
Role of MicroRNA-145 in DNA Damage Signalling and Senescence in Vascular Smooth Muscle Cells of Type 2 Diabetic Patients

 ,
,  ,
,  and
and 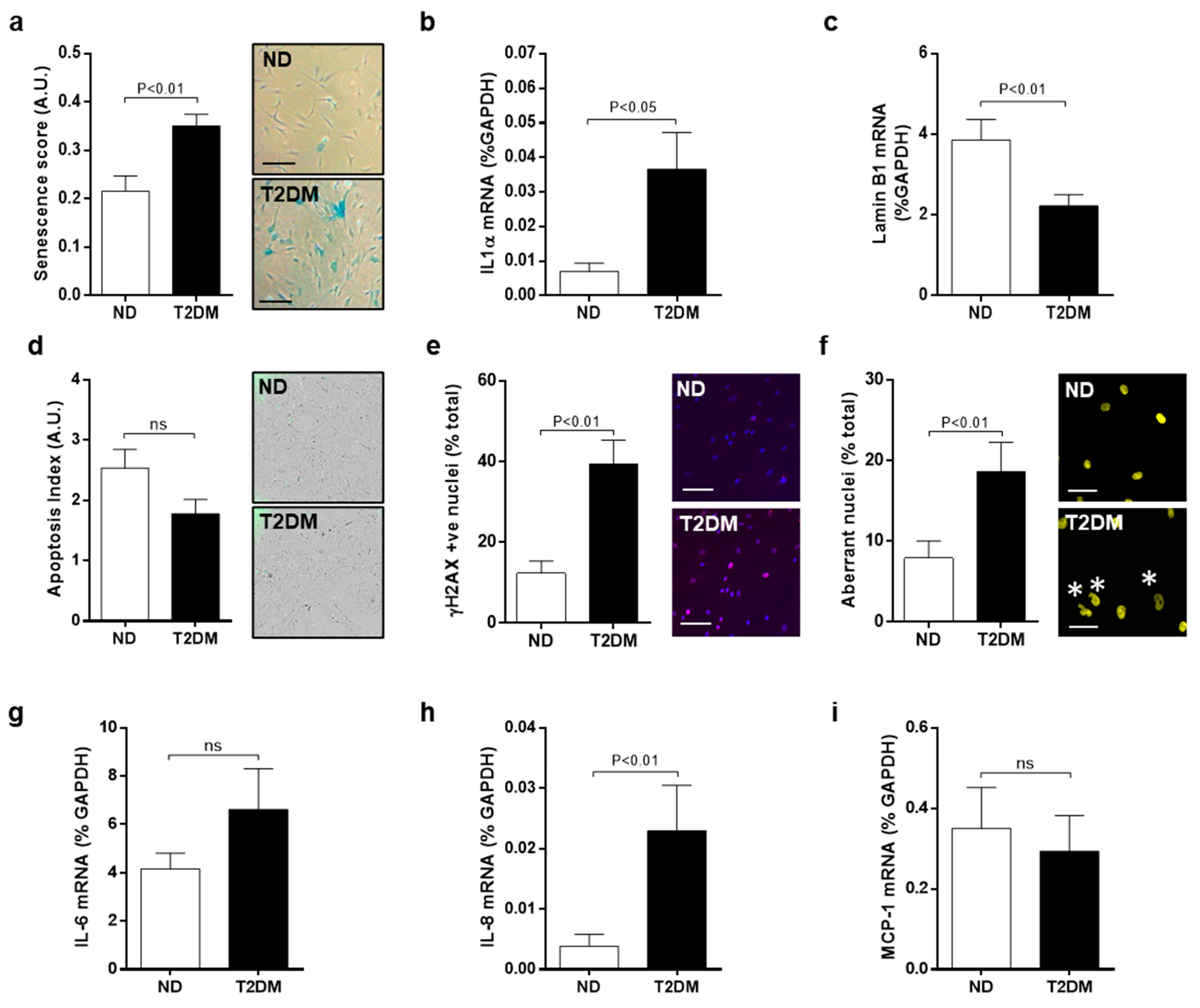{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Smooth Muscle Cell (SMC) Culture
2.2. Senescence Associated β-Galactosidase Staining
2.3. Quantitative Real-Time RT-PCR
2.4. Apoptosis Assay
2.5. Immunocytochemistry
2.6. Enzyme Llinked Immunosorbent Assays (ELISA)
2.7. Cell Proliferation
2.8. Immunoblotting
2.9. Quantification of miRNA-145 Levels
2.10. Inhibition of DDR Apical Kinases
2.11. siRNA Knockdown
2.12. miRNA-145 Overexpression
2.13. Statistical Analysis
3. Results
3.1. Inherent Characteristics of Senescence and DNA Damage in Native SV-SMC
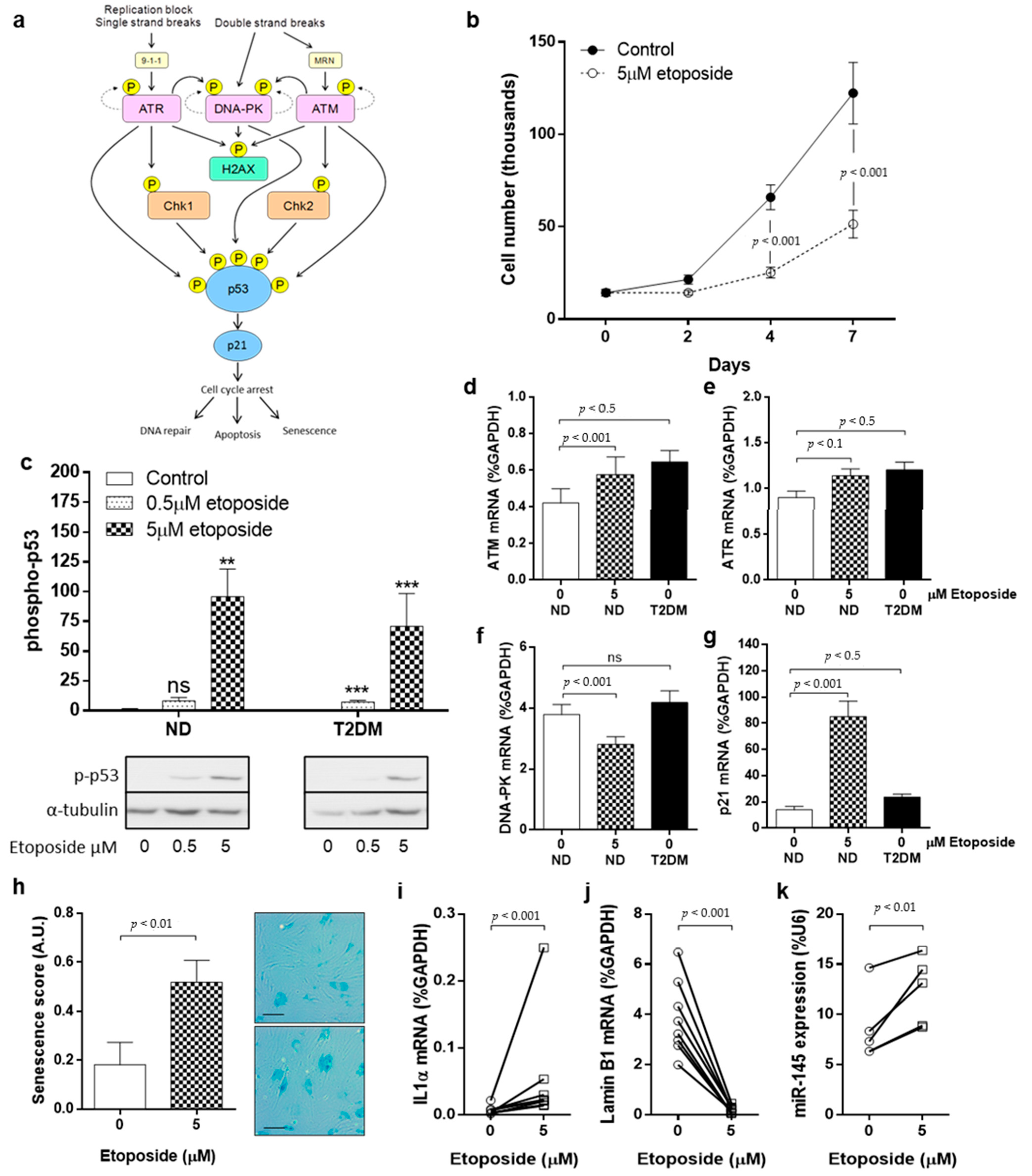
3.2. DNA Damage and DDR Pathway Activation in SV-SMC
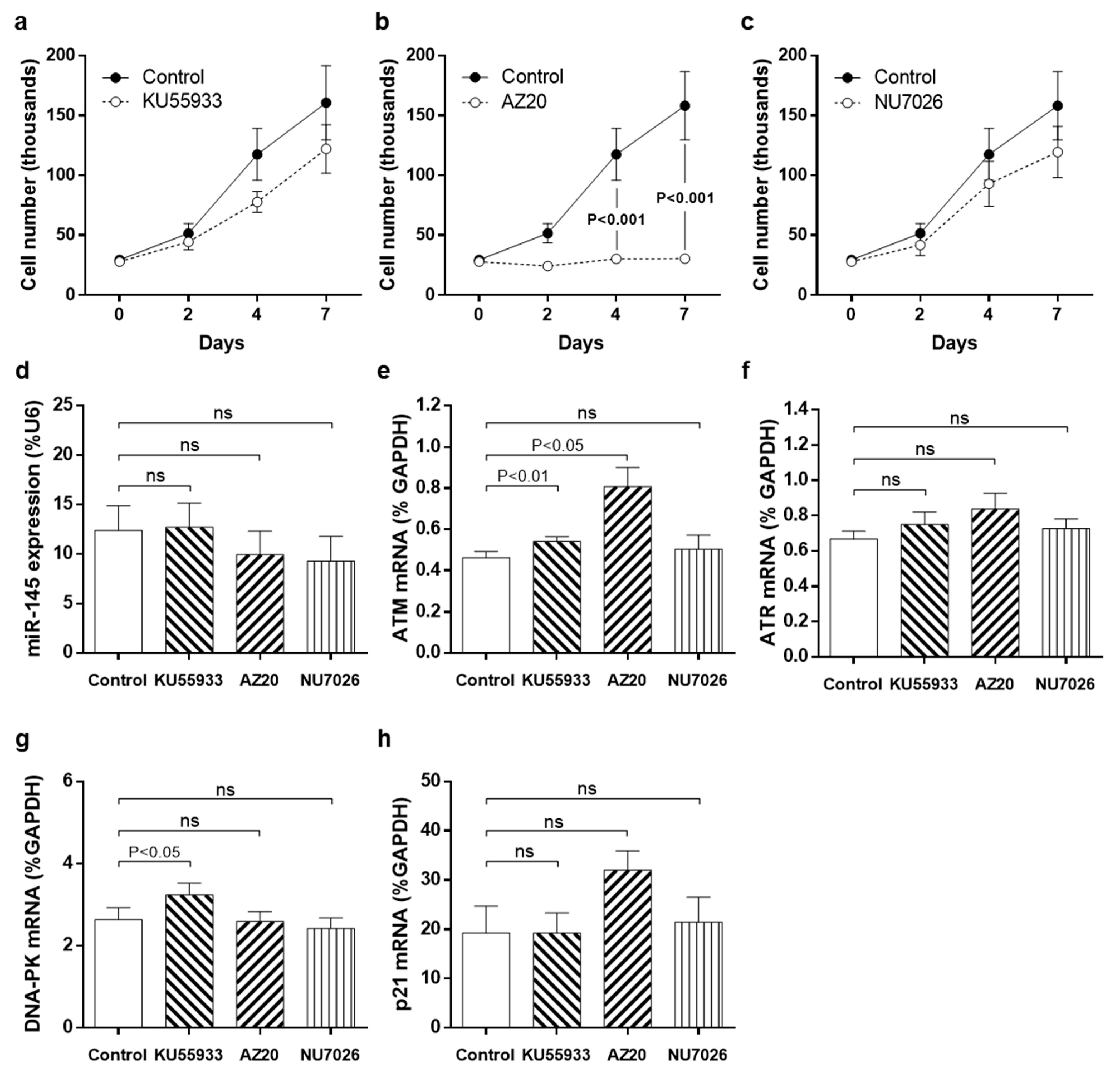
3.3. Pharmacological Inhibition of DDR Kinases
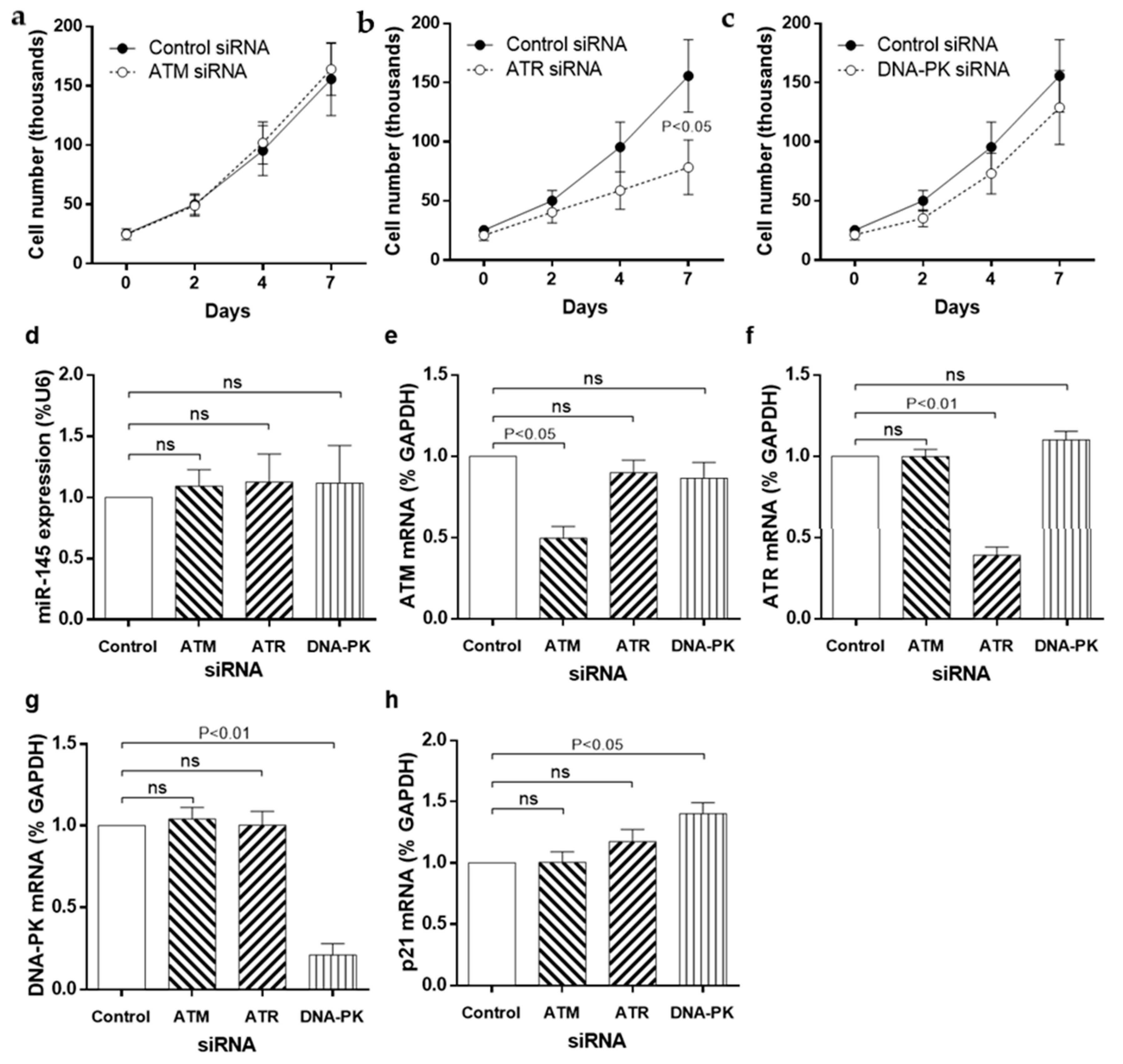
3.4. siRNA Knockdown of DDR Kinases
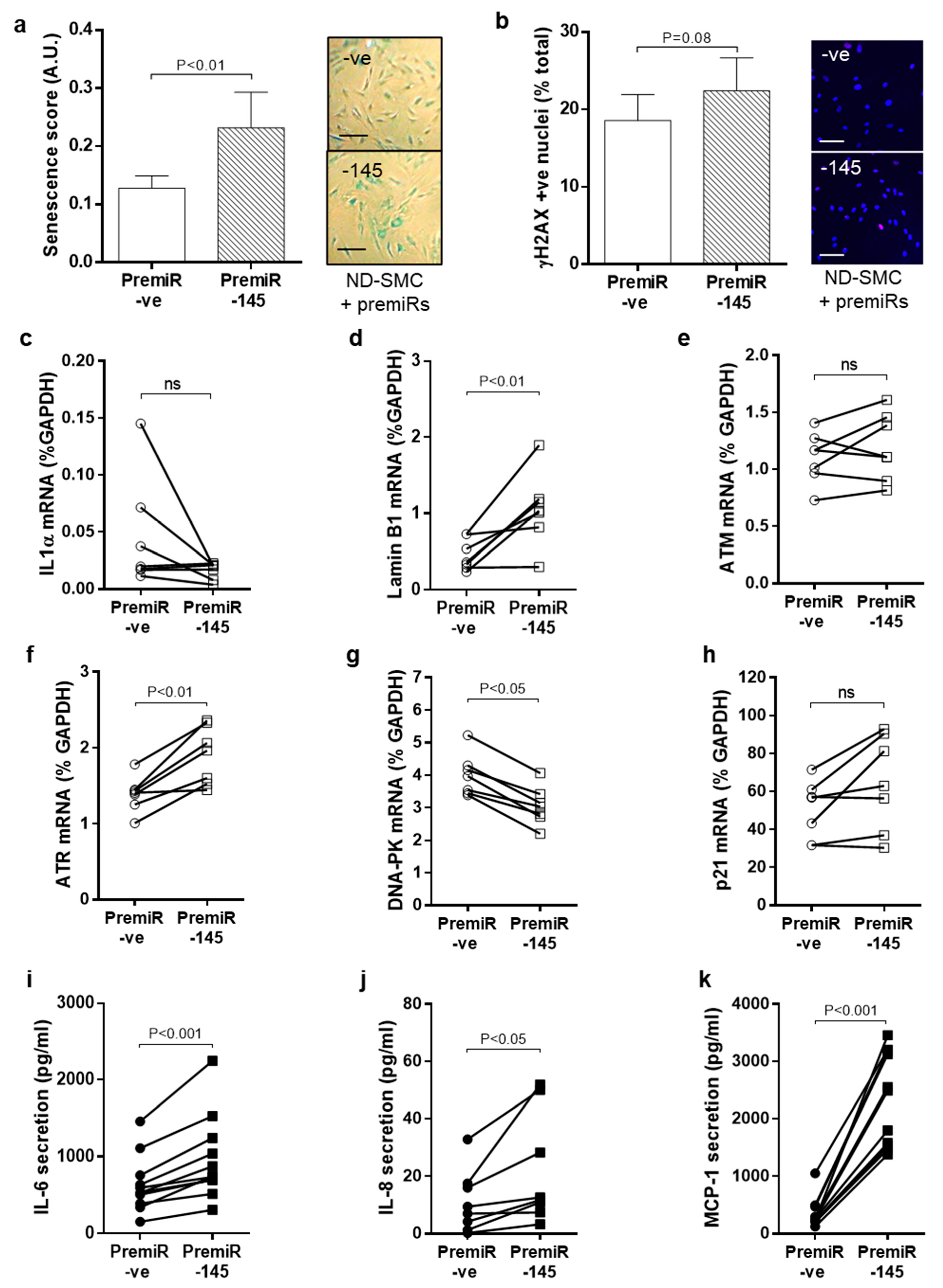
3.5. miRNA-145 Overexpression Modulates DNA Damage Signalling and Senescence
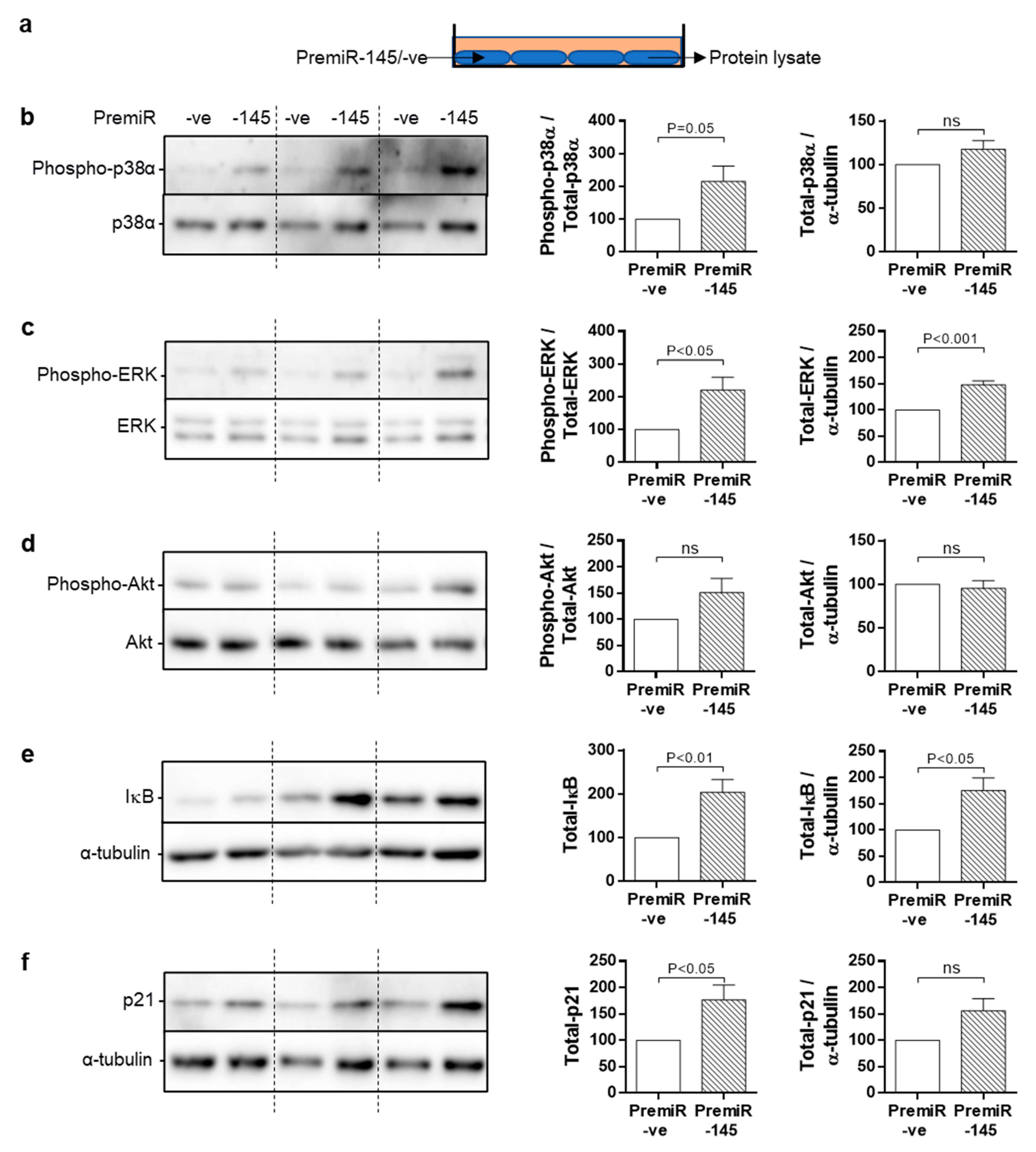
3.6. Chronic Signalling Pathway Activation in miRNA-145-Overexpressing SMC
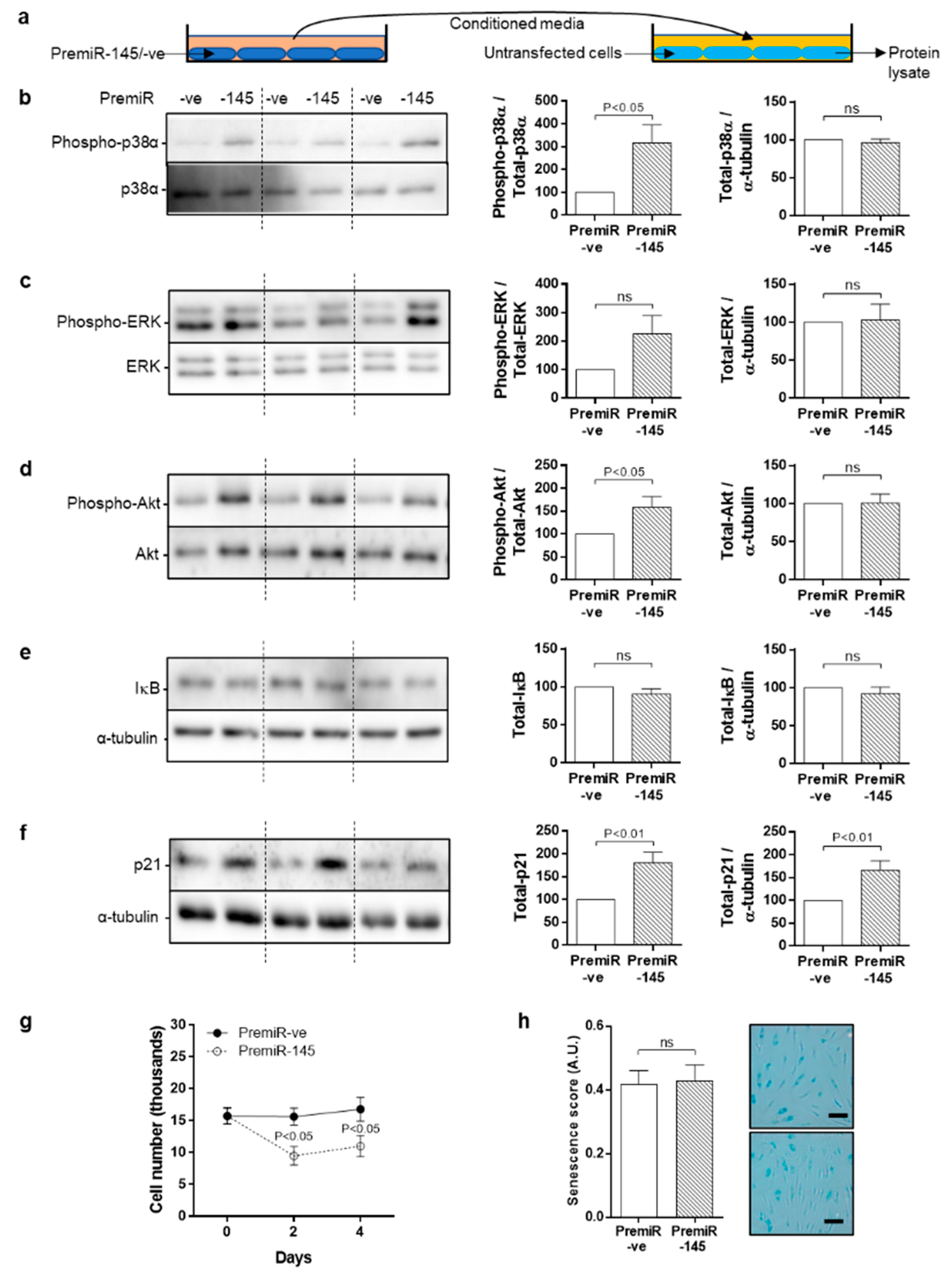
3.7. Bystander Effect of Conditioned Medium from miRNA-145 Overexpressing Cells
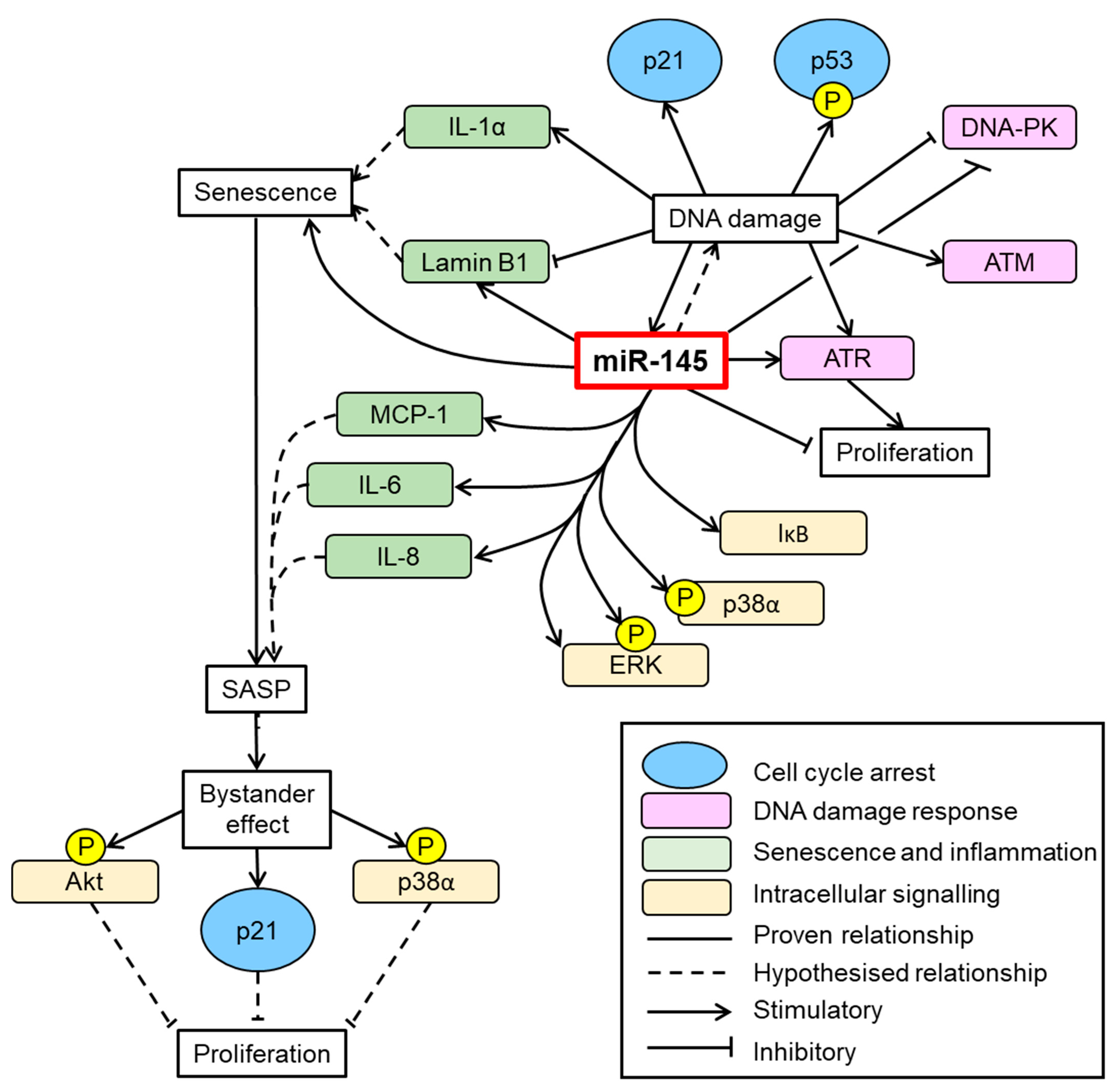
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Booth, G.L.; Kapral, M.K.; Fung, K.; Tu, J.V. Relation between age and cardiovascular disease in men and women with diabetes compared with non-diabetic people: A population-based retrospective cohort study. Lancet 2006, 368, 29–36. [Google Scholar] [CrossRef]
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Minamino, T. Vascular Senescence in Cardiovascular and Metabolic Diseases. Front. Cardiovasc. Med. 2018, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Hakala, T.; Pitkänen, O.; Halonen, P.; Mustonen, J.; Turpeinen, A.; Hippelainen, M. Early and late outcome after coronary artery bypass surgery in diabetic patients. Scand. Cardiovasc. J. 2005, 39, 177–181. [Google Scholar] [CrossRef]
- Kubal, C.; Srinivasan, A.K.; Grayson, A.D.; Fabri, B.M.; Chalmers, J.A. Effect of Risk-Adjusted Diabetes on Mortality and Morbidity After Coronary Artery Bypass Surgery. Ann. Thorac. Surg. 2005, 79, 1570–1576. [Google Scholar] [CrossRef] [PubMed]
- Owens, C.D. Adaptive changes in autogenous vein grafts for arterial reconstruction: Clinical implications. J. Vasc. Surg. 2010, 51, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Madi, H.A.; Riches, K.; Warburton, P.; O’Regan, D.J.; Turner, N.A.; Porter, K.E. Inherent differences in morphology, proliferation, and migration in saphenous vein smooth muscle cells cultured from nondiabetic and Type 2 diabetic patients. Am. J. Physiol. Cell Physiol. 2009, 297, C1307–C1317. [Google Scholar] [CrossRef]
- Riches, K.; Warburton, P.; O’Regan, D.J.; Turner, N.A.; Porter, K.E. Type 2 diabetes impairs venous, but not arterial smooth muscle cell function: Possible role of differential RhoA activity. Cardiovasc. Revasc. Med. 2014, 15, 141–148. [Google Scholar] [CrossRef]
- Riches, K.; Alshanwani, A.R.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Wood, I.C.; Turner, N.A.; Porter, K.E. Elevated expression levels of miR-143/5 in saphenous vein smooth muscle cells from patients with Type 2 diabetes drive persistent changes in phenotype and function. J. Mol. Cell Cardiol. 2014, 74, 240–250. [Google Scholar] [CrossRef]
- Hu, H.; Gatti, R.A. MicroRNAs: New players in the DNA damage response. J. Mol. Cell Biol. 2011, 3, 151–158. [Google Scholar] [CrossRef]
- Ugalde, A.P.; Ramsay, A.J.; De La Rosa, J.; Varela, I.; Marinño, G.; Cadinñanos, J.; Lu, J.; Freije, J.M.P.; López-Otín, C. Aging and chronic DNA damage response activate a regulatory pathway involving miR-29 and p53. EMBO J. 2011, 30, 2219–2232. [Google Scholar] [CrossRef]
- Iaconetti, C.; De Rosa, S.; Polimeni, A.; Sorrentino, S.; Gareri, C.; Carino, A.; Sabatino, J.; Colangelo, M.; Curcio, A.; Indolfi, C. Down-regulation of miR-23b induces phenotypic switching of vascular smooth muscle cells in vitro and in vivo. Cardiovasc. Res. 2015, 107, 522–533. [Google Scholar] [CrossRef]
- Santulli, G.; Wronska, A.; Uryu, K.; Diacovo, T.G.; Gao, M.; Marx, S.O.; Kitajewski, J.; Chilton, J.M.; Akat, K.M.; Tuschl, T.; et al. A selective microRNA-based strategy inhibits restenosis while preserving endothelial function. J. Clin. Investig. 2014, 124, 4102–4114. [Google Scholar] [CrossRef]
- Torella, D.; Iaconetti, C.; Catalucci, D.; Ellison, G.M.; Leone, A.; Waring, C.D.; Bochicchio, A.; Vicinanza, C.; Aquila, I.; Curcio, A.; et al. MicroRNA-133 Controls Vascular Smooth Muscle Cell Phenotypic Switch In Vitro and Vascular Remodeling In Vivo. Circ. Res. 2011, 109, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Taniguchi, T. MicroRNAs and DNA damage response: Implications for cancer therapy. Cell Cycle 2013, 12, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.E.; Naik, J.; Turner, N.A.; Dickinson, T.; Thompson, M.M.; London, N.J. Simvastatin inhibits human saphenous vein neointima formation via inhibition of smooth muscle cell proliferation and migration. J. Vasc. Surg. 2002, 36, 150–157. [Google Scholar] [CrossRef]
- Riches, K.; Angelini, T.G.; Mudhar, G.S.; Kaye, J.; Clark, E.; Bailey, M.A.; Sohrabi, S.; Korossis, S.; Walker, P.G.; Scott, D.J.A.; et al. Exploring smooth muscle phenotype and function in a bioreactor model of abdominal aortic aneurysm. J. Transl. Med. 2013, 11, 208. [Google Scholar] [CrossRef]
- Turner, N.A.; Mughal, R.S.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Mechanism of TNFα-induced IL-1α, IL-1β and IL-6 expression in human cardiac fibroblasts: Effects of statins and thiazolidinediones. Cardiovasc. Res. 2007, 76, 81–90. [Google Scholar] [CrossRef]
- Riches, K.; Clark, E.; Helliwell, R.J.; Angelini, T.G.; Hemmings, K.E.; Bailey, M.A.; Bridge, K.I.; Scott, D.J.A.; Porter, K.E. Progressive Development of Aberrant Smooth Muscle Cell Phenotype in Abdominal Aortic Aneurysm Disease. J. Vasc. Res. 2017, 55, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Clarke, M.C.H. Killing the old: Cell senescence in atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.-M.; DeMaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Gurung, R.; Choong, A.M.; Woo, C.C.; Foo, R.; Sorokin, V. Genetic and Epigenetic Mechanisms Underlying Vascular Smooth Muscle Cell Phenotypic Modulation in Abdominal Aortic Aneurysm. Int. J. Mol. Sci. 2020, 21, 6334. [Google Scholar] [CrossRef]
- Riches, K.; Huntriss, J.; Keeble, C.; Wood, I.C.; O’Regan, D.J.; Turner, N.A.; Porter, K.E. Mapping the methylation status of the miR-145 promoter in saphenous vein smooth muscle cells from individuals with type 2 diabetes. Diabetes Vasc. Dis. Res. 2017, 14, 122–129. [Google Scholar] [CrossRef]
- Riches-Suman, K. Diverse roles of microRNA-145 in regulating smooth muscle (dys)function in health and disease. Biochem. Soc. Trans. 2021, 49, 353–363. [Google Scholar] [CrossRef]
- Pashaei, E.; Guzel, E.; Ozgurses, M.E.; Demirel, G.; Aydin, N.; Ozen, M. A Meta-Analysis: Identification of Common Mir-145 Target Genes that have Similar Behavior in Different GEO Datasets. PLoS ONE 2016, 11, e0161491. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Hooten, N.N.; Evans, M.K. Techniques to Induce and Quantify Cellular Senescence. J. Vis. Exp. 2017, 123, e55533. [Google Scholar] [CrossRef]
- Chi, C.; Li, D.-J.; Jiang, Y.-J.; Tong, J.; Fu, H.; Wu, Y.-H.; Shen, F.-M. Vascular smooth muscle cell senescence and age-related diseases: State of the art. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1810–1821. [Google Scholar] [CrossRef] [PubMed]
- Bartoli-Leonard, F.; Wilkinson, F.L.; Schiro, A.; Inglott, F.S.; Alexander, M.Y.; Weston, R. Loss of SIRT1 in diabetes accelerates DNA damage-induced vascular calcification. Cardiovasc. Res. 2021, 117, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Yan, J.; Tang, D. ERK kinases modulate the activation of PI3 kinase related kinases (PIKKs) in DNA damage response. Histol. Histopathol. 2013, 28, 1547–1554. [Google Scholar]
- Job, A.; Schmitt, L.-M.; von Wenserski, L.; Lankat-Buttgereit, B.; Gress, T.M.; Buchholz, M.; Gallmeier, E. Inactivation of PRIM1 Function Sensitizes Cancer Cells to ATR and CHK1 Inhibitors. Neoplasia 2018, 20, 1135–1143. [Google Scholar] [CrossRef]
- Malhas, A.; Saunders, N.J.; Vaux, D.J. The nuclear envelope can control gene expression and cell cycle progression via miRNA regulation. Cell Cycle 2010, 9, 531–539. [Google Scholar] [CrossRef]
- Cioce, M.; Ganci, F.; Canu, V.; Sacconi, A.; Mori, F.; Canino, C.; Korita, E.; Casini, B.; Alessandrini, G.; Cambria, A.; et al. Protumorigenic effects of mir-145 loss in malignant pleural mesothelioma. Oncogene 2013, 33, 5319–5331. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yan, G.; Zhang, Q.; Jiang, Y.; Sun, H.; Hu, Y.; Sun, J.; Xu, B. miR-145 inhibits isoproterenol-induced cardiomyocyte hypertrophy by targeting the expression and localization of GATA6. FEBS Lett. 2013, 587, 1754–1761. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef]
- Malaquin, N.; Carrier-Leclerc, A.; Dessureault, M.; Rodier, F. DDR-mediated crosstalk between DNA-damaged cells and their microenvironment. Front. Genet. 2015, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Alspach, E.; Flanagan, K.C.; Luo, X.; Ruhland, M.K.; Huang, H.; Pazolli, E.; Donlin, M.J.; Marsh, T.; Piwnica-Worms, D.; Monahan, J.; et al. p38MAPK Plays a Crucial Role in Stromal-Mediated Tumorigenesis. Cancer Discov. 2014, 4, 716–729. [Google Scholar] [CrossRef] [PubMed]
- Farrokhnia, F.; Aplin, J.D.; Westwood, M.; Forbes, K. MicroRNA Regulation of Mitogenic Signaling Networks in the Human Placenta. J. Biol. Chem. 2014, 289, 30404–30416. [Google Scholar] [CrossRef]
- Hu, G.; Zhao, X.; Wang, C.; Geng, Y.; Zhao, J.; Xu, J.; Zuo, B.; Zhao, C.; Wang, C.; Zhang, X. MicroRNA-145 attenuates TNF-alpha-driven cartilage matrix degradation in osteoarthritis via direct suppression of MKK4. Cell Death Dis. 2017, 8, e3140. [Google Scholar] [CrossRef]
- Long, X.; Miano, J.M. Transforming growth factor-beta1 (TGF-β1) utilizes distinct pathways for the transcriptional activation of microRNA 143/145 in human coronary artery smooth muscle cells. J. Biol. Chem. 2011, 286, 30119–30129. [Google Scholar] [CrossRef]
- O’Leary, L.; Sevinç, K.; Papazoglou, I.M.; Tildy, B.; Detillieux, K.; Halayko, A.J.; Chung, K.F.; Perry, M.M. Airway smooth muscle inflammation is regulated by microRNA-145 in COPD. FEBS Lett. 2016, 590, 1324–1334. [Google Scholar] [CrossRef]
- Hong, S.; Noh, H.; Chen, H.; Padia, R.; Pan, Z.K.; Su, S.-B.; Jing, Q.; Ding, H.-F.; Huang, S. Signaling by p38 MAPK Stimulates Nuclear Localization of the Microprocessor Component p68 for Processing of Selected Primary MicroRNAs. Sci. Signal. 2013, 6, ra16. [Google Scholar] [CrossRef]
- Hu, B.; Wu, Z.; Jin, H.; Hashimoto, N.; Liu, T.; Phan, S.H. CCAAT/enhancer-binding protein β isoforms and the regulation of α-smooth muscle actin gene expression by IL-1β. J. Immunol. 2004, 173, 4661–4668. [Google Scholar] [CrossRef]
- Yin, Y.; Yan, Z.-P.; Lu, N.-N.; Xu, Q.; He, J.; Qian, X.; Yu, J.; Guan, X.; Jiang, B.-H.; Liu, L.-Z. Downregulation of miR-145 associated with cancer progression and VEGF transcriptional activation by targeting N-RAS and IRS1. Biochim. Biophys. Acta Bioenerg. 2013, 1829, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to Sustain G2 Arrest After DNA Damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Spizzo, R.; Nicoloso, M.S.; Lupini, L.; Lu, Y.; Fogarty, J.; Rossi, S.; Zagatti, B.; Fabbri, M.C.; Veronese, A.; Liu, X.; et al. miR-145 participates with TP53 in a death-promoting regulatory loop and targets estrogen receptor-α in human breast cancer cells. Cell Death Differ. 2009, 17, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘Bystander senescence’. Aging 2012, 4, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, V.; Vickneson, K.; Kofidis, T.; Woo, C.C.; Lin, X.Y.; Foo, R.; Shanahan, C.M. Role of Vascular Smooth Muscle Cell Plasticity and Interactions in Vessel Wall Inflammation. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.E.; Varty, K.; Jones, L.; Bell, P.R.; London, N.J. Human saphenous vein organ culture: A useful model of intimal hyperplasia? Eur. J. Vasc. Endovasc. Surg. 1996, 11, 48–58. [Google Scholar] [CrossRef][Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hemmings, K.E.; Riches-Suman, K.; Bailey, M.A.; O’Regan, D.J.; Turner, N.A.; Porter, K.E. Role of MicroRNA-145 in DNA Damage Signalling and Senescence in Vascular Smooth Muscle Cells of Type 2 Diabetic Patients. Cells 2021, 10, 919. https://doi.org/10.3390/cells10040919
Hemmings KE, Riches-Suman K, Bailey MA, O’Regan DJ, Turner NA, Porter KE. Role of MicroRNA-145 in DNA Damage Signalling and Senescence in Vascular Smooth Muscle Cells of Type 2 Diabetic Patients. Cells. 2021; 10(4):919. https://doi.org/10.3390/cells10040919
Chicago/Turabian StyleHemmings, Karen E., Kirsten Riches-Suman, Marc A. Bailey, David J. O’Regan, Neil A. Turner, and Karen E. Porter. 2021. "Role of MicroRNA-145 in DNA Damage Signalling and Senescence in Vascular Smooth Muscle Cells of Type 2 Diabetic Patients" Cells 10, no. 4: 919. https://doi.org/10.3390/cells10040919
APA StyleHemmings, K. E., Riches-Suman, K., Bailey, M. A., O’Regan, D. J., Turner, N. A., & Porter, K. E. (2021). Role of MicroRNA-145 in DNA Damage Signalling and Senescence in Vascular Smooth Muscle Cells of Type 2 Diabetic Patients. Cells, 10(4), 919. https://doi.org/10.3390/cells10040919