
Innate and Adaptive Immune Genes Associated with MERS-CoV Infection in Dromedaries

 , , , , , ,
, , , , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sampling
2.3. MERS-CoV Characterization
2.4. Camel DNA Extraction
2.5. Probe Design and In-Solution Hybridization Capture Target Enrichment
2.6. Variant Calling
2.7. Read-Based Imputation
2.8. Data Filtering
2.9. Heterozygosity Associated in the Immune Response Genes
2.10. Univariate Logistic Regression Analysis for Phenotype–Genotype Association
2.11. Linkage Disequilibrium (LD)-Based Gene-Set Test
3. Results
3.1. MERS-CoV Shedding and Antibody Prevalence in Dromedaries from the UAE
3.2. In-Solution Hybridization Capture and Variant Calls in Dromedary IR Genes
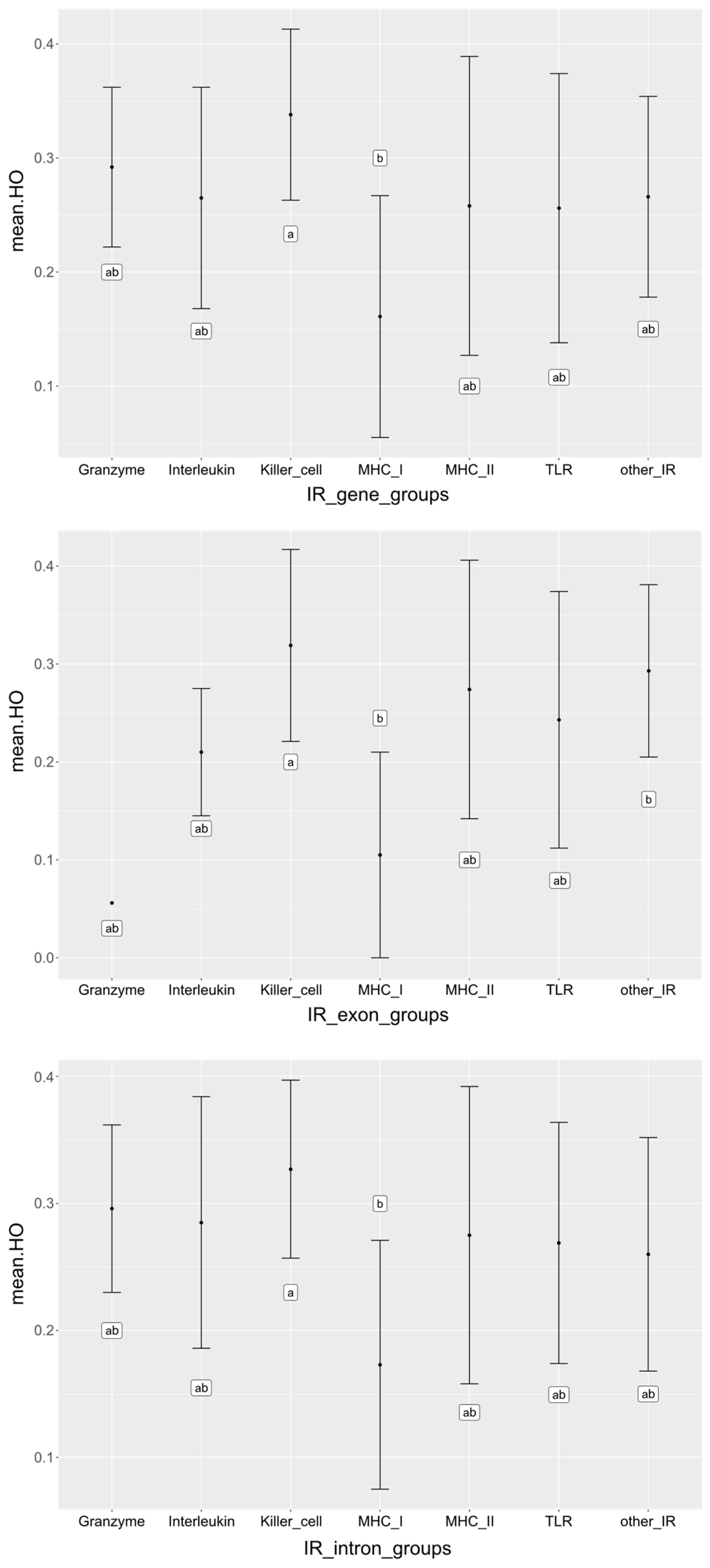
3.3. Diversity in the Targeted Immune Response (IR) Genes
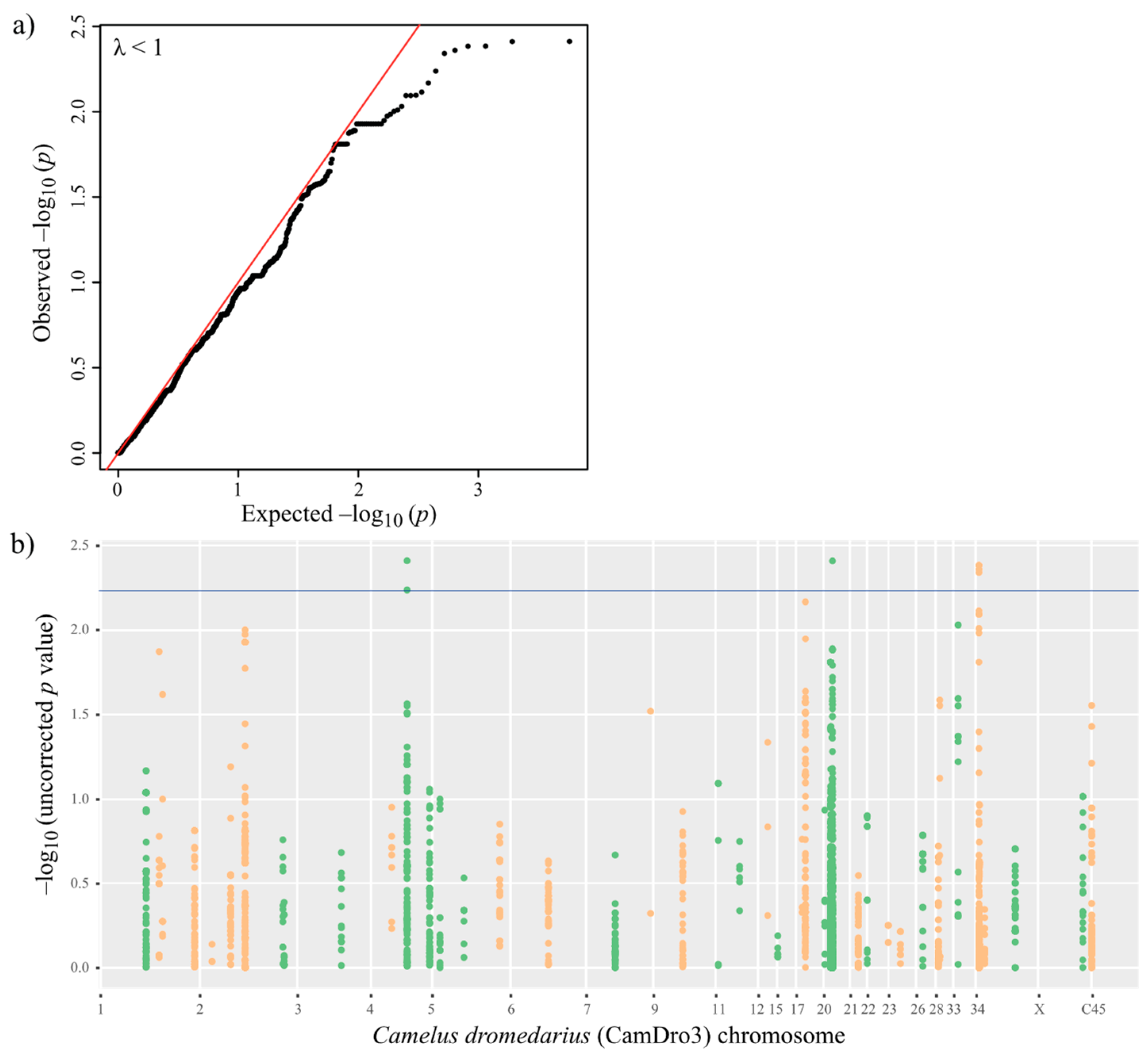
3.4. Phenotype–Genotype Association in MERS-CoV Antibody-Positive Dromedaries
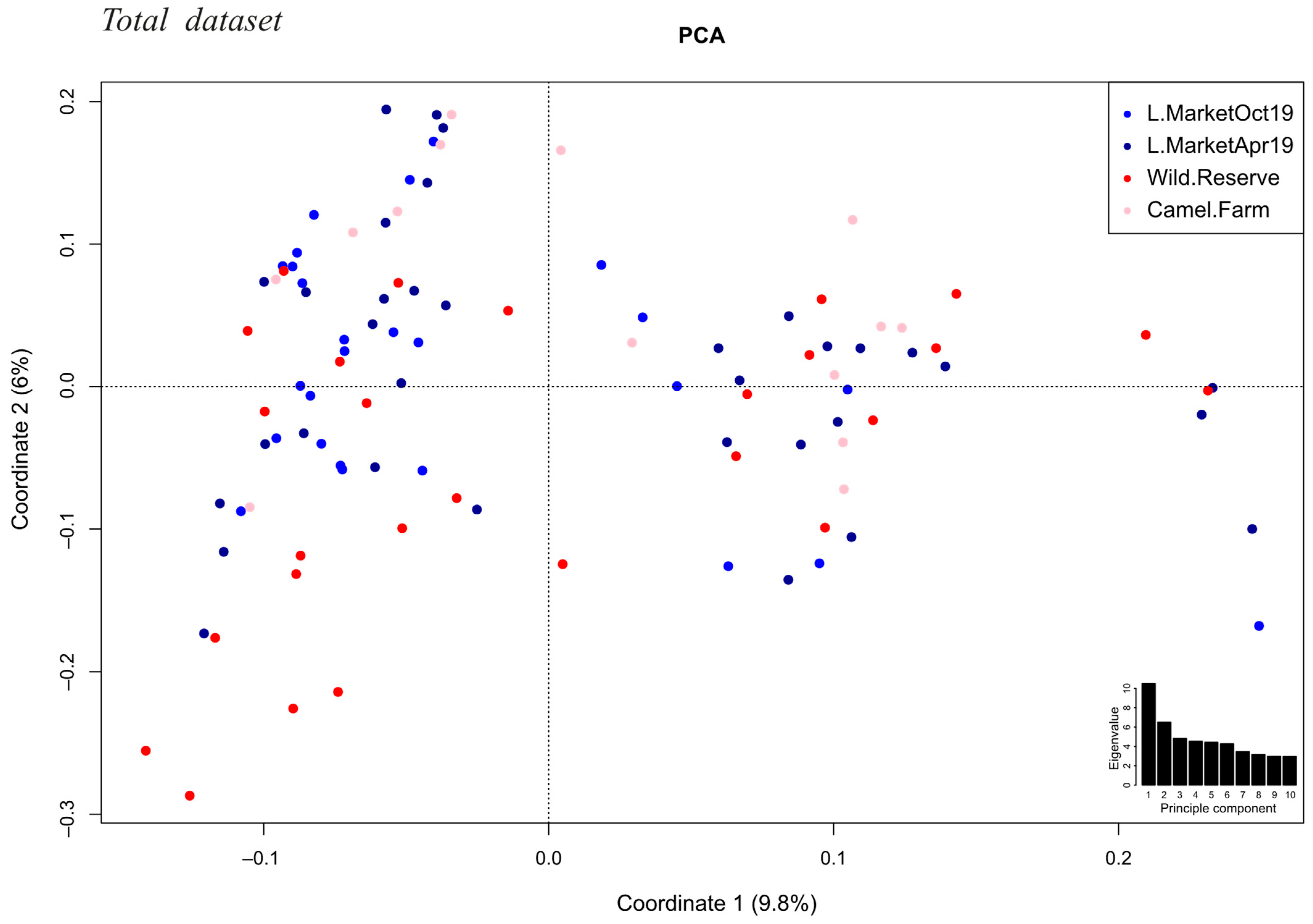
3.5. Accounting for Spatial and Temporal Sampling
3.6. Linkage Disequilibrium-Based Gene-Set Test
4. Discussion
4.1. High MERS-CoV Antibody Prevalence in Dromedaries from the UAE
4.2. Different Diversity in IR Gene Groups
4.3. Candidate IR Genes Associated with MERS-CoV Infection in Dromedaries
4.4. Challenges and Impact of IR Gene Associations with Betacoronavirus Infections
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Costagliola, A.; Liguori, G.; D’angelo, D.; Costa, C.; Ciani, F.; Giordano, A. Do animals play a role in the transmission of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2)? a commentary. Animals 2021, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; Van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Hijawi, B.; Abdallat, M.; Sayaydeh, A.; Alqasrawi, S.; Haddadin, A.; Jaarour, N.; Alsheikh, S.; Alsanouri, T. Novel coronavirus infections in Jordan, April 2012: Epidemiological findings from a retrospective investigation. East. Mediterr. Health J. 2013, 19 (Suppl. 1), 12–18. [Google Scholar] [CrossRef]
- Reusken, C.B.E.M.; Haagmans, B.L.; Müller, M.A.; Gutierrez, C.; Godeke, G.J.; Meyer, B.; Muth, D.; Raj, V.S.; De Vries, L.S.; Corman, V.M.; et al. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: A comparative serological study. Lancet Infect. Dis. 2013, 13, 859–866. [Google Scholar] [CrossRef]
- Nowotny, N.; Kolodziejek, J. Middle East Respiratory Syndrome coronavirus (MERS-CoV) in dromedary camels, Oman, 2013. Eurosurveillance 2014, 19, 1–5. [Google Scholar] [CrossRef]
- Dawson, P.; Malik, M.R.; Parvez, F.; Morse, S.S. What Have We Learned about Middle East Respiratory Syndrome Coronavirus Emergence in Humans? A Systematic Literature Review. Vector Borne Zoonotic Dis. 2019, 19, 174–192. [Google Scholar] [CrossRef]
- Gossner, C.; Danielson, N.; Gervelmeyer, A.; Berthe, F.; Faye, B.; Kaasik Aaslav, K.; Adlhoch, C.; Zeller, H.; Penttinen, P.; Coulombier, D. Human-Dromedary Camel Interactions and the Risk of Acquiring Zoonotic Middle East Respiratory Syndrome Coronavirus Infection. Zoonoses Public Health 2016, 63, 1–9. [Google Scholar] [CrossRef]
- Omrani, A.S.; Shalhoub, S. Middle East respiratory syndrome coronavirus (MERS-CoV): What lessons can we learn? J. Hosp. Infect. 2015, 91, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Hemida, M.G.; Elmoslemany, A.; Al-Hizab, F.; Alnaeem, A.; Almathen, F.; Faye, B.; Chu, D.K.W.; Perera, R.A.P.M.; Peiris, M. Dromedary Camels and the Transmission of Middle East Respiratory Syndrome Coronavirus (MERS-CoV). Transbound. Emerg. Dis. 2017, 64, 344–353. [Google Scholar] [CrossRef]
- Assiri, A.; McGeer, A.; Perl, T.M.; Price, C.S.; Al Rabeeah, A.A.; Cummings, D.A.T.; Alabdullatif, Z.N.; Assad, M.; Almulhim, A.; Makhdoom, H.; et al. Hospital outbreak of middle east respiratory syndrome coronavirus. N. Engl. J. Med. 2013, 369, 407–416. [Google Scholar] [CrossRef]
- Corman, V.M.; Jores, J.; Meyer, B.; Younan, M.; Liljander, A.; Said, M.Y.; Gluecks, I.; Lattwein, E.; Bosch, B.J.; Drexler, J.F.; et al. Antibodies against MERS coronavirus in dromedary camels, Kenya, 1992–2013. Emerg. Infect. Dis. 2014, 20, 1319–1322. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Li, K.S.M.; Luk, H.K.H.; He, Z.; Teng, J.L.L.; Yuen, K.-Y.; Wernery, U.; Woo, P.C.Y. Middle East Respiratory Syndrome Coronavirus Antibodies in Bactrian and Hybrid Camels from Dubai. mSphere 2020, 5, e00898-19. [Google Scholar] [CrossRef]
- Reusken, C.B.E.M.; Schilp, C.; Raj, V.S.; De Bruin, E.; Kohl, R.H.G.; Farag, E.A.B.A.; Haagmans, B.L.; Al-Romaihi, H.; Le Grange, F.; Bosch, B.J.; et al. MERS-CoV infection of alpaca in a region where MERS-CoV is endemic. Emerg. Infect. Dis. 2016, 22, 1129–1131. [Google Scholar] [CrossRef]
- Ciccarese, S.; Burger, P.A.; Ciani, E.; Castelli, V.; Linguiti, G.; Plasil, M.; Massari, S.; Horin, P.; Antonacci, R. The camel adaptive immune receptors repertoire as a singular example of structural and functional genomics. Front. Genet. 2019, 10, 997. [Google Scholar] [CrossRef]
- Muyldermans, S.; Baral, T.N.; Retamozzo, V.C.; De Baetselier, P.; De Genst, E.; Kinne, J.; Leonhardt, H.; Magez, S.; Nguyen, V.K.; Revets, H.; et al. Camelid immunoglobulins and nanobody technology. Vet. Immunol. Immunopathol. 2009, 128, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Ming, L.; Wang, Z.; Yi, L.; Batmunkh, M.; Liu, T.; Siren, D.; He, J.; Juramt, N.; Jambl, T.; Li, Y.; et al. Chromosome-level assembly of wild Bactrian camel genome reveals organization of immune gene loci. Mol. Ecol. Resour. 2020, 20, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Jovčevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26. [Google Scholar] [CrossRef]
- Hanke, L.; Perez, L.V.D.J.S.; Das, H.; Schulte, T.; Morro, A.M.; Corcoran, M.; Achour, A.; Hedestam, G.K.; Hällberg, B.M.; Murrell, B.; et al. An alpaca nanobody neutralizes SARS-CoV-2 by blocking receptor interaction. Nat. Commun. 2020, 11, 4420. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; De Vlieger, D.; Corbett, K.S.; Torres, G.M.; Wang, N.; Van Breedam, W.; Roose, K.; van Schie, L.; Hoffmann, M.; Pöhlmann, S.; et al. Structural Basis for Potent Neutralization of Betacoronaviruses by Single-Domain Camelid Antibodies. Cell 2020, 181, 1004–1015.e15. [Google Scholar] [CrossRef]
- Koenig, P.-A.; Das, H.; Liu, H.; Kümmerer, B.M.; Gohr, F.N.; Jenster, L.-M.; Schiffelers, L.D.J.; Tesfamariam, Y.M.; Uchima, M.; Wuerth, J.D.; et al. Structure-guided multivalent nanobodies block SARS-CoV-2 infection and suppress mutational escape. Science 2021, 371, eabe6230. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Hui, K.P.Y.; Perera, R.A.P.M.; Miguel, E.; Niemeyer, D.; Zhao, J.; Channappanavar, R.; Dudas, G.; Oladipo, J.O.; Traoré, A.; et al. MERS coronaviruses from camels in Africa exhibit region-dependent genetic diversity. Proc. Natl. Acad. Sci. USA 2018, 115, 3144–3149. [Google Scholar] [CrossRef] [PubMed]
- Adney, D.R.; Clancy, C.S.; Bowen, R.A.; Munster, V.J. Camelid Inoculation with Middle East Respiratory Syndrome Coronavirus: Experimental Models of Reservoir Host Infection. Viruses 2020, 12, 1370. [Google Scholar] [CrossRef] [PubMed]
- Elbers, J.P.; Rogers, M.F.; Perelman, P.L.; Proskuryakova, A.A.; Serdyukova, N.A.; Johnson, W.E.; Horin, P.; Corander, J.; Murphy, D.; Burger, P.A. Improving Illumina assemblies with Hi-C and long reads: An example with the North African dromedary. Mol. Ecol. Resour. 2019, 19, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Lado, S.; Elbers, J.P.; Rogers, M.F.; Melo-Ferreira, J.; Yadamsuren, A.; Corander, J.; Horin, P.; Burger, P.A. Nucleotide diversity of functionally different groups of immune response genes in Old World camels based on newly annotated and reference-guided assemblies. BMC Genom. 2020, 21, 606. [Google Scholar] [CrossRef]
- Pedersen, A.B.; Babayan, S.A. Wild immunology. Mol. Ecol. 2011, 20, 872–880. [Google Scholar] [CrossRef]
- Fitak, R.R.; Mohandesan, E.; Corander, J.; Yadamsuren, A.; Chuluunbat, B.; Abdelhadi, O.; Raziq, A.; Nagy, P.; Walzer, C.; Faye, B.; et al. Genomic signatures of domestication in Old World. Commun. Biol. 2020, 3, 316. [Google Scholar] [CrossRef]
- Guo, F.; Ming, L.; Si, R.; Yi, L.; He, J.; Ji, R. A genome-wide association study identifies quantitative trait loci affecting hematological traits in camelus bactrianus. Animals 2020, 10, 96. [Google Scholar] [CrossRef] [PubMed]
- Bahbahani, H.; Musa, H.H.; Wragg, D.; Shuiep, E.S.; Almathen, F.; Hanotte, O. Genome Diversity and Signatures of Selection for Production and Performance Traits in Dromedary Camels. Front. Genet. 2019, 10, 893. [Google Scholar] [CrossRef]
- Plasil, M.; Mohandesan, E.; Fitak, R.R.; Musilova, P.; Kubickova, S.; Burger, P.A.; Horin, P. The major histocompatibility complex in Old World camelids and low polymorphism of its class II genes. BMC Genom. 2016, 17, 117. [Google Scholar] [CrossRef]
- Plasil, M.; Wijkmark, S.; Elbers, J.P.; Oppelt, J.; Burger, P.A.; Horin, P. The major histocompatibility complex of Old World camelids: Class I and class I-related genes. Hla 2019, 93, 203–215. [Google Scholar] [CrossRef]
- Futas, J.; Oppelt, J.; Jelinek, A.; Elbers, J.P.; Wijacki, J.; Knoll, A.; Burger, P.A.; Horin, P. Natural killer cell receptor genes in camels: Another mammalian model. Front. Genet. 2019, 10, 620. [Google Scholar] [CrossRef] [PubMed]
- Elbers, J.P.; Brown, M.B.; Taylor, S.S. Identifying genome-wide immune gene variation underlying infectious disease in wildlife populations—A next generation sequencing approach in the gopher tortoise. BMC Genom. 2018, 19, 64. [Google Scholar] [CrossRef]
- Yu, F.; Zhang, X.; Tian, S.; Geng, L.; Xu, W.; Ma, N.; Wang, M.; Jia, Y.; Liu, X.; Ma, J.; et al. Comprehensive investigation of cytokine- and immune-related gene variants in HBV-associated hepatocellular carcinoma patients. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Griffin, G.K.; Sholl, L.M.; Lindeman, N.I.; Fletcher, C.D.M.; Hornick, J.L. Targeted genomic sequencing of follicular dendritic cell sarcoma reveals recurrent alterations in NF-κB regulatory genes. Mod. Pathol. 2016, 29, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Müller, M.A.; Costabel, U.; Timm, J.; Binger, T.; Meyer, B.; Kreher, P.; Lattwein, E.; Eschbach-Bludau, M.; Nitsche, A.; et al. Assays for laboratory confirmation of novel human coronavirus (HCOV-EMC) infections. Eurosurveillance 2012, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- De Volo, S.B.; Reynolds, R.T.; Douglas, M.R.; Antolin, M.F. An improved extraction method to increase DNA yield from molted feathers. Condor 2008, 110, 762–766. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Davies, R.W.; Flint, J.; Myers, S.; Mott, R. Rapid genotype imputation from sequence without reference panels. Nat. Genet. 2016, 48, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Lindenbaum, P. JVarkit: Java-based utilities for Bioinformatics. FigShare 2015, 10, m9. [Google Scholar]
- Purcell, S.; Neale, B.; Todd-brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Gremme, G.; Steinbiss, S.; Kurtz, S. Genome tools: A comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013, 10, 645–656. [Google Scholar] [CrossRef]
- Goudet, J. HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef]
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 3rd ed.; Sage Publications Inc.: New York, NY, USA, 2019. [Google Scholar]
- Hothorn, T.; Bretz, F.; Westfall, P. Simultaneous Inference in General Parametric Models. Biom. J. 2008, 50, 346–363. [Google Scholar] [CrossRef]
- Turner, S.D. qqman: An R package for visualizing GWAS results using QQ and manhattan plots. J. Open Source Softw. 2018, 3, 731. [Google Scholar] [CrossRef]
- Yin, T.; Cook, D.; Lawrence, M. ggbio: An R package for extending the grammar of graphics for genomic data. Genome Biol. 2012, 13, R77. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Narum, S.R. Beyond Bonferroni: Less conservative analyses for conservation genetics. Conserv. Genet. 2006, 7, 783–787. [Google Scholar] [CrossRef]
- Purcell, S.; Cherny, S.S.; Sham, P.C. Genetic power calculator: Design of linkage and association genetic mapping studies of complex traits. Bioinformatics 2003, 19, 149–150. [Google Scholar] [CrossRef]
- VanRaden, P.M.; Sun, C.; O’Connell, J.R. Fast imputation using medium or low-coverage sequence data. BMC Genet. 2015, 16, 82. [Google Scholar] [CrossRef]
- Engering, A.; Hogerwerf, L.; Slingenbergh, J. Pathogen-host-environment interplay and disease emergence. Emerg. Microbes Infect. 2013, 2, 1–7. [Google Scholar] [CrossRef]
- Alagaili, A.N.; Briese, T.; Mishra, N.; Kapoor, V.; Sameroff, S.C.; de Wit, E.; Munster, V.J.; Hensley, L.E.; Zalmout, I.S.; Kapoor, A.; et al. Middle east respiratory syndrome coronavirus infection in dromedary camels in Saudi Arabia. MBio 2014, 5. [Google Scholar] [CrossRef]
- Adney, D.R.; van Doremalen, N.; Brown, V.R.; Bushmaker, T.; Scott, D.; de Wit, E.; Bowen, R.A.; Munster, V.J. Replication and shedding of MERS-CoV in upper respiratory tract of inoculated dromedary camels. Emerg. Infect. Dis. 2014, 20, 1999–2005. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zimmerman, D.; Deem, S.L. A Review of Zoonotic Pathogens of Dromedary Camels. Ecohealth 2019, 16, 356–377. [Google Scholar] [CrossRef] [PubMed]
- Mohd, H.A.; Al-Tawfiq, J.A.; Memish, Z.A. Middle East Respiratory Syndrome Coronavirus (MERS-CoV) origin and animal reservoir. Virol. J. 2016, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.F.; Shastri, A.J.; Ye, C.; Weldon, C.H.; Filshtein-, T.; Coker, D.; Symons, A.; Esparza-gordillo, J.; The 23andMe COVID-19 Team; Aslibekyan, S.; et al. Trans-ethnic analysis reveals genetic and non-genetic associations with COVID-19 susceptibility and severity. medRxiv 2020. [Google Scholar] [CrossRef]
- Ovsyannikova, I.G.; Haralambieva, I.H.; Crooke, S.N.; Poland, G.A.; Kennedy, R.B. The role of host genetics in the immune response to SARS-CoV-2 and COVID-19 susceptibility and severity. Immunol. Rev. 2020, 296, 205–219. [Google Scholar] [CrossRef]
- Hajeer, A.H.; Balkhy, H.; Johani, S.; Yousef, M.Z.; Arabi, Y. Association of human leukocyte antigen class II alleles with severe Middle East respiratory syndrome-coronavirus infection. Ann. Thorac. Med. 2016, 11, 211–213. [Google Scholar] [CrossRef]
- Harton, J.; Jin, L.; Hahn, A.; Drake, J. Immunological Functions of the Membrane Proximal Region of MHC Class II Molecules. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.N. Guest Editorial Functional intronic polymorphisms: Buried treasure awaiting discovery within our genes. Hum. Genom. 2010, 4, 284–288. [Google Scholar] [CrossRef]
- Huai, W.; Song, H.; Wang, L.; Li, B.; Zhao, J.; Han, L.; Gao, C.; Jiang, G.; Zhang, L.; Zhao, W. Phosphatase PTPN4 Preferentially Inhibits TRIF-Dependent TLR4 Pathway by Dephosphorylating TRAM. J. Immunol. 2015, 194, 4458–4465. [Google Scholar] [CrossRef] [PubMed]
- Brandao, S.C.S.; Ramos, J.d.O.X.; Dompieri, L.T.; Godoi, E.T.A.M.; Figueiredo, J.L.; Sarinho, E.S.C.; Chelvanambi, S.; Aikawa, M. Is Toll-like receptor 4 involved in the severity of COVID-19 pathology in patients with cardiometabolic comorbidities? Cytokine Growth Factor Rev. 2020, 5, 135–143. [Google Scholar] [CrossRef]
- Mubarak, A.; Alturaiki, W.; Hemida, M.G. Middle east respiratory syndrome coronavirus (MERS-CoV): Infection, immunological response, and vaccine development. J. Immunol. Res. 2019, 2019, 6491738. [Google Scholar] [CrossRef] [PubMed]
- Haddad, H.; Al-Zyoud, W. miRNA target prediction might explain the reduced transmission of SARS-CoV-2 in Jordan, Middle East. Non Coding RNA Res. 2020, 5, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; O’Neal, W.K.; Randell, S.H.; Blackburn, K.; Moyer, M.B.; Boucher, R.C.; Ostrowski, L.E. Identification of dynein heavy chain 7 as an inner arm component of human cilia that is synthesized but not assembled in a case of primary ciliary dyskinesia. J. Biol. Chem. 2002, 277, 17906–17915. [Google Scholar] [CrossRef]
- Nunnari, G.; Sanfilippo, C.; Castrogiovanni, P.; Imbesi, R.; Li Volti, G.; Barbagallo, I.; Musumeci, G.; Di Rosa, M. Network perturbation analysis in human bronchial epithelial cells following SARS-CoV2 infection. Exp. Cell Res. 2020, 395, 112204. [Google Scholar] [CrossRef]
- Tao, X.; Hill, T.E.; Morimoto, C.; Peters, C.J.; Ksiazek, T.G.; Tseng, C.-T.K. Bilateral Entry and Release of Middle East Respiratory Syndrome Coronavirus Induces Profound Apoptosis of Human Bronchial Epithelial Cells. J. Virol. 2013, 87, 9953–9958. [Google Scholar] [CrossRef]
- Yuen, K.M.; Chan, R.W.; Mok, C.K.; Wong, A.C.; Kang, S.S.; Nicholls, J.M.; Chan, M.C. Differential onset of apoptosis in avian influenza H5N1 and seasonal H1N1 virus infected human bronchial and alveolar epithelial cells: An in vitro and ex vivo study. Influenza Other Respir. Viruses 2011, 5, 437–438. [Google Scholar]
- Dendrou, C.A.; Petersen, J.; Rossjohn, J.; Fugger, L. HLA variation and disease. Nat. Rev. Immunol. 2018, 18, 325–339. [Google Scholar] [CrossRef]
- Wang, S.F.; Chen, K.H.; Chen, M.; Li, W.Y.; Chen, Y.J.; Tsao, C.H.; Yen, M.Y.; Huang, J.C.; Chen, Y.M.A. Human-Leukocyte Antigen Class I Cw 1502 and Class II DR 0301 Genotypes Are Associated with Resistance to Severe Acute Respiratory Syndrome (SARS) Infection Sheng-Fan. Viral Immunol. 2011, 24, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.L.; Birol, I. HLA Predictions from the Bronchoalveolar Lavage Fluid Samples of Five Patients at the Early Stage of the Wuhan Seafood Market COVID-19 Outbreak. Bioinformatics. 2020, 36, 5271–5273. [Google Scholar] [CrossRef]
- Park, J.H.; Gail, M.H.; Weinberg, C.R.; Carroll, R.J.; Chung, C.C.; Wang, Z.; Chanock, S.J.; Fraumeni, J.F.; Chatterjee, N. Distribution of allele frequencies and effect sizes and their interrelationships for common genetic susceptibility variants. Proc. Natl. Acad. Sci. USA 2011, 108, 18026–18031. [Google Scholar] [CrossRef]
- Liu, J.; Wu, P.; Gao, F.; Qi, J.; Kawana-Tachikawa, A.; Xie, J.; Vavricka, C.J.; Iwamoto, A.; Li, T.; Gao, G.F. Novel Immunodominant Peptide Presentation Strategy: A Featured HLA-A*2402-Restricted Cytotoxic T-Lymphocyte Epitope Stabilized by Intrachain Hydrogen Bonds from Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Protein. J. Virol. 2010, 84, 11849–11857. [Google Scholar] [CrossRef] [PubMed]
- Novelli, A.; Andreani, M.; Biancolella, M.; Liberatoscioli, L.; Passarelli, C.; Colona, V.L.; Rogliani, P.; Leonardis, F.; Campana, A.; Carsetti, R.; et al. HLA allele frequencies and susceptibility to COVID-19 in a group of 99 Italian patients. Hla 2020, 96, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Wachsmuth, L.; Kulozik, A.E.; Gehring, N.H. Two mammalian MAGOH genes contribute to exon junction complex composition and nonsense-mediated decay. RNA Biol. 2013, 10, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- McAleer, J.P.; Vella, A.T. Understanding how lipopolysaccharide impacts CD4 T-cell immunity. Crit. Rev. Immunol. 2008, 28, 281–299. [Google Scholar]
- Zhu, Z.; Qi, Y.; Ge, A.; Zhu, Y.; Xu, K.; Ji, H.; Shi, Z.; Cui, L.; Zhou, M. Comprehensive characterization of serum microRNA profile in response to the emerging avian influenza A (H7N9) virus infection in humans. Viruses 2014, 6, 1525–1539. [Google Scholar] [CrossRef]
- Leon-Icaza, S.A.; Zeng, M.; Rosas-Taraco, A.G. microRNAs in viral acute respiratory infections: Immune regulation, biomarkers, therapy, and vaccines. ExRNA 2019, 1, 1. [Google Scholar] [CrossRef]
- Sardar, R.; Satish, D.; Gupta, D. Identification of Novel SARS-CoV-2 Drug Targets by Host MicroRNAs and Transcription Factors Co-regulatory Interaction Network Analysis. Front. Genet. 2020, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hoober, J.K.; Eggink, L.L.; Cote, R. Stories from the Dendritic Cell Guardhouse. Front. Immunol. 2019, 10, 2880. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| IR Gene Group | Genes a | Exons a | Introns a | ||||||
|---|---|---|---|---|---|---|---|---|---|
| SNPs | HO | HE | SNPs | HO | HE | SNPs | HO | HE | |
| Granzyme | 54 | 0.292 | 0.311 | 5 | 0.056 | 0.066 | 50 | 0.296 | 0.315 |
| Interleukin | 121 | 0.265 | 0.284 | 21 | 0.210 | 0.218 | 107 | 0.285 | 0.305 |
| Killer cells | 282 | 0.338 | 0.342 | 38 | 0.319 | 0.313 | 253 | 0.327 | 0.336 |
| MHC class I | 435 | 0.161 | 0.193 | 96 | 0.155 | 0.185 | 353 | 0.173 | 0.205 |
| MHC class II | 596 | 0.258 | 0.277 | 140 | 0.274 | 0.290 | 456 | 0.275 | 0.294 |
| TLR | 124 | 0.256 | 0.298 | 55 | 0.243 | 0.272 | 74 | 0.269 | 0.311 |
| Other IR genes | 1253 | 0.266 | 0.280 | 235 | 0.293 | 0.307 | 1074 | 0.260 | 0.273 |
| Chr | Position (Minor/Major Allele) | Gene | Association Test p-Value | Allele Freq. Minor Allele | Genotype Counts | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cases | Controls | Exact p-Value | Odds Ratio | Cases | Controls | Exact p-Value | ||||
| 5 | T8508361C intron 25 | PTPN4 | 0.003873 ** | 0.08824 | 0.2 | 0.06096 | 0.3871 | 0/6/28 | 2/20/38 | 0.1241 |
| 20 | A23100696G intron 4 | HLA-A-24-like | 0.003881 ** | 0.25 | 0.08475 | 0.00278 * | 3.6 | 1/16/19 | 0/10/49 | 0.002554 * |
| 34 | G15362634A intron 1 | MAGOHB | 0.004123 ** | 0.3 | 0.4308 | 0.09343 | 0.5663 | 1/19/15 | 16/24/25 | 0.01201 * |
| 34 | A15363451G intron 2 | MAGOHB | 0.004129 ** | 0.2941 | 0.4308 | 0.06634 | 0.5506 | 1/18/15 | 16/24/25 | 0.01392 * |
| 34 | 15367780 | INTERGENIC | 0.004364 ** | - | - | - | - | - | - | - |
| 34 | G15361800A intron 1 | MAGOHB | 0.004553 ** | 0.3 | 0.4206 | 0.1239 | 0.5903 | 1/19/15 | 15/23/25 | 0.01467 * |
| 5 | C8506434T intron 26 | PTPN4 | 0.005774 ** | 0.1 | 0.2097 | 0.07213 | 0.4188 | 0/7/28 | 2/22/38 | 0.1349 |
| 17 | T23840747C intron 6 | DNAH7 | 0.006791 | 0.1806 | 0.1154 | 0.2087 | 1.689 | 1/11/24 | 1/13/51 | 0.3252 |
| 34 | C15363470T intron 2 | MAGOHB | 0.008005 | 0.3286 | 0.4385 | 0.173 | 0.6267 | 2/19/14 | 16/25/24 | 0.04715* |
| 20 | A20676706C intron 4 | HLA-DPB1-like | 0.01548 | 0.04167 | 0.09231 | 0.2651 | 0.4275 | 0/3/33 | 2/8/55 | 0.6674 |
| 20 | T20677126C exon 4 | HLA-DPB1-like | 0.01548 | 0.04167 | 0.09231 | 0.2651 | 0.4275 | 0/3/33 | 2/8/55 | 0.6674 |
| 20 | G20678240T intron 2 | HLA-DPB1-like | 0.01548 | 0.04167 | 0.09231 | 0.2651 | 0.4275 | 0/3/33 | 2/8/55 | 0.6674 |
| 20 | T20679052C exon 2 | HLA-DPB1-like | 0.01548 | 0.04167 | 0.09231 | 0.2651 | 0.4275 | 0/3/33 | 2/8/55 | 0.6674 |
| 20 | A20679884C intron 1 | HLA-DPB1-like | 0.01548 | 0.04167 | 0.09231 | 0.2651 | 0.4275 | 0/3/33 | 2/8/55 | 0.6674 |
| 20 | G20680467A intron 1 | HLA-DPB1-like | 0.01548 | 0.04167 | 0.09231 | 0.2651 | 0.4275 | 0/3/33 | 2/8/55 | 0.6674 |
| 20 | T20680474C intron 1 | HLA-DPB1-like | 0.01548 | 0.04167 | 0.09231 | 0.2651 | 0.4275 | 0/3/33 | 2/8/55 | 0.6674 |
| 20 | T20680741C intron 1 | HLA-DPB1-like | 0.01548 | 0.04167 | 0.08594 | 0.3868 | 0.4625 | 0/3/33 | 2/7/55 | 0.6561 |
| 20 | G20681619C intron 1 | HLA-DPB1-like | 0.01548 | 0.04167 | 0.09231 | 0.2651 | 0.4275 | 0/3/33 | 2/8/55 | 0.6674 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lado, S.; Elbers, J.P.; Plasil, M.; Loney, T.; Weidinger, P.; Camp, J.V.; Kolodziejek, J.; Futas, J.; Kannan, D.A.; Orozco-terWengel, P.; et al. Innate and Adaptive Immune Genes Associated with MERS-CoV Infection in Dromedaries. Cells 2021, 10, 1291. https://doi.org/10.3390/cells10061291
Lado S, Elbers JP, Plasil M, Loney T, Weidinger P, Camp JV, Kolodziejek J, Futas J, Kannan DA, Orozco-terWengel P, et al. Innate and Adaptive Immune Genes Associated with MERS-CoV Infection in Dromedaries. Cells. 2021; 10(6):1291. https://doi.org/10.3390/cells10061291
Chicago/Turabian StyleLado, Sara, Jean P. Elbers, Martin Plasil, Tom Loney, Pia Weidinger, Jeremy V. Camp, Jolanta Kolodziejek, Jan Futas, Dafalla A. Kannan, Pablo Orozco-terWengel, and et al. 2021. "Innate and Adaptive Immune Genes Associated with MERS-CoV Infection in Dromedaries" Cells 10, no. 6: 1291. https://doi.org/10.3390/cells10061291
APA StyleLado, S., Elbers, J. P., Plasil, M., Loney, T., Weidinger, P., Camp, J. V., Kolodziejek, J., Futas, J., Kannan, D. A., Orozco-terWengel, P., Horin, P., Nowotny, N., & Burger, P. A. (2021). Innate and Adaptive Immune Genes Associated with MERS-CoV Infection in Dromedaries. Cells, 10(6), 1291. https://doi.org/10.3390/cells10061291