
THAP9 Transposase Cleaves DNA via Conserved Acidic Residues in an RNaseH-Like Domain


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Multiple Sequence Alignment and Secondary Structure Prediction
2.2.2. Homology Modeling, Docking and MD Simulations
2.2.3. Site-Directed Mutagenesis
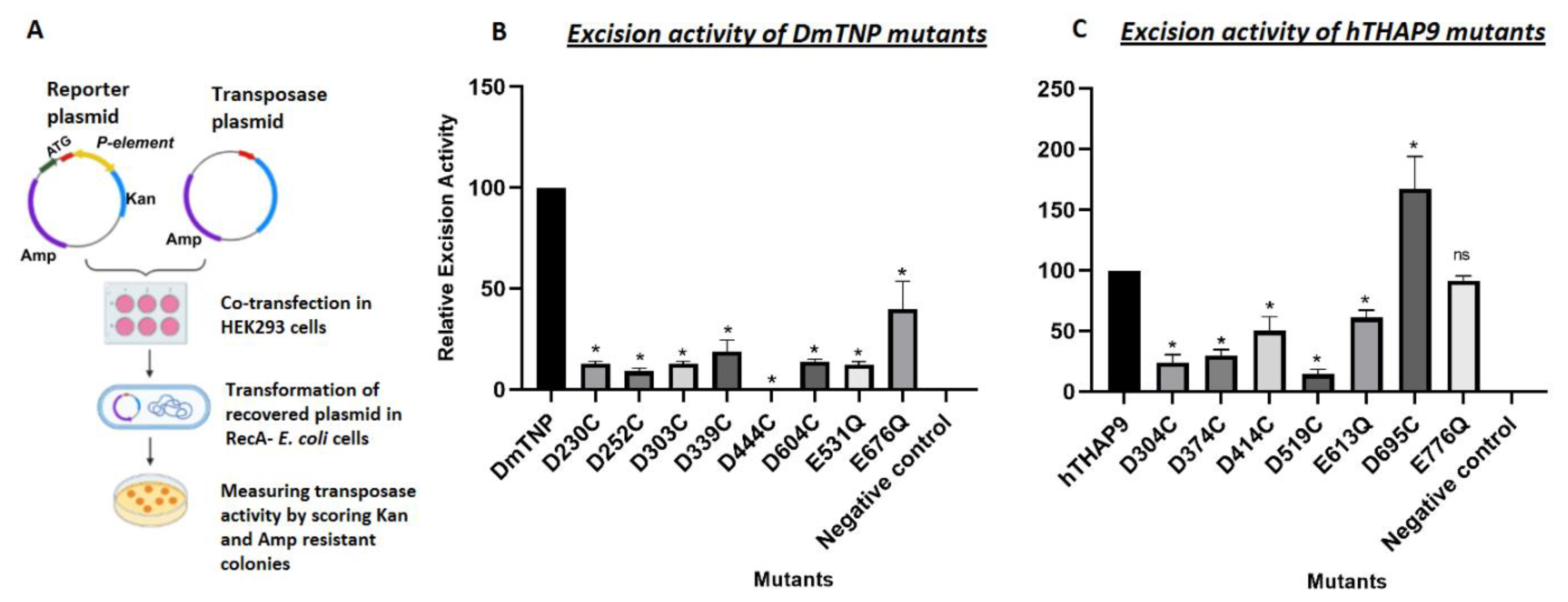
2.2.4. In Vivo P Element Excision Assay
Relative Excision activity= (Excision activity of mutant × 100)/Excision activity of wild type transposase
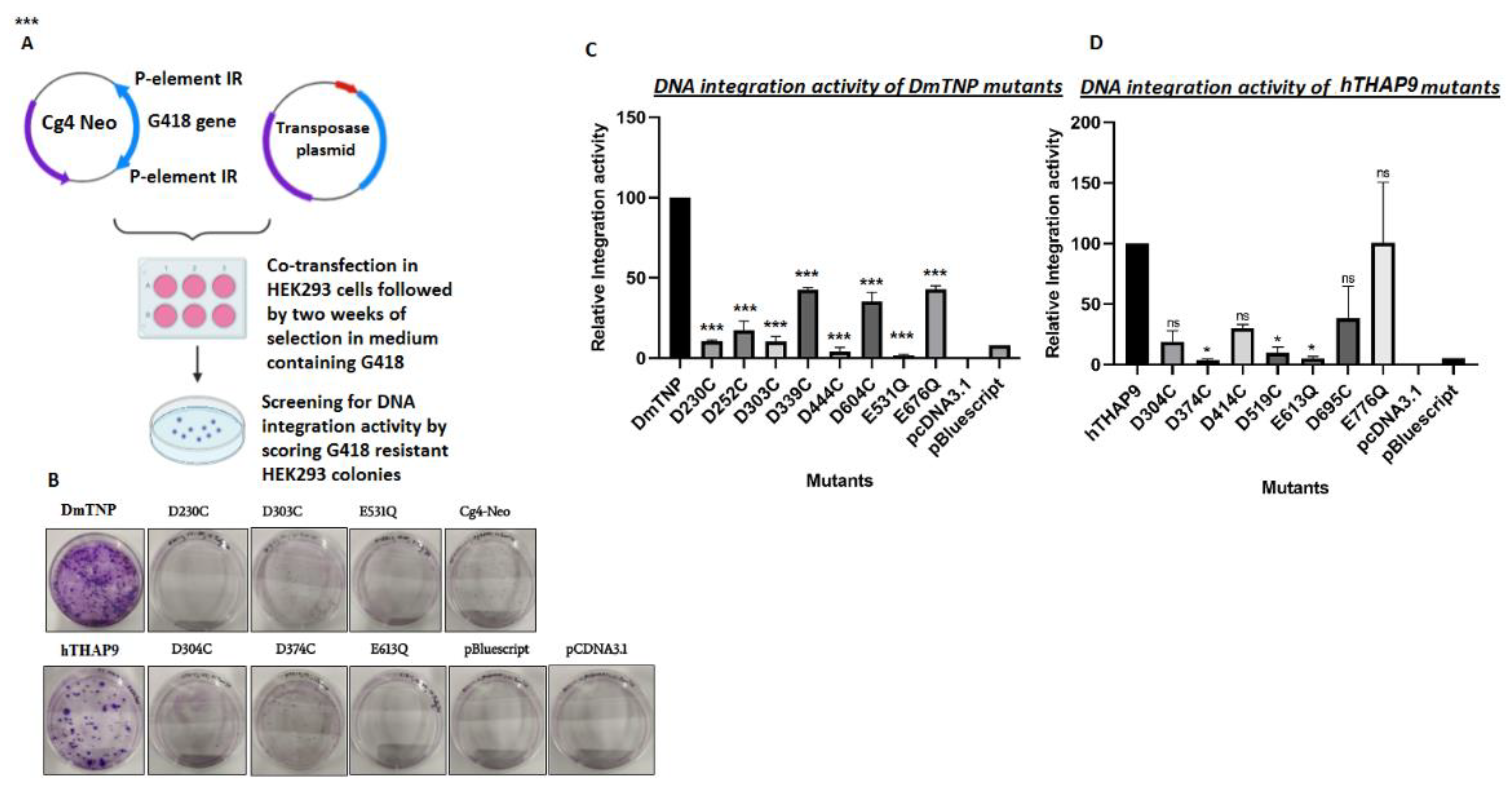
2.2.5. In Vivo Integration Assay
2.2.6. Protein Immunoblotting
3. Results
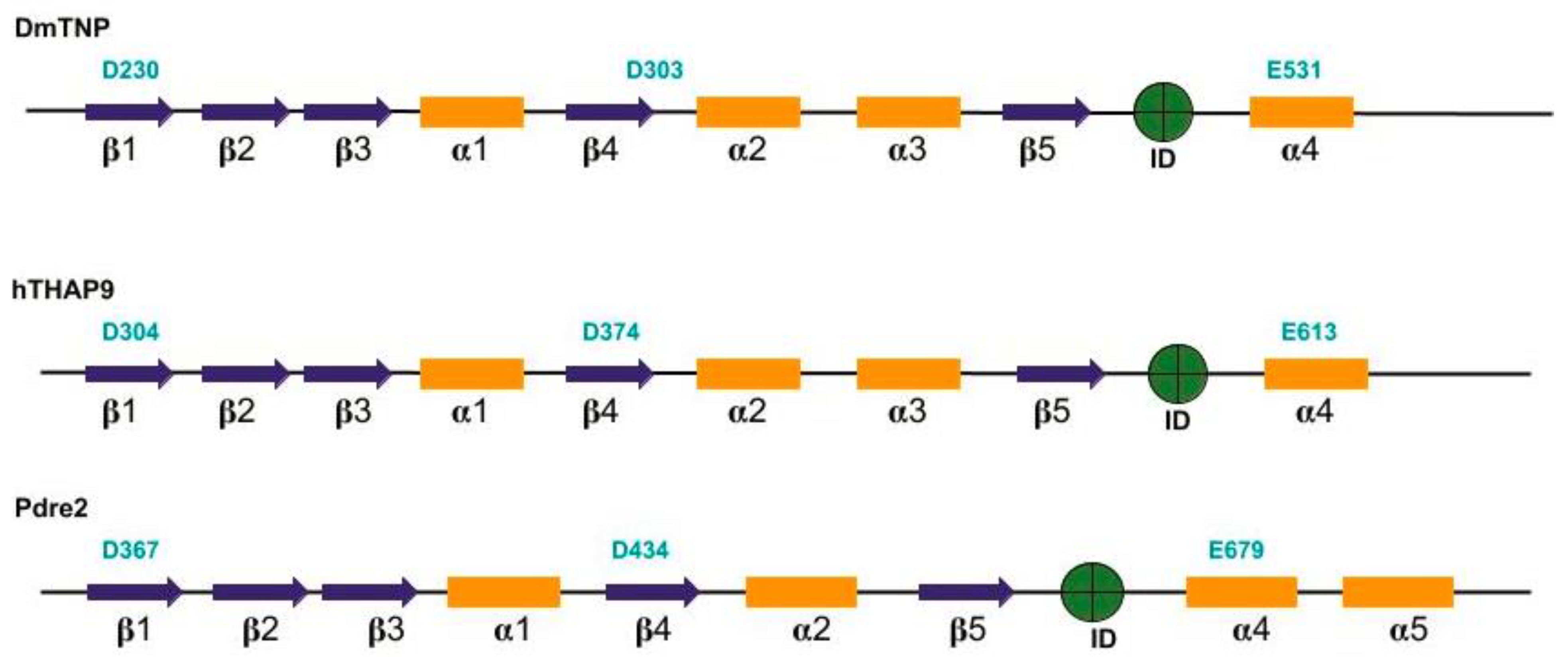
3.1. Prediction of Secondary Structure Elements and RNase-H domain in hTHAP9 and Its Homologs
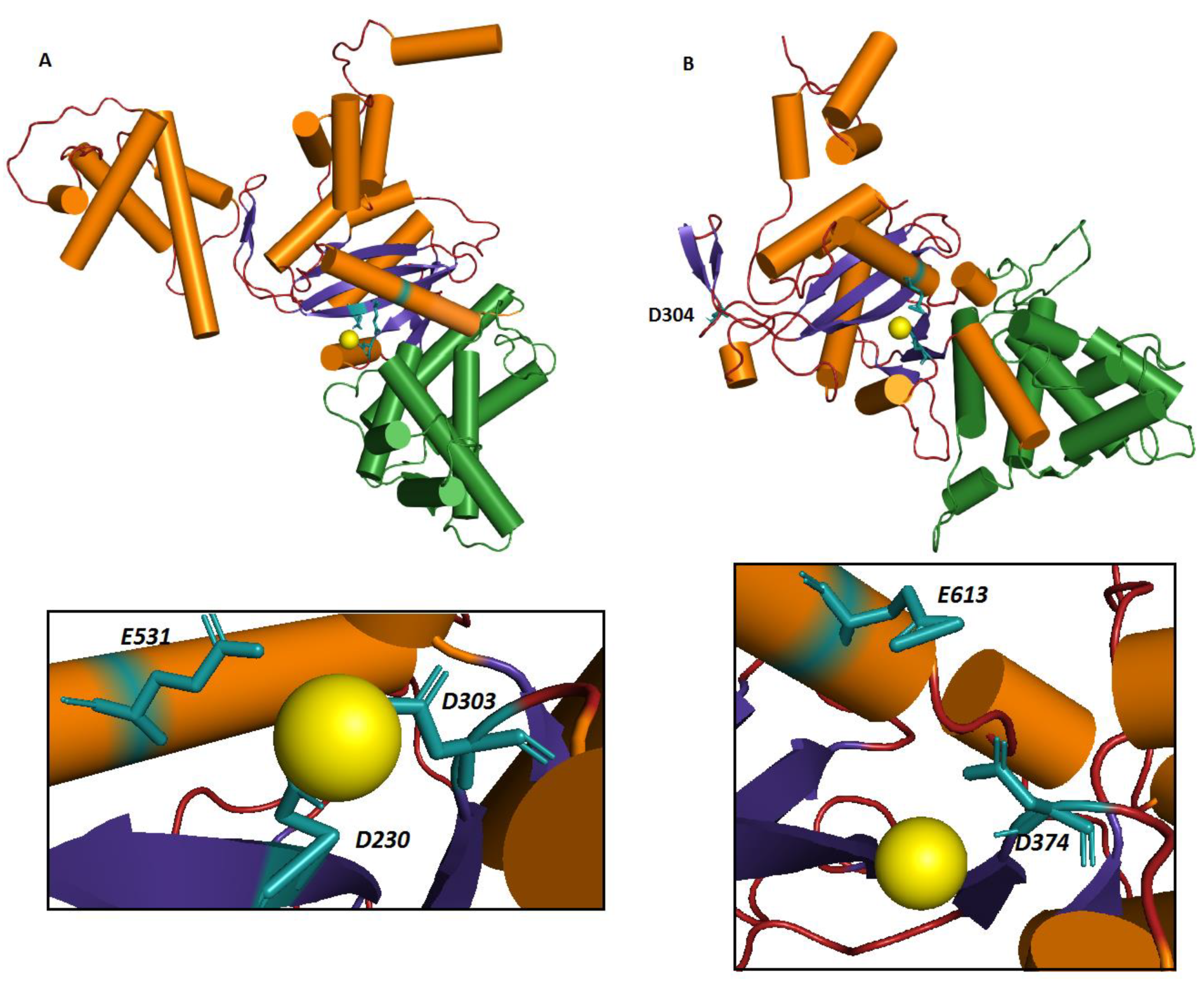
3.2. Homology Modeling of hTHAP9 Identified a Putative RNaseH-Like Domain
3.3. Identification of Other Conserved Acidic Residues in the Catalytic Domain of hTHAP9 and Its Homologs
3.4. Signature Stringin the RNaseH-like catalytic domain
3.5. Site-Directed Mutagenesis of Conserved Acidic Residues in the Catalytic Domain of hTHAP9 and Its Homologs
3.6. Many Conserved Acidic Residues in the Catalytic Domain of hTHAP9 and Its Homologs Are Important for DNA Excision
3.7. Many Conserved Acidic Residues in the Catalytic Domain of hTHAP9 and Its Homologs Are Important for DNA Integration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Craig, N.L.; Chandler, M.; Gellert, M.; Lambowitz, A.M.; Rice, P.A.; Sandmeyer, S.B. (Eds.) Mobile DNA III; American Society of Microbiology: Washington, DC, USA, 2015. [Google Scholar] [CrossRef]
- Sinzelle, L.; Izsvák, Z.; Ivics, Z. Molecular domestication of transposable elements: From detrimental parasites to useful host genes. Cell. Mol. Life Sci. CMLS 2009, 66, 1073–1093. [Google Scholar] [CrossRef]
- Feschotte, C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008, 9, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Jangam, D.; Feschotte, C.; Betrán, E. Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet. TIG 2017, 33, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C.; Pritham, E.J. DNA Transposons and the Evolution of Eukaryotic Genomes. Annu. Rev. Genet. 2007, 41, 331–368. [Google Scholar] [CrossRef]
- Hickman, A.B.; Chandler, M.; Dyda, F. Integrating prokaryotes and eukaryotes: DNA transposases in light of structure. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 50–69. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed]
- Yuan, Y.-W.; Wessler, S.R. The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc. Natl. Acad. Sci. USA 2011, 108, 7884–7889. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, M.; Gaidamakov, S.A.; Crouch, R.J.; Yang, W. Crystal structures of RNase H bound to an RNA/DNA hybrid: Substrate specificity and metal-dependent catalysis. Cell 2005, 121, 1005–1016. [Google Scholar] [CrossRef]
- Asante-Appiah, E.; Skalka, A.M. HIV-1 integrase: Structural organization, conformational changes, and catalysis. Adv. Virus Res. 1999, 52, 351–369. [Google Scholar]
- Rice, P.A.; Baker, T.A. Comparative architecture of transposase and integrase complexes. Nat. Struct. Biol. 2001, 8, 302–307. [Google Scholar] [CrossRef]
- Ghanim, G.E.; Kellogg, E.H.; Nogales, E.; Rio, D.C. Structure of a P element transposase–DNA complex reveals unusual DNA structures and GTP-DNA contacts. Nat. Struct. Mol. Biol. 2019, 26, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Kofler, R.; Hill, T.; Nolte, V.; Betancourt, A.J.; Schlötterer, C. The recent invasion of natural Drosophila simulans populations by the P-element. Proc. Natl. Acad. Sci. USA 2015, 112, 6659–6663. [Google Scholar] [CrossRef] [PubMed]
- Kidwell, M.G. Evolution of hybrid dysgenesis determinants in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1983, 80, 1655–1659. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-López, M.; García-Pérez, J.L. DNA Transposons: Nature and Applications in Genomics. Curr. Genom. 2010, 11, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Rio, D.C. P Transposable Elements in Drosophila and other Eukaryotic Organisms. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.A. Structure of the P element transpososome reveals new twists on the DD(E/D) theme. Nat. Struct. Mol. Biol. 2019, 26, 989–990. [Google Scholar] [CrossRef]
- Hammer, S.E.; Strehl, S.; Hagemann, S. Homologs of Drosophila P Transposons Were Mobile in Zebrafish but Have Been Domesticated in a Common Ancestor of Chicken and Human. Mol. Biol. Evol. 2005, 22, 833–844. [Google Scholar] [CrossRef]
- Hagemann, S.; Hammer, S.E. The implications of DNA transposons in the evolution of P elements in zebrafish (Danio rerio). Genomics 2006, 88, 572–579. [Google Scholar] [CrossRef]
- Majumdar, S.; Singh, A.; Rio, D.C. The Human THAP9 Gene Encodes an Active P-Element DNA Transposase. Science 2013, 339, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.P.; Leroy, C.; Sander, C. MView: A web-compatible database search or multiple alignment viewer. Bioinformatics 1998, 14, 380–381. [Google Scholar] [CrossRef] [PubMed]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, E.; Abraham, M.J.; Hess, B.; van der Spoel, D. GROMACS 2020.4 Manual. 2020. Available online: https://zenodo.org/record/4054996/export/xm#.YK78tKERWUk (accessed on 15 July 2020). [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- PyMOL (v2.0.4). The PyMOL Molecular Graphics System, Schrödinger, LLC. Available online: https://pymol.org/2/ (accessed on 18 July 2020).
- SAVES (v6.0). Available online: https://servicesn.mbi.ucla.edu/SAVES/ (accessed on 20 July 2020).
- Hickman, A.B.; Dyda, F. DNA Transposition at Work. Chem. Rev. 2016, 116, 12758–12784. [Google Scholar] [CrossRef]
- Kim, M.-S.; Lapkouski, M.; Yang, W.; Gellert, M. Crystal structure of the V(D)J recombinase RAG1-RAG2. Nature 2015, 518, 507–511. [Google Scholar] [CrossRef]
- Wright, J.D.; Lim, C. A fast method for predicting amino acid mutations that lead to unfolding. Protein Eng. Des. Sel. 2001, 14, 479–486. [Google Scholar] [CrossRef]
- Kim, H.-S.; Chen, Q.; Kim, S.K.; Nickoloff, J.A.; Hromas, R.; Georgiadis, M.M.; Lee, S.-H. The DDN catalytic motif is required for Metnase functions in non-homologous end joining (NHEJ) repair and replication restart. J. Biol. Chem. 2014, 289, 10930–10938. [Google Scholar] [CrossRef]
- Fugmann, S.D.; Villey, I.J.; Ptaszek, L.M.; Schatz, D.G. Identification of Two Catalytic Residues in RAG1 that Define a Single Active Site within the RAG1/RAG2 Protein Complex. Mol. Cell 2000, 5, 97–107. [Google Scholar] [CrossRef]
- Zhou, L.; Mitra, R.; Atkinson, P.W.; Hickman, A.B.; Dyda, F.; Crai, N.L. Transposition of hAT elements links transposable elements and V(D)J recombination. Nature 2004, 432, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Beall, E.L.; Rio, D.C. Drosophila P-element transposase is a novel site-specific endonuclease. Genes Dev. 1997, 11, 2137–2151. [Google Scholar] [CrossRef]
- Ma, B.-G.; Chen, L.; Ji, H.-F.; Chen, Z.-H.; Yang, F.-R.; Wang, L.; Qu, G.; Jiang, Y.-Y.; Ji, C.; Zhang, H.-Y. Characters of very ancient proteins. Biochem. Biophys. Res. Commun. 2008, 366, 607–611. [Google Scholar] [CrossRef]
- Moelling, K.; Broecker, F.; Russo, G.; Sunagawa, S. RNase H as Gene Modifier, Driver of Evolution and Antiviral Defense. Front. Microbiol. 2017, 8, 1745. [Google Scholar] [CrossRef]
- Hickman, A.B.; Perez, Z.N.; Zhou, L.; Musingarimi, P.; Ghirlando, R.; Hinshaw, J.E.; Craig, N.L.; Dyda, F. Molecular architecture of a eukaryotic DNA transposase. Nat. Struct. Mol. Biol. 2005, 12, 715–721. [Google Scholar] [CrossRef]
- Robertson, H.M. The Tcl-mariner superfamily of transposons in animals. J. Insect Physiol. 1995, 41, 99–105. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, N.; Feschotte, C.; Wessler, S.R. PIF- and Pong-Like Transposable Elements: Distribution, Evolution and Relationship with Tourist-Like Miniature Inverted-Repeat Transposable Elements. Genetics 2004, 166, 971–986. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C. Merlin, a New Superfamily of DNA Transposons Identified in Diverse Animal Genomes and Related to Bacterial IS1016 Insertion Sequences. Mol. Biol. Evol. 2004, 21, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Kapitonov, V.V.; Jurka, J. Helitrons on a roll: Eukaryotic rolling-circle transposons. Trends Genet. TIG 2007, 23, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Kapitonov, V.V.; Jurka, J. RAG1 Core and V(D)J Recombination Signal Sequences Were Derived from Transib Transposons. PLoS Biol. 2005, 3, e181. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Steitz, T.A. Recombining the structures of HIV integrase, RuvC and RNase H. Struct. Lond. Engl. 1995, 3, 131–134. [Google Scholar] [CrossRef]
- Iv, N.; Pb, H. DDE transposases: Structural similarity and diversity. Adv. Drug Deliv. Rev. 2010, 62, 1187–1195. [Google Scholar]
- Muszewska, A.; Hoffman-Sommer, M.; Grynberg, M. LTR Retrotransposons in Fungi. PLoS ONE 2011, 6, e29425. [Google Scholar] [CrossRef] [PubMed]
- Capy, P.; Langin, T.; Higuet, D.; Maurer, P.; Bazin, C. Do the integrases of LTR-retrotransposons and class II element transposases have a common ancestor? Genetica 1997, 100, 63–72. [Google Scholar] [CrossRef]
- Rice, P.; Craigie, R.; Davies, D.R. Retroviral integrases and their cousins. Curr. Opin. Struct. Biol. 1996, 6, 76–83. [Google Scholar] [CrossRef]
- Montaño, S.P.; Pigli, Y.Z.; Rice, P.A. The μ transpososome structure sheds light on DDE recombinase evolution. Nature 2012, 491, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Majorek, K.A.; Dunin-Horkawicz, S.; Steczkiewicz, K.; Muszewska, A.; Nowotny, M.; Ginalski, K.; Bujnicki, J.M. The RNase H-like superfamily: New members, comparative structural analysis and evolutionary classification. Nucleic Acids Res. 2014, 42, 4160–4179. [Google Scholar] [CrossRef]
- Sarnovsky, R.J.; May, E.W.; Craig, N.L. The Tn7 transposase is a heteromeric complex in which DNA breakage and joining activities are distributed between different gene products. EMBO J. 1996, 15, 6348–6361. [Google Scholar] [CrossRef]
- Mátés, L.; Chuah, M.K.L.; Belay, E.; Jerchow, B.; Manoj, N.; Acosta-Sanchez, A.; Grzela, D.P.; Schmitt, A.; Becker, K.; Matrai, J. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 2009, 41, 753–761. [Google Scholar] [CrossRef]
- Yusa, K.; Zhou, L.; Li, M.A.; Bradley, A.; Craig, N.L. A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. USA 2011, 108, 1531–1536. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DmTNP | hTHAP9 |
|---|---|
| D230 | |
| D252 | D304 |
| D303 | D374 |
| D339 | D414 |
| D444 | D519 |
| E531 | E613 |
| D604 | D695 |
| E676 | E776 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, V.; Thakore, P.; Majumdar, S. THAP9 Transposase Cleaves DNA via Conserved Acidic Residues in an RNaseH-Like Domain. Cells 2021, 10, 1351. https://doi.org/10.3390/cells10061351
Sharma V, Thakore P, Majumdar S. THAP9 Transposase Cleaves DNA via Conserved Acidic Residues in an RNaseH-Like Domain. Cells. 2021; 10(6):1351. https://doi.org/10.3390/cells10061351
Chicago/Turabian StyleSharma, Vasudha, Prachi Thakore, and Sharmistha Majumdar. 2021. "THAP9 Transposase Cleaves DNA via Conserved Acidic Residues in an RNaseH-Like Domain" Cells 10, no. 6: 1351. https://doi.org/10.3390/cells10061351
APA StyleSharma, V., Thakore, P., & Majumdar, S. (2021). THAP9 Transposase Cleaves DNA via Conserved Acidic Residues in an RNaseH-Like Domain. Cells, 10(6), 1351. https://doi.org/10.3390/cells10061351