 2002, 135, 855–875. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, T.; Mitoma, H.; Harashima, S.; Tsukamoto, H.; Shimoda, T. Transmembrane TNF-alpha: Structure, function and interaction with anti-TNF agents. Rheumatology 2010, 49, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grell, M.; Douni, E.; Wajant, H.; Lohden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [Green Version]
- Faustman, D.L.; Davis, M. TNF receptor 2 and disease: Autoimmunity and regenerative medicine. Front. Immunol. 2003, 4, 478. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Oppenheim, J.J. Resolving the identity myth: Key markers of functional CD4+FoxP3+ regulatory T cells. Int. Immunopharmacol. 2011, 11, 1489–1496. [Google Scholar] [CrossRef] [Green Version]
- Nie, Y.; He, J.; Shirota, H.; Trivett, A.L.; Yang Klinman, D.M.; Oppenheim, J.J.; Chen, X. Blockade of TNFR2 signaling enhances the immunotherapeutic effect of CpG ODN in a mouse model of colon cancer. Sci. Signal. 2018, 11, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Subleski, J.J.; Kopf, H.; Howard, O.M.; Mannel, D.N.; Oppenheim, J.J. Cutting edge: Expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: Applicability to tumor-infiltrating T regulatory cells. J. Immunol. 2008, 180, 6467–6471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, F.; Du, R.; Wei, F.; Zhao, H.; Yu, J.; Wang, C.; Zhan, Z.; Ding, T.; Ren, X.; Chen, X.; et al. Expression of TNFR2 by regulatory T cells in peripheral blood is correlated with clinical pathology of lung cancer patients. Cancer Immunol. Immunother. 2015, 64, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.Y.; Lin, Y.C.; Chiang, J.M.; Mahalingam, J.; Su, S.H.; Huang, C.T.; Chen, W.T.; Huang, C.H.; Jeng, W.J.; Chen, Y.C.; et al. Blockade of TNF-alpha signaling benefits cancer therapy by suppressing effector regulatory T cell expansion. Oncoimmunology 2015, 4, e1040215. [Google Scholar] [CrossRef]
- Ham, B.; Wang, N.; D'Costa, Z.; Fernandez, M.C.; Bourdeau, F.; Auguste, P.; Illemann, M.; Eefsen, R.L.; Hoyer-Hansen, G.; Vainer, B.; et al. TNF Receptor-2 facilitates an immunosuppressive microenvironment in the liver to promote the colonization and growth of hepatic metastases. Cancer Res. 2015, 75, 5235–5247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Chen, J.; Bi, E.; Zhao, Y.; Qin, T.; Wang, Y.; Wang, A.; Gao, S.; Yi, Q.; Wang, S. TNF-alpha enhances Th9 cell differentiation and antitumor immunity via TNFR2-dependent pathways. J. Immunother. Cancer 2019, 7, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wajant, H.; Beilhack, A. Targeting regulatory t cells by addressing tumor necrosis factor and its receptors in allogeneic hematopoietic cell transplantation and cancer. Front. Immunol. 2019, 10, 2040. [Google Scholar] [CrossRef] [PubMed]
- Torrey, H.; Butterworth, J.; Mera, T.; Okubo, Y.; Wang, L.; Baum, D.; Defusco, A.; Plager, S.; Warden, S.; Huang, D.; et al. Targeting TNFR2 with antagonistic antibodies inhibits proliferation of ovarian cancer cells and tumor-associated Tregs. Sci. Signal. 2017, 10, 462. [Google Scholar] [CrossRef]
- Al-Hatamleh, M.A.I.; Ahmad, S.; Boer, J.C.; Lim, J.; Chen, X.; Plebanski, M.; Mohamud, R. A perspective review on the role of nanomedicine in the modulation of TNF-TNFR2 axis in breast cancer immunotherapy. J. Oncol. 2019, 2019, 6313242. [Google Scholar] [CrossRef]
- Vanamee, E.S.; Faustman, D.L. TNFR2: A novel target for cancer immunotherapy. Trends. Mol. Med. 2017, 23, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Oppenheim, J.J. Targeting TNFR2, an immune checkpoint stimulator and oncoprotein, is a promising treatment for cancer. Sci. Signal. 2017, 10, 462. [Google Scholar] [CrossRef]
- Sheng, Y.; Li, F.; Qin, Z. TNF receptor 2 makes tumor necrosis factor a friend of tumors. Front. Immunol. 2018, 9, 1170. [Google Scholar] [CrossRef] [Green Version]
- Case, K.; Tran, L.; Yang, M.; Zheng, H.; Kuhtreiber, W.M.; Faustman, D.L. TNFR2 blockade alone or in combination with PD-1 blockade shows therapeutic efficacy in murine cancer models. J. Leukoc. Biol. 2020, 107, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Dadiani, M.; Necula, D.; Kahana-Edwin, S.; Oren, N.; Baram, T.; Marin, I.; Morzaev-Sulzbach, D.; Pavlovski, A.; Balint-Lahat, N.; Anafi, L.; et al. Ben-Baruch, A. TNFR2+ TILs are significantly associated with improved survival in triple-negative breast cancer patients. Cancer Immunol. Immunother. 2020, 69, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat. Rev. Clin. Oncol. 2011, 8, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dermawan, K.; Jin, M.; Liu, R.; Zheng, H.; Xu, L.; Zhang, Y.; Cai, Y.; Chu, Y.; Xiong, S. Differential impairment of regulatory T cells rather than effector T cells by paclitaxel-based chemotherapy. Clin. Immunol. 2008, 129, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Duan, X.F.; Wang, L.P.; Xu, Y.J.; Huang, L.; Zhang, T.F.; Liu, J.Y.; Li, F.; Zhang, Z.; Yue, D.L.; et al. Selective depletion of regulatory T cell subsets by docetaxel treatment in patients with nonsmall cell lung cancer. J. Immunol. Res. 2014, 2014, 286170. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Zielinski, C.C. Optimal strategies for the treatment of metastatic triple-negative breast cancer with currently approved agents. Ann. Oncol. 2012, 23, 46–51. [Google Scholar] [CrossRef]
- Park, J.H.; Ahn, J.H.; Kim, S.B. How shall we treat early triple-negative breast cancer (TNBC): From the current standard to upcoming immuno-molecular strategies. ESMO Open 2018, 3, e000357. [Google Scholar] [CrossRef] [Green Version]
- Tam, E.M.; Fulton, R.B.; Sampson, J.F.; Muda, M.; Camblin, A.; Richards, J.; Koshkaryev, A.; Tang, J.; Kurella, V.; Jiao, Y.; et al. Antibody-mediated targeting of TNFR2 activates CD8(+) T cells in mice and promotes antitumor immunity. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Ye, L.L.; Wei, X.S.; Zhang, M.; Niu, Y.R.; Zhou, Q. The significance of tumor necrosis factor receptor type II in CD8(+) Regulatory T cells and CD8(+) effector T cells. Front. Immunol. 2018, 9, 583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.Y.; Teh, S.J.; Yang, J.; Chow, M.T.; Teh, H.S. TNFR2-deficient memory CD8 T cells provide superior protection against tumor cell growth. J. Immunol. 2009, 183, 6051–6057. [Google Scholar] [CrossRef] [Green Version]
- DeBerge, M.P.; Ely, K.H.; Wright, P.F.; Thorp, E.B.; Enelow, R.I. Shedding of TNF receptor 2 by effector CD8(+) T cells by ADAM17 is important for regulating TNF-alpha availability during influenza infection. J. Leukoc. Biol. 2015, 98, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Yeong, J.; Lim, J.C.T.; Lee, B.; Li, H.; Ong, C.C.H.; Thike, A.A.; Yeap, W.H.; Yang, Y.; Lim, A.Y.H.; Tay, T.K.Y.; et al. Prognostic value of CD8 + PD-1+ immune infiltrates and PDCD1 gene expression in triple negative breast cancer. J. Immunother. Cancer 2019, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Dill, E.A.; Dillon, P.M.; Bullock, T.N.; Mills, A.M. IDO expression in breast cancer: An assessment of 281 primary and metastatic cases with comparison to PD-L1. Mod. Pathol. 2018, 31, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Zarour, H.M. Reversing T-cell dysfunction and exhaustion in cancer. Clin. Cancer Res. 2016, 22, 1856–1864. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-cell exhaustion: Characteristics, causes and conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Becht, E.; Vano, Y.; Petitprez, F.; Lacroix, L.; Validire, P.; Sanchez-Salas, R.; Ingels, A.; Oudard, S.; Moatti, A.; et al. Tumor-infiltrating and peripheral blood t-cell immunophenotypes predict early relapse in localized clear cell renal cell carcinoma. Clin. Cancer Res. 2017, 23, 4416–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef]
- DuPre, S.A.; Hunter, K.W., Jr. Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: Association with tumor-derived growth factors. Exp. Mol. Pathol. 2007, 82, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, M.R.; Marone, G.; Mantovani, A. Cancer inflammation and cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Reza, I.; Diaz, L.; Garcia-Becerra, R. Preclinical and clinical aspects of TNF-alpha and its receptors TNFR1 and TNFR2 in breast cancer. J. Biomed. Sci. 2017, 24, 90. [Google Scholar] [CrossRef] [Green Version]
- Cruceriu, D.; Baldasici, O.; Balacescu, O.; Berindan-Neagoe, I. The dual role of tumor necrosis factor-alpha (TNF-alpha) in breast cancer: Molecular insights and therapeutic approaches. Cell Oncol. 2020, 43, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ben-Baruch, A. (2020) Partners in crime: TNFalpha-based networks promoting cancer progression. Cancer Immunol. Immunother. 2020, 69, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Chavey, C.; Bibeau, F.; Gourgou-Bourgade, S.; Burlinchon, S.; Boissiere, F.; Laune, D.; Roques, S.; Lazennec, G. Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 2007, 9, R15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, G.; Ofri-Shahak, M.; Haas, I.; Yaal-Hahoshen, N.; Leider-Trejo, L.; Leibovich-Rivkin, T.; Weitzenfeld, P.; Meshel, T.; Shabtai, E.; Gutman, M.; et al. Inflammatory mediators in breast cancer: Coordinated expression of TNFalpha & IL-1beta with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer 2011, 11, 130. [Google Scholar]
- Jin, L.; Yuan, R.Q.; Fuchs, A.; Yao, Y.; Joseph, A.; Schwall, R.; Schnitt, S.J.; Guida, A.; Hastings, H.M.; Andres, J.; et al. Expression of interleukin-1beta in human breast carcinoma. Cancer 1997, 80, 421–434. [Google Scholar] [CrossRef]
- Emens, L.A.; Middleton, G. The interplay of immunotherapy and chemotherapy: Harnessing potential synergies. Cancer Immunol. Res. 2015, 3, 436–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladoire, S.; Arnould, L.; Apetoh, L.; Coudert, B.; Martin, F.; Chauffert, B.; Fumoleau, P.; Ghiringhelli, F. Pathologic complete response to neoadjuvant chemotherapy of breast carcinoma is associated with the disappearance of tumor-infiltrating foxp3+ regulatory T cells. Clin. Cancer Res. 2008, 14, 2413–2420. [Google Scholar] [CrossRef] [Green Version]
- Kaewkangsadan, V.; Verma, C.; Eremin, J.M.; Cowley, G.; Ilyas, M.; Eremin, O. Tumour-draining axillary lymph nodes in patients with large and locally advanced breast cancers undergoing neoadjuvant chemotherapy (NAC): The crucial contribution of immune cells (effector, regulatory) and cytokines (Th1, Th2) to immune-mediated tumour cell death induced by NAC. BMC Cancer 2018, 18, 123. [Google Scholar]
- Gonda, K.; Shibata, M.; Ohtake, T.; Matsumoto, Y.; Tachibana, K.; Abe, N.; Ohto, H.; Sakurai, K.; Takenoshita, S. Myeloid-derived suppressor cells are increased and correlated with type 2 immune responses, malnutrition, inflammation, and poor prognosis in patients with breast cancer. Oncol. Lett. 2017, 14, 1766–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathological Characteristics of TNBC Cases (n = 41) | ||
|---|---|---|
| Median Age at Dx (Range), Years: 53 (30–84) | ||
| Number (n) | Percent (%) | |
| Tumor stage | ||
| pT1 | 26 | 63.4 |
| pT2 | 14 | 34.1 |
| pT3 | 1 | 2.4 |
| Lymph node involvement | ||
| pN0 | 28 | 68.3 |
| pN1 | 11 | 26.8 |
| pNx | 2 | 4.9 |
| Tumor grade | ||
| 2 | 5 | 12.2 |
| 3 | 35 | 85.4 |
| NA | 1 | 2.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baram, T.; Erlichman, N.; Dadiani, M.; Balint-Lahat, N.; Pavlovski, A.; Meshel, T.; Morzaev-Sulzbach, D.; Gal-Yam, E.N.; Barshack, I.; Ben-Baruch, A. Chemotherapy Shifts the Balance in Favor of CD8+ TNFR2+ TILs in Triple-Negative Breast Tumors. Cells 2021, 10, 1429. https://doi.org/10.3390/cells10061429
Baram T, Erlichman N, Dadiani M, Balint-Lahat N, Pavlovski A, Meshel T, Morzaev-Sulzbach D, Gal-Yam EN, Barshack I, Ben-Baruch A. Chemotherapy Shifts the Balance in Favor of CD8+ TNFR2+ TILs in Triple-Negative Breast Tumors. Cells. 2021; 10(6):1429. https://doi.org/10.3390/cells10061429
Chicago/Turabian StyleBaram, Tamir, Nofar Erlichman, Maya Dadiani, Nora Balint-Lahat, Anya Pavlovski, Tsipi Meshel, Dana Morzaev-Sulzbach, Einav Nili Gal-Yam, Iris Barshack, and Adit Ben-Baruch. 2021. "Chemotherapy Shifts the Balance in Favor of CD8+ TNFR2+ TILs in Triple-Negative Breast Tumors" Cells 10, no. 6: 1429. https://doi.org/10.3390/cells10061429
APA StyleBaram, T., Erlichman, N., Dadiani, M., Balint-Lahat, N., Pavlovski, A., Meshel, T., Morzaev-Sulzbach, D., Gal-Yam, E. N., Barshack, I., & Ben-Baruch, A. (2021). Chemotherapy Shifts the Balance in Favor of CD8+ TNFR2+ TILs in Triple-Negative Breast Tumors. Cells, 10(6), 1429. https://doi.org/10.3390/cells10061429