
Repositioning Trimebutine Maleate as a Cancer Treatment Targeting Ovarian Cancer Stem Cells

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Analysis
2.3. Cell Sorting
2.4. Sphere Cell Proliferation Assay
2.5. Apoptosis (AV/PI staining) and Cell Cycle (PI Staining) Analysis
2.6. Quantitative RT-PCR
2.7. Whole Cell Patch Clamp Recording
2.8. CSCs Culture Condition and Western Blot Analysis
2.9. Drug Sensitivity of Ovarian CSCs in a Xenograft Tumor Model
2.10. Statistical Analysis
3. Results
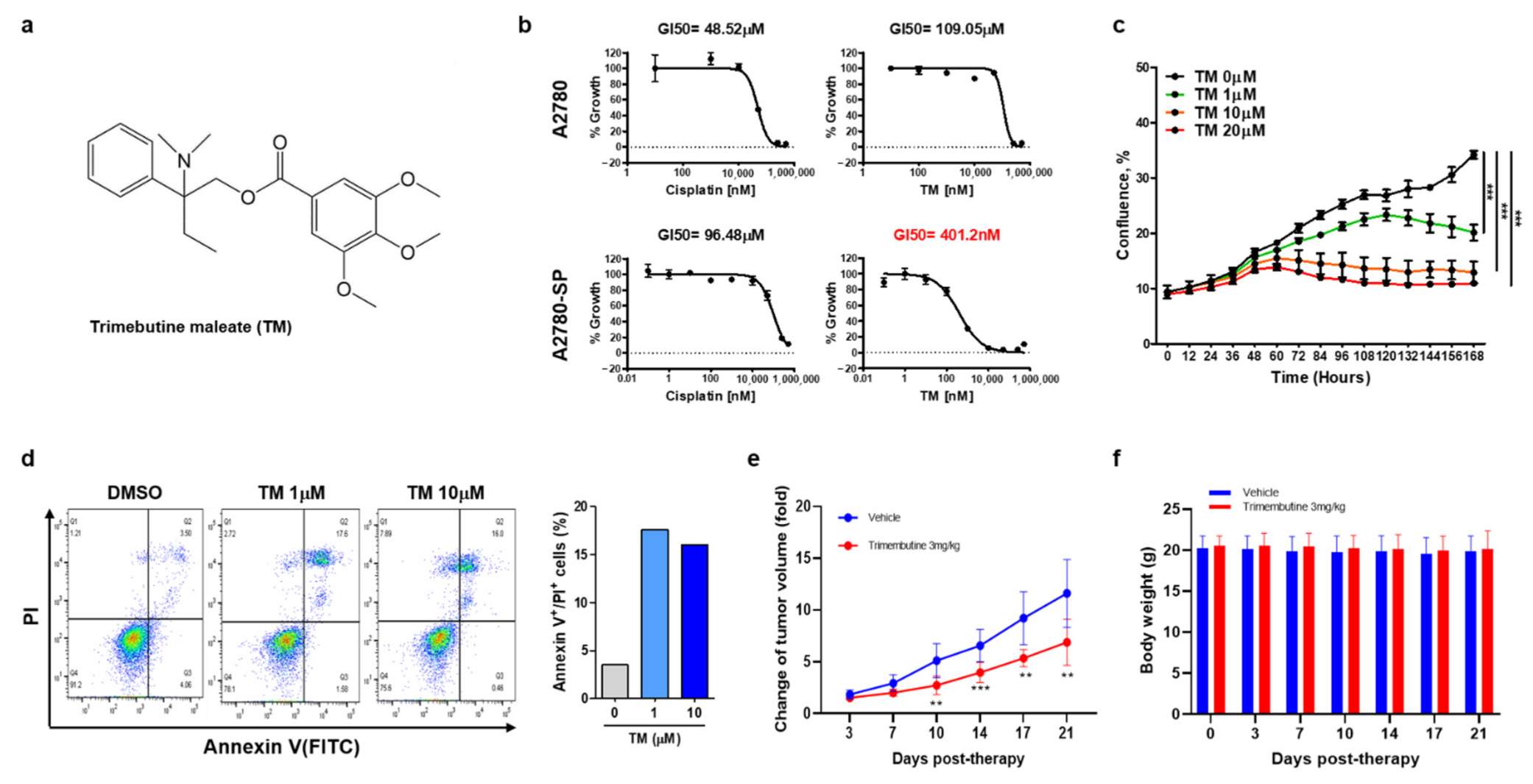
3.1. TM Specifically Inhibits Ovarian CSC Growth and Induces Cell Death
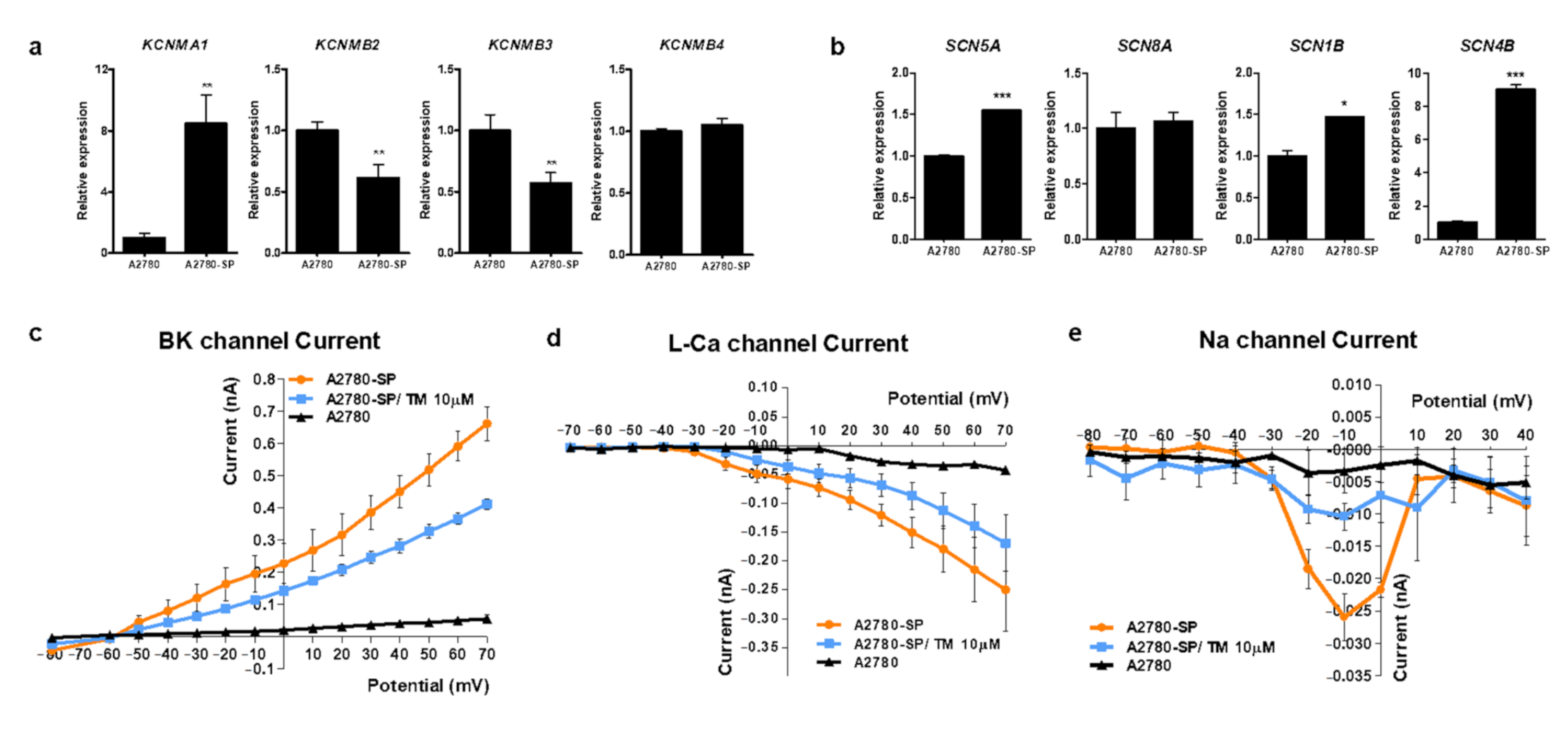
3.2. BKCa and Sodium Channel Subunits Are Overexpressed on Ovarian CSCs
3.3. TM Can Inhibit Ca2+, Na+ and BKCa Channel Currents in Ovarian CSCs
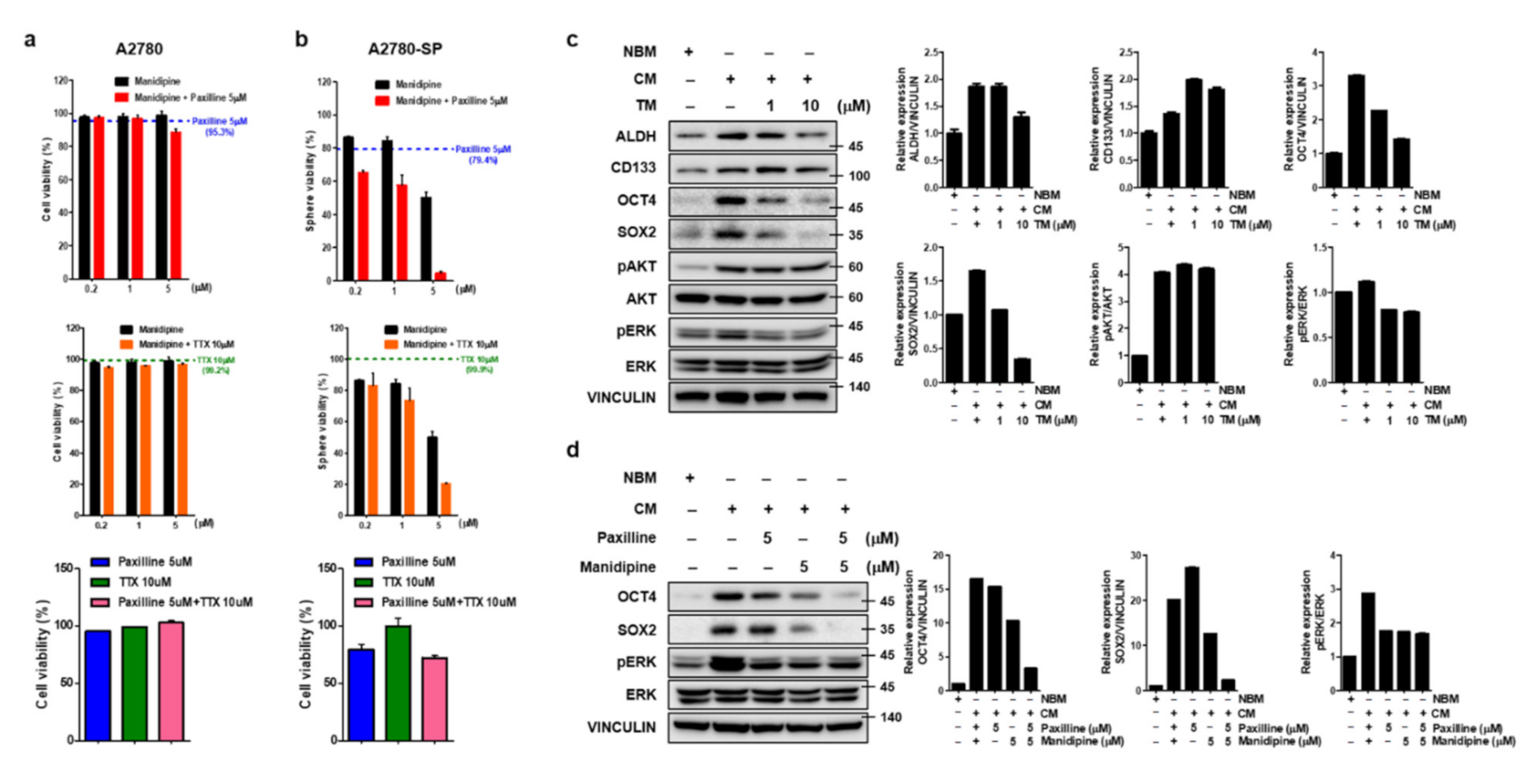
3.4. Simultaneous Inhibition of BK and Calcium Channels Amplifies Cell Growth Inhibition in Ovarian CSCs
3.5. TM Further Reduces Stemness-Related Transcription Factors by the Simultaneous Inhibition of BK and Calcium Channel in Ovarian CSCs
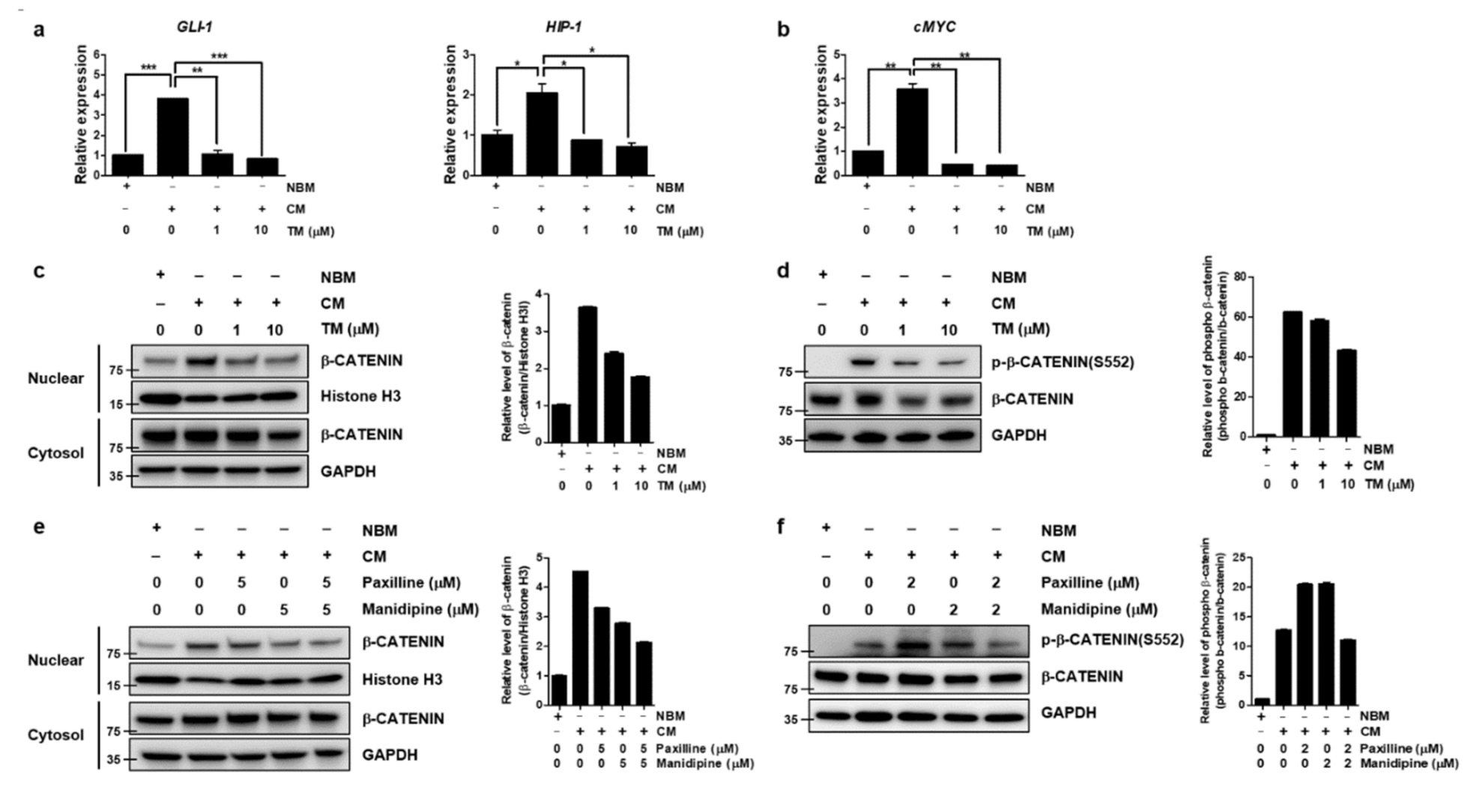
3.6. TM Further Reduces Wnt/β-Catenin Signaling by Dual Inhibition of BK and Calcium Channels in Ovarian CSCs
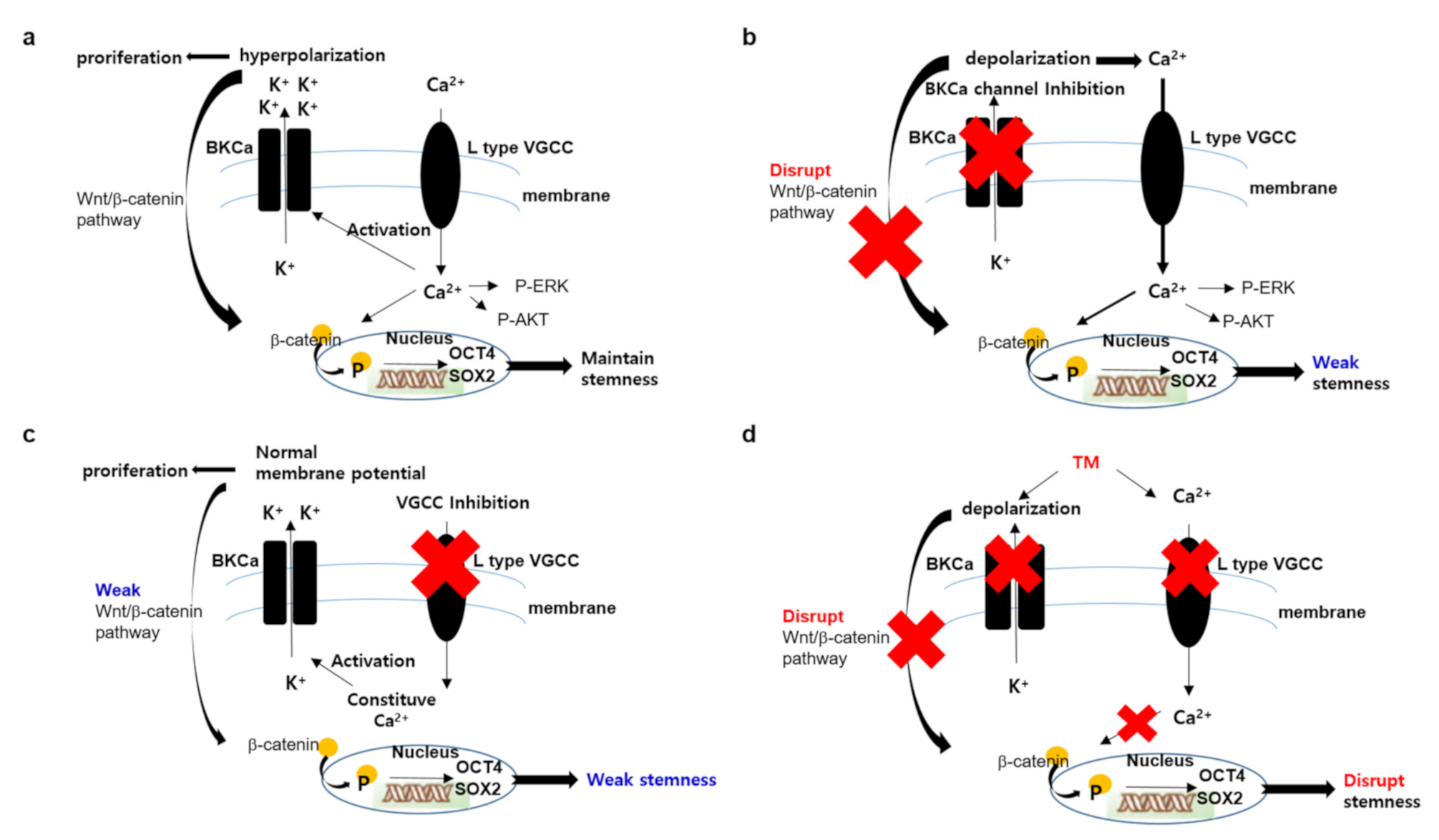
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Peres, L.C.; Cushing-Haugen, K.L.; Köbel, M.; Harris, H.R.; Berchuck, A.; Rossing, M.A.; Schildkraut, J.M.; Doherty, J.A. Invasive Epithelial Ovarian Cancer Survival by Histotype and Disease Stage. J. Natl. Cancer Inst. 2019, 111, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.J.; Armstrong, D.K.; Alvarez, R.D.; Bakkum-Gamez, J.N.; Behbakht, K.; Chen, L.-M.; Copeland, L.; Crispens, M.A.; DeRosa, M.; Dorigo, O.; et al. Ovarian Cancer, Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2016, 14, 1134–1163. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Han, Y.; Kim, S.I.; Kim, H.S.; Kim, S.J.; Song, Y.S. Tumor evolution and chemoresistance in ovarian cancer. NPJ Precis Oncol. 2018, 2, 20. [Google Scholar] [CrossRef]
- Bamford, J.; Webster, R.M. The ovarian cancer drug market. Nat. Rev. Drug Discov. 2017, 16, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Cortez, A.J.; Tudrej, P.; Kujawa, K.A.; Lisowska, K.M. Advances in ovarian cancer therapy. Cancer Chemother. Pharmacol. 2018, 81, 17–38. [Google Scholar] [CrossRef]
- Lisowska, K.M.; Olbryt, M.; Student, S.; Kujawa, K.A.; Cortez, A.J.; Simek, K.; Dansonka-Mieszkowska, A.; Rzepecka, I.K.; Tudrej, P.; Kupryjańczyk, J. Unsupervised analysis reveals two molecular subgroups of serous ovarian cancer with distinct gene expression profiles and survival. J. Cancer Res. Clin. Oncol. 2016, 142, 1239–1252. [Google Scholar] [CrossRef]
- Chien, J.; Kuang, R.; Landen, C.; Shridhar, V. Platinum-Sensitive Recurrence in Ovarian Cancer: The Role of Tumor Microenvironment. Front. Oncol. 2013, 3, 251. [Google Scholar] [CrossRef]
- Frank, N.Y.; Schatton, T.; Frank, M.H. The therapeutic promise of the cancer stem cell concept. J. Clin. Investig. 2010, 120, 41–50. [Google Scholar] [CrossRef]
- Salem, M.L.; El-Badawy, A.S.; Li, Z. Immunobiology and signaling pathways of cancer stem cells: Implication for cancer therapy. Cytotechnology 2014, 67, 749–759. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.G.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nat. Cell Biol. 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Haygood, C.L.W.; Arend, R.C.; Straughn, J.M.; Buchsbaum, D.J. Ovarian cancer stem cells: Can targeted therapy lead to improved progression-free survival? World J. Stem Cells 2014, 6, 441–447. [Google Scholar] [CrossRef]
- Lee, H.; Kim, J.W.; Kim, D.K.; Choi, D.K.; Lee, S.; Yu, J.H.; Kwon, O.-B.; Lee, J.; Lee, D.-S.; Kim, J.H.; et al. Calcium Channels as Novel Therapeutic Targets for Ovarian Cancer Stem Cells. Int. J. Mol. Sci. 2020, 21, 2327. [Google Scholar] [CrossRef]
- Delvaux, M.; Wingate, D. Trimebutine: Mechanism of Action, Effects on Gastrointestinal Function and Clinical Results. J. Int. Med. Res. 1997, 25, 225–246. [Google Scholar] [CrossRef]
- Frexinos, J.; Fioramonti, J.; Bueno, L. Effect of trimebutine on colonic myoelectrical activity in IBS patients. Eur. J. Clin. Pharmacol. 1985, 28, 181–185. [Google Scholar] [CrossRef]
- Schang, J.-C.; Devroede, G.; Pilote, M. Effects of trimebutine on colonic function in patients with chronic idiopathic constipation. Dis. Colon Rectum 1993, 36, 330–336. [Google Scholar] [CrossRef]
- Takenaga, H.; Magaribuchi, T.; Tamaki, H. Effects of trimebutine maleate (TM-906) on the spontaneous contraction of isolated guinea pig colon. Jpn. J. Pharmacol. 1984, 34, 177–181. [Google Scholar] [CrossRef]
- Roman, F.; Pascaud, X.; Taylor, J.E.; Junien, J.-L. Interactions of trimebutine with guinea-pig opioid receptors. J. Pharm. Pharmacol. 1987, 39, 404–407. [Google Scholar] [CrossRef]
- Allescher, H.D.; Ahmad, S.; Classen, M.; Daniel, E.E. Interaction of trimebutine and Jo-1196 (fedotozine) with opioid receptors in the canine ileum. J. Pharmacol. Exp. Ther. 1991, 257, 836–842. [Google Scholar]
- Pascaud, X.; Roman, F.; Petoux, F.; Vauche, D.; Junien, J.L. Participation des récepteurs opioïdes dans le mode d’action de la trimébutine [Involvement of opiate receptors in the mode of action of trimebutine]. Gastroenterol. Clin. Biol. 1987, 11, 77B–81B. [Google Scholar]
- Tan, W.; Zhang, H.; Luo, H.-S.; Xia, H. Effects of trimebutine maleate on colonic motility through Ca2+-activated K+ channels and L-type Ca2+ channels. Arch. Pharmacal Res. 2011, 34, 979–985. [Google Scholar] [CrossRef]
- Masaaki, N.; Tsutomu, K.; Hajime, T. Effects of trimebutine on cytosolic Ca2+ and force transitions in intestinal smooth muscle. Eur. J. Pharmacol. 1991, 195, 317–321. [Google Scholar] [CrossRef]
- Nagasaki, M.; Komori, S.; Tamaki, H.; Ohashi, H. Effect of trimebutine on K+ current in rabbit ileal smooth muscle cells. Eur. J. Pharmacol. 1993, 235, 197–203. [Google Scholar] [CrossRef]
- Roman, F.J.; Lanet, S.; Hamon, J.; Brunelle, G.; Maurin, A.; Champeroux, P.; Richard, S.; Alessandri, N.; Gola, M. Pharmacological properties of trimebutine and N-monodesmethyltrimebutine. J. Pharmacol. Exp. Ther. 1999, 289, 1391–1397. [Google Scholar]
- Seo, E.J.; Kwon, Y.W.; Jang, I.H.; Kim, D.K.; Lee, S.I.; Choi, E.J.; Kim, K.-H.; Suh, D.-S.; Lee, J.H.; Choi, K.U.; et al. Autotaxin Regulates Maintenance of Ovarian Cancer Stem Cells through Lysophosphatidic Acid-Mediated Autocrine Mechanism. Stem Cells 2016, 34, 551–564. [Google Scholar] [CrossRef]
- Lee, H.; Kim, J.W.; Choi, D.K.; Yu, J.H.; Kim, J.H.; Lee, D.-S.; Min, S.-H. Poziotinib suppresses ovarian cancer stem cell growth via inhibition of HER4-mediated STAT5 pathway. Biochem. Biophys. Res. Commun. 2020, 526, 158–164. [Google Scholar] [CrossRef]
- Annett, S.; Robson, T. Targeting cancer stem cells in the clinic: Current status and perspectives. Pharmacol. Ther. 2018, 187, 13–30. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, W.; Li, Y. Stemness-related markers in cancer. Cancer Transl. Med. 2017, 3, 87–95. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 1–28. [Google Scholar] [CrossRef]
- Wang, Y.K.; Zhu, Y.L.; Qiu, F.M.; Zhang, T.; Chen, Z.G.; Zheng, S.; Huang, J. Activation of Akt and MAPK pathways enhances the tumorigenicity of CD133+ primary colon cancer cells. Carcinogenesis 2010, 31, 1376–1380. [Google Scholar] [CrossRef]
- Morgensztern, D.; McLeod, H.L. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anti. Cancer Drugs 2005, 16, 797–803. [Google Scholar] [CrossRef]
- Dierks, C.; Beigi, R.; Guo, G.-R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-Positive Leukemic Stem Cells Is Dependent on Hedgehog Pathway Activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef]
- Merchant, A.A.; Matsui, W. Targeting Hedgehog—A Cancer Stem Cell Pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef]
- Wei, P.; Walls, M.; Qiu, M.; Ding, R.; Denlinger, R.H.; Wong, A.; Tsaparikos, K.; Jani, J.P.; Hosea, N.A.; Sands, M.; et al. Evaluation of Selective γ-Secretase Inhibitor PF-03084014 for Its Antitumor Efficacy and Gastrointestinal Safety to Guide Optimal Clinical Trial Design. Mol. Cancer Ther. 2010, 9, 1618–1628. [Google Scholar] [CrossRef]
- Vermeulen, L.; Melo, F.D.S.E.; Van Der Heijden, M.; Cameron, K.; De Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Donahue, R.N.; McLaughlin, P.J.; Zagon, I.S. Cell proliferation of human ovarian cancer is regulated by the opioid growth factor-opioid growth factor receptor axis. Am. J. Physiol. Integr. Comp. Physiol. 2009, 296, R1716–R1725. [Google Scholar] [CrossRef]
- Avella, D.M.; Kimchi, E.T.; Donahue, R.N.; Tagaram, H.R.S.; McLaughlin, P.J.; Zagon, I.S.; Staveley-O’Carroll, K.F. The opioid growth factor-opioid growth factor receptor axis regulates cell proliferation of human hepatocellular cancer. Am. J. Physiol. Integr. Comp. Physiol. 2010, 298, R459–R466. [Google Scholar] [CrossRef]
- Fan, Y.-P.; Liu, P.; Xue, W.-K.; Zhao, W.-J.; Pan, H.-C. Trimebutine Promotes Glioma Cell Apoptosis as a Potential Anti-tumor Agent. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Guéguinou, M.; Chantôme, A.; Fromont, G.; Bougnoux, P.; Vandier, C.; Potier-Cartereau, M. KCa and Ca2+ channels: The complex thought. Biochim. Biophys. Acta Bioenerg. 2014, 1843, 2322–2333. [Google Scholar] [CrossRef]
- Contreras, G.F.; Castillo, K.; Enrique, N.; Carrasquel-Ursulaez, W.; Castillo, J.P.; Milesi, V.; Neely, A.; Alvarez, O.; Ferreira, G.; González, C.; et al. A BK (Slo1) channel journey from molecule to physiology. Channels 2013, 7, 442–458. [Google Scholar] [CrossRef] [PubMed]
- Oeggerli, M.; Tian, Y.; Ruiz, C.; Wijker, B.; Sauter, G.; Obermann, E.; Güth, U.; Zlobec, I.; Sausbier, M.; Kunzelmann, K.; et al. Role of KCNMA1 in Breast Cancer. PLoS ONE 2012, 7, e41664. [Google Scholar] [CrossRef] [PubMed]
- Khaitan, D.; Sankpal, U.T.; Weksler, B.; Meister, E.A.; Romero, I.A.; Couraud, P.-O.; Ningaraj, N.S. Role of KCNMA1gene in breast cancer invasion and metastasis to brain. BMC Cancer 2009, 9, 258. [Google Scholar] [CrossRef] [PubMed]
- Bloch, M.; Ousingsawat, J.; Simon, R.; Schraml, P.; Gasser, T.C.; Mihatsch, M.J.; Kunzelmann, K.; Bubendorf, L. KCNMA1 gene amplification promotes tumor cell proliferation in human prostate cancer. Oncogene 2006, 26, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Sundelacruz, S.; Levin, M.; Kaplan, D.L. Role of Membrane Potential in the Regulation of Cell Proliferation and Differentiation. Stem Cell Rev. Rep. 2009, 5, 231–246. [Google Scholar] [CrossRef]
- Mazar, J.; Deyoung, K.; Khaitan, D.; Meister, E.; Almodovar, A.; Goydos, J.; Ray, A.; Perera, R.J. The Regulation of miRNA-211 Expression and Its Role in Melanoma Cell Invasiveness. PLoS ONE 2010, 5, e13779. [Google Scholar] [CrossRef]
- Wondergem, R.; Bartley, J.W. Menthol increases human glioblastoma intracellular Ca2+, BK channel activity and cell migration. J. Biomed. Sci. 2009, 16, 90. [Google Scholar] [CrossRef]
- Schickling, B.M.; England, S.K.; Aykin-Burns, N.; Norian, L.A.; Leslie, K.K.; Frieden-Korovkina, V.P. BKCa channel inhibitor modulates the tumorigenic ability of hormone-independent breast cancer cells via the Wnt pathway. Oncol. Rep. 2014, 33, 533–538. [Google Scholar] [CrossRef][Green Version]
- Yi, F.; Pereira, L.; Hoffman, J.A.; Shy, B.R.; Yuen, C.M.; Liu, D.R.; Merrill, B.J. Opposing effects of Tcf3 and Tcf1 control Wnt stimulation of embryonic stem cell self-renewal. Nat. Cell Biol. 2011, 13, 762–770. [Google Scholar] [CrossRef]
- Yong, X.; Tang, B.; Xiao, Y.-F.; Xie, R.; Qin, Y.; Luo, G.; Hu, C.-J.; Dong, H.; Yang, S.-M. Helicobacter pylori upregulates Nanog and Oct4 via Wnt/β-catenin signaling pathway to promote cancer stem cell-like properties in human gastric cancer. Cancer Lett. 2016, 374, 292–303. [Google Scholar] [CrossRef]
- Gackière, F.; Warnier, M.; Katsogiannou, M.; Derouiche, S.; Delcourt, P.; Dewailly, E.; Slomianny, C.; Humez, S.; Prevarskaya, N.; Roudbaraki, M.; et al. Functional coupling between large-conductance potassium channels and Cav3.2 voltage-dependent calcium channels participates in prostate cancer cell growth. Biol. Open 2013, 2, 941–951. [Google Scholar] [CrossRef]
- Lheureux, S.; Msc, M.B.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA A Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef]
- Li, N.; Zhan, X. Identification of clinical trait–related lncRNA and mRNA biomarkers with weighted gene co-expression network analysis as useful tool for personalized medicine in ovarian cancer. EPMA J. 2019, 10, 273–290. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Forward 5′-3′ | Reverse 5′-3′ |
|---|---|---|
| GAPDH | GGAGCCAAAAGGGTCATCAT | GTGATGGCATGGACTGTGGT |
| KCNMA1 | TCTTTGCTCTCAGCATCGGTG | CCGCAAGCCGAAGTAGAGAAG |
| KCNMB2 | GAGGACCGAGCTATTCTCCTG | TGTTTCCGTGATGGACGCATT |
| KCNMB3 | TTCTTGCTCGGAACAACCATT | GATGGCAGTGCAGGTCGATT |
| KCNMB4 | GGTCTACGTGAACAACTCTGAG | GGAGGGATATAGGAGCACTTGG |
| SCN5A | TCTCTATGGCAATCCACCCCA | GAGGACATACAAGGCGTTGGT |
| SCN8A | CTCCTGACTGGTCGAAGAATGT | CATGGGTCCCGTAAAAAGGTAA |
| SCN1B | CTGCTGGCCTTAGTGGTCG | GTGAAGGTCTCAGCGTTGGTC |
| SCN4B | CAACAGCAGTGACGCATTCAA | CTCCTTAGTAGAGCCTACCAGAG |
| OPRM1 | GCCCTTCCAGAGTGTGAATTAC | GTGCAGAGGGTGAATATGCTG |
| OPRK1 | ATCATCACGGCGGTCTACTC | ACTCTGAAAGGGCATGGTTGTA |
| CACNA1C | ATGAACATGCCTCTGAACAGCG | TTCAGAAGCAGCGGACACAGC |
| CACNA1D | TGTGATTTGCAAGATGACGAGCC | TGAGATTGGCATTTGTTGAGGTG |
| CACNA1F | ATTTCTGTGTCTCTGCCTGTCG | TAGGTAGGAAAGCCGATCAGG |
| CACNA1G | TGTCTCCGCACGGTCTGTAA | AAGCCGGTTCCAAGTGTCTC |
| CACNA1H | TTGGGTTCCGGTCGGTTCT | ATGCCCGTAGCCATCTTCA |
| CACNA1I | ATCGGTTATGCTTAGGATTGTCA | TGCTCCCGTTGCTTGGTCTC |
| GLI-1 | AGCGTGAGCCTGAATCTGTG | CAGCATGTACTGGGCTTTGAA |
| HIP-1 | ACACGCCAGAACGTGCATA | CACTGCGTTGCTAGACAGAG |
| c-MYC | GGCTCCTGGCAAAAGGTCA | CTGCGTAGTTGTGCTGATGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.; Kwon, O.-B.; Lee, J.-E.; Jeon, Y.-H.; Lee, D.-S.; Min, S.-H.; Kim, J.-W. Repositioning Trimebutine Maleate as a Cancer Treatment Targeting Ovarian Cancer Stem Cells. Cells 2021, 10, 918. https://doi.org/10.3390/cells10040918
Lee H, Kwon O-B, Lee J-E, Jeon Y-H, Lee D-S, Min S-H, Kim J-W. Repositioning Trimebutine Maleate as a Cancer Treatment Targeting Ovarian Cancer Stem Cells. Cells. 2021; 10(4):918. https://doi.org/10.3390/cells10040918
Chicago/Turabian StyleLee, Heejin, Oh-Bin Kwon, Jae-Eon Lee, Yong-Hyun Jeon, Dong-Seok Lee, Sang-Hyun Min, and Jun-Woo Kim. 2021. "Repositioning Trimebutine Maleate as a Cancer Treatment Targeting Ovarian Cancer Stem Cells" Cells 10, no. 4: 918. https://doi.org/10.3390/cells10040918
APA StyleLee, H., Kwon, O.-B., Lee, J.-E., Jeon, Y.-H., Lee, D.-S., Min, S.-H., & Kim, J.-W. (2021). Repositioning Trimebutine Maleate as a Cancer Treatment Targeting Ovarian Cancer Stem Cells. Cells, 10(4), 918. https://doi.org/10.3390/cells10040918