Abstract
Our study assesses the effects of anti-VEGF (Vascular Endothelial Growth Factor) drugs and Trichostatin A (TSA), an inhibitor of histone deacetylase (HDAC) activity, on cultured ARPE-19 (Adult Retinal Pigment Epithelial-19) cells that are immortalized human retinal pigment epithelial cells. ARPE-19 cells were treated with the following anti-VEGF drugs: aflibercept, ranibizumab, or bevacizumab at 1× and 2× concentrations of the clinical intravitreal dose (12.5 μL/mL and 25 μL/mL, respectively) and analyzed for transcription profiles of genes associated with the pathogenesis age-related macular degeneration (AMD). HDAC activity was measured using the Fluorometric Histone Deacetylase assay. TSA downregulated HIF-1α and IL-1β genes, and upregulated BCL2L13, CASPASE-9, and IL-18 genes. TSA alone or bevacizumab plus TSA showed a significant reduction of HDAC activity compared to untreated ARPE-19 cells. Bevacizumab alone did not significantly alter HDAC activity, but increased gene expression of SOD2, BCL2L13, CASPASE-3, and IL-18 and caused downregulation of HIF-1α and IL-18. Combination of bevacizumab plus TSA increased gene expression of SOD2, HIF-1α, GPX3A, BCL2L13, and CASPASE-3, and reduced CASPASE-9 and IL-β. In conclusion, we demonstrated that anti-VEGF drugs can: (1) alter expression of genes involved in oxidative stress (GPX3A and SOD2), inflammation (IL-18 and IL-1β) and apoptosis (BCL2L13, CASPASE-3, and CASPASE-9), and (2) TSA-induced deacetylation altered transcription for angiogenesis (HIF-1α), apoptosis, and inflammation genes.
1. Introduction
Pathological angiogenesis, which subsequently leads to choroidal neovascularization, subretinal fibrosis, and exudative hemorrhage, is an underlying cause of the severe, late-stage, wet form of AMD (Age-related Macular Degeneration) [1].
Wet or neovascular AMD, which accounts for 10–20% of cases, is the less common of the two types of AMD. However, 90% of AMD-associated irreversible vision loss is attributed to wet AMD [2]. Dry AMD is characterized by degeneration of Retinal Pigment Epithelial (RPE) cells and accounts for 80% of AMD cases.
VEGF (Vascular Endothelial Growth Factor) is a signaling growth factor for vascular endothelial cells and a critical angiogenic factor that stimulates ocular neovascularization. Therefore, the most widely used wet AMD treatment targets the pro-angiogenic activity of VEGF to inhibit ocular neovascularization [3]. Administration of intravitreal injections of anti-VEGF drugs, such as ranibizumab (Lucentis), bevacizumab (Avastin), and aflibercept (Eylea), to wet AMD patients is successfully and routinely being used as a wet AMD therapy worldwide [4,5]. Despite the widespread use of anti-VEGF drugs, 10–15% of patients fail to respond to rigorous treatment protocols in clinical trial settings [6,7,8,9,10]. This variability in AMD patients’ response to therapy has been attributed to several clinical, behavioral, and genetic factors [11]. Pharmacogenetic studies have identified VEGFA, VEGFR2 (VEGF Receptor 2), CFH (Complement Factor H), and ARMS2 (Age-Related Maculopathy Susceptibility 2) as potential biomarkers for response to anti-VEGF drugs [12]. Many investigators, including the CATT (Comparison of Age-related Macular Degeneration Treatments) and IVAN (Inhibition of VEGF in Age-related choroidal Neovascularisation) research groups, did not find any significant association between genes polymorphism and visual or anatomic responses to treatment [13,14,15,16,17]. This inconsistency in findings by pharmacogenetic studies could be explained in part by possible gene–gene or gene–environmental interactions [18,19].
Genetic and environmental factors contribute to the development and progression of AMD. Genome-wide association studies (GWAS) have identified 52 genetic variants distributed across 34 loci associated with AMD [20]. Furthermore, epigenetic modifications, which include DNA methylation, histone acetylation/deacetylation, non-coding RNA-mediated gene silencing, and chromatin remodeling [21] have been implicated in the pathogenesis of AMD by selective transcription of genes involved in angiogenesis, inflammation, and oxidative stress pathways [22,23]. Epigenetic mechanisms result in covalent modifications in the DNA and regulate gene transcription either by activation or repression, in response to environmental stimuli and are often heritable [24]. Epigenetics can elucidate gene–environment interactions and explain why a certain genotype frequently results in different phenotypes [25]. Histone acetylation is catalyzed by histone transferases (HATs) and acts to destabilize nucleosomes and unwrap DNA to make it accessible to transcription factors. Conversely, histone deacetylation, carried out by histone deacetylases (HDACs), stabilizes nucleosomes and represses DNA transcription [26]. Histone acetylation is known to regulate the expression of 2–10% of genes. Other non-histone proteins, particularly transcription factors, are also regulated by acetylation/deacetylation. This could explain the fact that gene expression is not always silenced by deacetylation [27].
AMD being a leading cause of blindness in the United States and the third major cause of visual impairment worldwide, [28] poses a major health risk to the elderly population, and AMD risk is projected to increase by 54% in the United States in the next five year [29]. Therefore, we speculate that delving into the mechanisms of action of the currently used anti-VEGF drugs might contribute to the design of more effective therapeutic strategies for wet AMD. To this end, the current in vitro study was designed to examine the effects of anti-VEGF drugs on epigenetic regulation in immortalized human ARPE-19 cell lines. The ARPE-19 cell line used in this study was originally developed from the retinal pigment epithelium (RPE) of a human donor eye and resembles the phenotype and properties characteristic of aged native human RPE cells, lack of pigmentation, weak tight junctions, reduced expression of all-trans retinol, Pigment-Epithelium-Derived Factor (PEDF), and RPE markers, and hypersensitivity to VEGF activity, thereby making the ARPE-19 cell line an ideal in vitro AMD model [30]. However, it should be emphasized that the ARPE-19 cell line loses some of the aging RPE characteristics, especially with increasing passages, such as morphology, retinoid metabolism, and VEGF secretion. Furthermore, it should be mentioned that depigmentation in the RPE of AMD eyes is different from that of ARPE-19 cells. Melanosome density in the RPE decreases significantly with normal aging and more evidently in AMD, but melanin is not completely lost. More importantly, RPE melanin in AMD loses its antioxidant properties [31].
RPE cells in vivo form the outer blood-brain barrier and support photoreceptor cells and enable phototransduction. The outer blood-brain barrier is formed of a continuous layer of tight junctions that enable transepithelial transport and phototransduction. Pigment-Epithelium-Derived Factor (PEDF), which maintains barrier integrity and is endogenously secreted by RPE in vivo in large amounts, shows only basal levels in aged eyes. The in vitro ARPE-19 cell line is a rapidly growing immortalized human cell line derived from primary RPE cells from the globes of a 19-year-old male donor. ARPE-cell lines express RPE cell-specific markers CRALBP and RPE65 and form a viable cuboidal to columnar epithelium monolayer in culture media. Our recent study characterized and confirmed the expression of the following RPE-specific markers in the ARPE-19 cell line used in our lab: Cellular retinaldehyde binding protein-1 (CRALBP), Bestrophin1 (BEST1), and Keratin-18 (KRT18) [32]. Although ARPE-19 cells retain the characteristic features of RPE, including defined cell borders and pigmentation, they require considerable time for differentiation and are unable to completely differentiate into RPE-like layers found in vivo. ARPE-19 cells show partial polarization as some of the cells in the monolayer resemble the morphology of differentiated RPE cells such as apical microvilli, polarized distribution of organelles, basolateral infoldings, and junctional complexes on the apical plasma membrane. ARPE-19 cells exhibit low transepithelial resistance (TER) that reaches a maximum value of 50–100 Ω cm2 after 28 days of culture in low-serum media in laminin-coated Transwell-COL filters. It is speculated that the low TER might be due to heterogeneity of the cell line since some of the cells show polarization [33].
The present study demonstrated that treatment of ARPE-19 cells with anti-VEGF drugs altered the total HDAC protein activity and the gene expression levels of apoptotic, inflammatory, and oxidative stress-related genes. Moreover, addition of Trichostatin-A (TSA), an HDAC inhibitor, along with an anti-VEGF drug modulated the gene expression of VEGF, apoptotic and inflammatory markers. These results suggest that epigenetics modulation in ARPE-19 cells is strongly influenced by anti-VEGF drug treatment.
2. Materials and Methods
2.1. ARPE-19 Cell Culture
Human RPE cells (ARPE-19 cells, ATCC, Manassas, VA, USA) were cultured till confluent in 175 cm2-flasks containing DMEM/F-12 culture medium (Dulbecco’s Modification of Eagle’s Medium, Mediatech, Inc., Manassas, VA, USA), 10% dialyzed fetal bovine serum, antibiotics (penicillin G 100 U/mL, streptomycin sulfate 0.1 mg/mL, gentamicin 10 μg/mL, amphotericin B 2.5 μg/mL) and 17.5 mM glucose. All ARPE-19 cells were at passage 10 and cultured side-by-side under identical standard conditions of 37 °C in 5% CO2 and 95% relative humidity, in order to avoid any potential technical variability.
ARPE-19 cells are a spontaneously arising RPE cell line derived by Amy Aotaki-Keen from the normal eyes of a 19-year-old male who died from head trauma in a motor vehicle accident in 1986. The ARPE-19 cell line, established using the cuboidal basal cell layer cultured in specific culture media, expresses the RPE-specific markers cellular retinaldehyde binding protein and RPE-65.
In our study we utilized ARPE-19 cells at passage 10 in all our experiments to ensure the cells retained acceptable fidelity.
2.2. Drug Treatment of ARPE-19 Cell Cultures
ARPE-19 cells were plated in triplicate for 24 h in 6-well plates at a density of 500,000 cells per well, culture media were removed and replaced with the same media containing anti-VEGF drugs: aflibercept, ranibizumab or bevacizumab at 1× and 2× concentrations of the clinical intravitreal dose (12.5 μL/mL and 25 μL/mL, respectively). The clinical dose was calculated by assuming that the amount of each drug clinically used in intravitreal injections distributes equally throughout the 4 mL human vitreous. Untreated cells were used as control.
In order to further explore the potential relationship between anti-angiogenic treatment and the acetylation status of the target genes expression, a subset of ARPE-19 cells was treated with trichostatin A (TSA), an inhibitor of histone deacetylase (HDAC) activity, at 0.3 μM concentration, or a combination of 1X bevacizumab plus 0.3 μM TSA.
The control and anti-VEGF treated cultures were incubated for an additional 24 h, then RNA was isolated to be used for Quantitative Real-Time PCR (qRT-PCR) analyses.
Proteins extracted from cultures of untreated ARPE-19 cells, as well as cells treated with 1X bevacizumab, 0.3 μM TSA, and a combination of both drugs were analyzed for HDAC activity as described below.
2.3. RNA Extraction, Amplification of cDNA, and Quantitative Real-Time PCR (qRT-PCR) Analysis
ARPE-19 cells were pelleted for RNA isolation using a PureLink RNA Mini Kit (Ambion, Thermo Fisher Scientific, Waltham, MA, USA). For qRT-PCR analyses, 100 ng of individual RNA samples were reverse transcribed into complementary DNA (cDNA) using SuperScript VILO Master Mix (Thermo Fisher Scientific, Waltham, MA, USA).
We investigated transcription profiles of downstream genes known to play a role in AMD pathogenesis. RNA samples were isolated from cells that were (a) untreated; (b) treated with 1× or 2× concentrations of the three anti-VEGF drugs; (c) treated with 0.3μM TSA alone; and (d) treated with 1× bevacizumab plus TSA (bevacizumab/TSA). qRT-PCR was performed using primers for downstream pathway genes, including angiogenesis (VEGF-A and HIF-1α), apoptosis (BCL2L13, CASPASE-3, and CASPASE-9), inflammation (IL-18 and IL-1β) and oxidative stress (GPX3A and SOD2) (Table 1). The qRT-PCR was performed on individual samples using QuantiFast SYBR Green PCR Kit (Qiagen, Inc., Germantown, MD, USA) on StepOne Plus Real-Time PCR system (Thermo Fisher Scientific, Waltham, MA, USA). For the various target genes, housekeeping genes that had comparable amplification efficiencies were chosen in order to maximize the accuracy of our ΔΔCT values. The housekeeper genes were either hypoxanthine phosphorbosyltransferase 1 (HPRT1) or hydroxymethylbilane synthase (HMBS). Untreated samples were used as control. ΔΔCts (differences in cycle thresholds) were obtained and folds calculated using the formula 2ΔΔCt.

Table 1.
Gene symbols, names, gene bank accession numbers, and functions.
2.4. Protein Extraction and HDAC Activity Assay
ARPE-19 cell samples were lysed using RIPA buffer (Cat. #89900, Life Technologies Corp., Calsbad, CA, USA), supernatants were transferred to a new microfuge tube and protein concentrations were measured using the Bio-Rad DC Protein Assay system (Bio-Rad Laboratories, Richmond, CA, USA) according to the manufacturer’s instructions. Protein samples were kept in a −80 °C freezer until time of use in the HDAC activity assay.
HDAC activity in the protein samples was measured using the Fluorometric Histone Deacetylase Assay Kit (Sigma–Aldrich, St. Louis, MO, USA) according to the manufacturer’s protocol. Briefly, protein samples, assay buffer and HDAC substrate solution were added to the wells of a 96-well plate. Each well contained 20 μL of the protein sample, 30 μL of assay buffer and 50 μL of HDAC substrate solution. The plate was incubated at 30 °C for 30 min, then 10 μL of Developer solution added to each well. The plate was incubated 10 min at room temperature, and fluorescence measured with a microplate spectrofluorometer (Gemini XPS, Molecular Devices, Sunnyvale, CA, USA) using an excitation wavelength of 350 nm and an emission wavelength of 440 nm. Samples were plated in triplicate and Hela cell lysate used as a positive control for HDAC activity. Experiments were performed on three replicates i.e., in triplicate. The plate in which the cells were plated was read three times and the fluorescence intensity was averaged. The entire experiment, i.e., treatment and reading the fluorescence was repeated three times to ensure reproducibility. Protein samples from untreated ARPE-19 cells were used as control and were normalized to 100%.
2.5. Statistical Analyses
Statistical analyses of the data were performed by unpaired t-test using GraphPad Prism, Version 5 (GraphPad Software, Inc., La Jolla, CA, USA). p < 0.05 was considered statistically significant. Untreated samples (controls) were normalized to a value of 100% for comparison to treated samples.
3. Results
3.1. Measurement of HDAC Activity in ARPE-19 Cells before and after Treatment with Anti-VEGF and TSA
Treatment of ARPE-19 cells with 0.3 μM TSA resulted in significant reduction of HDAC activity (p = 0.0003), as did the combination of bevacizumab 1× plus TSA (p < 0.0001) (Figure 1). Although treatment with bevacizumab 1X alone did not significantly alter HDAC activity (p = 0.15), its addition to TSA significantly potentiated its inhibition of HDAC activity (p = 0.0001).
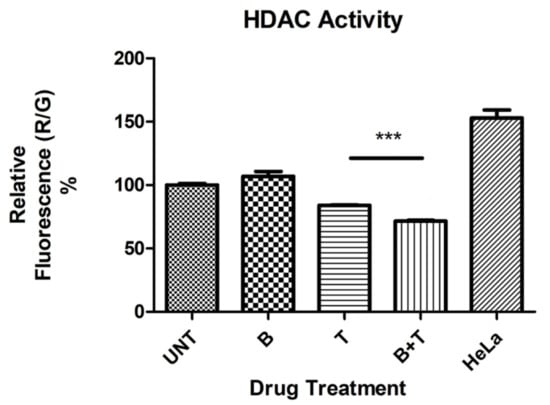
Figure 1.
Histone deacetylase (HDAC) activity as determined by relative fluorescence (%) in untreated and treated ARPE-19 cultures. *** p < 0.001; Bars with no asterisk represent nonsignificant difference. UNT: Untreated; B: Bevacizumab 1× conc.; T: Trichostatin A 0.3 μM; B+T: Bevacizumab 1× conc. + Trichostatin A 0.3 μM. HeLa: Hela cell lysate positive control for HDAC activity. Error bars represent ‘Mean ± SEM’.
3.2. Effect of Anti-VEGF Treatment and HDAC Inhibition on the Expression Profiles of Downstream Genes
Aflibercept 1×-treated ARPE-19 cells showed upregulation of VEGF-A (1.12-fold, p = 0.67), HIF-1α (1.03-fold, p = 0.63), GPX3A (1.13-fold, p = 0.49), BCL2L13 (1.26-fold, p = 0.05), CASPASE-3 (1.21-fold, p = 0.04), CASPASE-9 (1.32-fold, p = 0.003) and IL-1β (1.38-fold, p = 0.22); and downregulation of SOD2 (0.76-fold, p = 0.14) and IL-18 (0.95-fold, p = 0.051), compared to untreated cells (Table 2, Figure 2).
Table 2.
Expression folds for downstream genes in untreated and anti-vascular endothelial growth factor (anti-VEGF)-treated ARPE-19 cultures *.
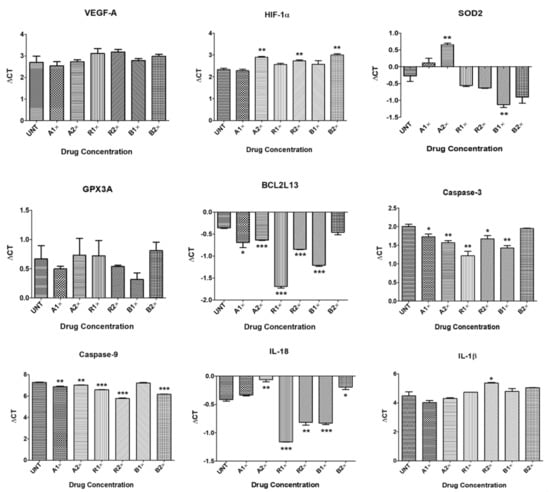
Figure 2.
Quantitative Real-Time PCR (qPCR) data showing Delta Cts for downstream genes in untreated and anti-VEGF treated ARPE-19 cultures. * p < 0.05; ** p < 0.01; *** p < 0.001; Bars with no asterisk represent nonsignificant difference. UNT: Untreated; A1×: Aflibercept 1× conc.; A2×: Aflibercept 2× conc.; R1×: Ranibizumab 1× conc.; R2×: Ranibizumab 2× conc.; B1×: Bevacizumab 1× conc.; B2×: Bevacizumab 2× conc. Error bars represent ‘Mean ± SEM’.
Treatment with aflibercept 2× downregulated the expression of VEGF-A (0.98-fold, p = 0.94), HIF-1α (0.67-fold, p = 0.002), SOD2 (0.52-fold, p = 0.005), GPX3A (0.96-fold, p = 0.87), and IL-18 (0.78-fold, p = 0.002), and led to upregulation of BCL2L13 (1.21-fold, p < 0.0001), CASPASE-3 (1.36-fold, p = 0.006), CASPASE-9 (1.19-fold, p = 0.004) and IL-1β (1.14-fold, p = 0.55) (Table 2, Figure 2).
ARPE-19 cells treated with ranibizumab 1× showed downregulation of VEGF-A (0.75-fold, p = 0.40), HIF-1α (0.85-fold, p = 0.05), GPX3A (0.96-fold, p = 0.89), and IL-1β (0.84-fold, p = 0.43) and upregulation of SOD2 (1.22-fold, p = 0.16), BCL2L13 (1.21-fold, p > 0.0001), CASPASE-3 (1.73-fold, p = 0.004), CASPASE-9 (1.61-fold, p < 0.0001) and IL-18 (1.67-fold, p < 0.0001) (Table 2, Figure 2).
Ranibizumab 2×-treated cells showed downregulation of VEGF-A (0.72-fold, p = 0.20), HIF-1α (0.76-fold, p = 0.008) and IL-1β (0.54-fold, p = 0.04), and upregulation of SOD2 (1.28-fold, p = 0.09), GPX3A (1.09-fold, p = 0.60), BCL2L13 (1.4-fold, p < 0.0001), CASPASE-3 (1.26-fold, p = 0.04), CASPASE-9 (2.82-fold, p < 0.0001) and IL-18 (1.32-fold, p = 0.002) (Table 2, Figure 2).
Treatment with bevacizumab 1× decreased the expression of VEGF-A (0.95-fold, p = 0.80), HIF-1α (0.85-fold, p = 0.26) and IL-1β (0.81-fold, p = 0.42), and increased the expression of SOD2 (1.8-fold, p = 0.009), GPX3A (1.28-fold, p = 0.23), BCL2L13 (1.8-fold, p < 0.0001), CASPASE-3 (1.5-fold, p = 0.003), CASPASE-9 (1.04-fold, p = 0.52) and IL-18 (1.33-fold, p = 0.0003) (Table 2, Figure 2).
Bevacizumab 2× resulted in downregulation of VEGF-A (0.82-fold, p = 0.41), HIF-1α (0.63-fold, p = 0.001), GPX3A (0.91-fold, p = 0.63), IL-18 (0.86-fold, p = 0.02), and IL-1β (0.68-fold, p = 0.12); and upregulation of SOD2 (1.54-fold, p = 0.06), BCL2L13 (1.07-fold, p = 0.16), CASPASE-3 (1.04-fold, p = 0.39), and CASPASE-9 (2.16-fold, p < 0.0001) (Table 2, Figure 2).
TSA treated (0.3 μM) ARPE-19 cultures resulted in downregulation of VEGF-A (0.75-fold, p = 0.24), HIF-1α (0.66-fold, p = 0.001) and IL-1β (0.2-fold, p = 0.001), and caused upregulation of SOD2 (1.69-fold, p = 0.11), GPX3A (2.11-fold, p = 0.06), BCL2L13 (1.13-fold, p = 0.0003), CASPASE-3 (1.14-fold, p = 0.07), CASPASE-9 (1.49-fold, p = 0.003) and IL-18 (1.69-fold, p < 0.0001) compared to untreated cells (Table 3, Figure 3).
Table 3.
Expression folds of downstream genes after treatment with Trichostatin A alone and in combination with Bevacizumab 1× *.
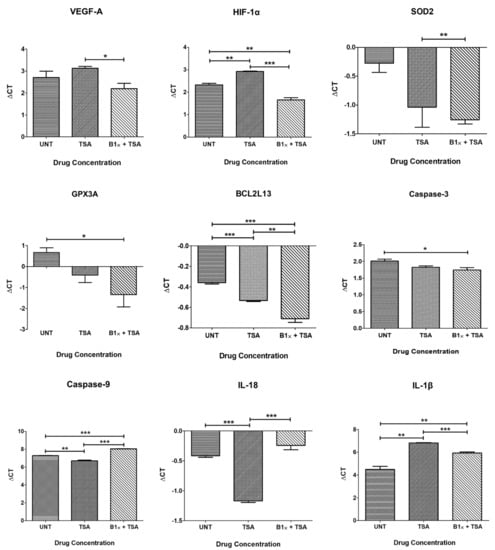
Figure 3.
Quantitative Real-Time PCR (qPCR) data showing Delta Cts for downstream genes after treatment with Trichostatin A and Bevacizumab. * p < 0.05; ** p < 0.01; *** p < 0.001; Bars with no asterisk represent nonsignificant difference. UNT: Untreated; TSA: Trichostatin A 0.3 μM; B1×+TSA: Bevacizumab 1× conc. + Trichostatin A 0.3 μM. Error bars represent ‘Mean ± SEM’.
The combination of bevacizumab 1× plus TSA resulted in upregulation of VEGF-A (1.42-fold, p = 0.25), HIF-1α (1.60-fold, p = 0.006) SOD2 (1.97-fold, p = 0.005), GPX3A (4.03-fold, p = 0.03), BCL2L13 (1.28-fold, p = 0.0007) and CASPASE-3 (1.2-fold, p = 0.05), and decreased expression of CASPASE-9 (0.59-fold, p = 0.09), IL-18 (0.89-fold, p = 0.09) and IL-1β (0.37-fold, p = 0.008) compared to untreated cells (Table 3, Figure 3).
When comparing cells treated with both drugs to cells treated with TSA alone, the addition of bevacizumab 1× significantly reversed the effect of TSA on the expression of VEGF-A (p = 0.02), HIF-1α (p = 0.0003), CASPASE-9 (p < 0.0001), and IL-18 (p = 0.0003), reduced its effect on an IL-1β (p = 0.0006), and potentiated its effect on BCL2L13 (p = 0.008) (Figure 3).
4. Discussion
In this study, we demonstrated differential HDAC total protein activity in response to anti-VEGF drug treatment in ARPE-19 cells. Treatment of ARPE-19 cells with anti-VEGF drugs alone in varying concentrations as well as additive effects of TSA and anti-VEGF drug significantly modulated the gene expression profiles of apoptotic, inflammatory, and oxidative stress markers. Therefore, our results suggest that addition of anti-VEGF drugs to cultured ARPE-19 cells strongly influence the regulation of epigenetic markers and downstream molecular markers.
Environmental stimuli are thought to induce epigenetic changes that accumulate in the cell with increasing age, as evidenced by age being the major risk factor for AMD, as well as the discordance of disease phenotype in identical twins that possess similar risk profiles for AMD [34]. Hypermethylation of genes encoding for reactive oxygen species (ROS) scavengers, namely GSTM1 and GSTM5, was demonstrated in AMD RPE and choroid, downregulating their expression and rendering the cells more susceptible to oxidative damage. Additionally, several micro RNAs (miRNAs) have been implicated in AMD pathogenesis through their contribution to aberrant angiogenesis, inflammation, and apoptosis in response to oxidative stress both in vitro and in vivo [22]. Hypomethylation of the promoter region of interleukin 17 receptor C (IL17RC), a pro-inflammatory gene, promotes its expression and increases inflammation in AMD patients [35]. Oliver et al. conducted the first genome-wide epigenetic study in AMD and found hypomethylation at HTRA/ARMS2 locus, which is a major susceptibility locus for AMD. They also observed hypermethylation of the PRSS50 locus which had not been previously associated with AMD [23].
Over the last decade, intravitreal injection of anti-angiogenic drugs has been the mainstay of therapy for neovascular AMD as well as for macular edema associated with diabetic retinopathy and retinal vein occlusion [36]. The three drugs commonly used in clinical practice are bevacizumab, ranibizumab and aflibercept. Bevacizumab is a full-length monoclonal antibody that has a molecular weight of 149 kilodalton (kDa) and binds all isoforms of VEGF-A rendering them inactive. Ranibizumab is an antigen-binding Fab fragment of the same parent antibody as bevacizumab. It lacks the Fc domain and has a molecular weight of 48 kDa. Aflibercept, a 96.9 kDa recombinant fusion protein, contains immunoglobulin fragments from both VEGF receptors: VEGFR1 and 2, combined with an Fc antibody fragment. It acts as a decoy receptor that not only binds VEGF-A, but also, unlike the former two drugs, can bind VEGF-B and placental-like growth factor (PlGF) [37,38,39]. The different molecular weights and structures of the three drugs may have an effect on their ocular and systemic pharmacokinetics and pharmacodynamics [40]. All three drugs have been shown to accumulate intracellularly. Uptake of bevacizumab and aflibercept is mediated through their Fc portion, but ranibizumab is likely internalized through a VEGF/VEGFR2-mediated mechanism [30].
RPE dysfunction is central to the pathogenesis of AMD, and RPE cells are the main source of VEGF in the retina [41]. Many investigators have tested the safety of anti-VEGF agents on RPE cells both in vitro and in animal models. Our group previously demonstrated a good safety profile of the three anti-angiogenic drugs on ARPE-19 cell cultures at the clinical dose, but mild cytotoxic effects were found at higher doses [42,43]. Other studies have shown similar results [44,45].
A study by Dinc et al. in which APRE-19 cells were subjected to H2O2-induced oxidative stress and levels of miRNA expression were evaluated, suggested an epigenetic role for anti-VEGF drugs [46]. In that study, several miRNAs were dysregulated in response to oxidative stress compared to untreated samples. Preincubation of cells with any of the three anti-VEGF drugs before H2O2 treatment significantly altered the miRNA dysregulation induced by H2O2. The authors suggested that anti-VEGF drugs could protect RPE cells from oxidative stress through their effect on miRNAs.
We tried to determine whether the changes in HDAC expression would affect the activity of HDAC enzymes in ARPE-19 cells using an assay kit designed to measure the collective activity of all HDACs. Bevacizumab treatment alone failed to alter HDAC activity to a significant extent compared to untreated cultures. Our inability to observe a significant net effect on HDAC enzyme activity may be because bevacizumab had opposite effects on the expression of both HDAC1 and HDAC6 genes, so the overall HDAC activity after treatment was neutral. As expected, Trichostatin A (TSA) significantly inhibited HDAC activity. TSA is known to inhibit both class I and II HDACs. Interestingly, the combination of TSA plus bevacizumab increased significantly the frank inhibitory effect on HDAC activity, but the mechanisms for additive effects are not clear and need to be further investigated.
Next, the expression levels for genes known to regulate important pathogenetic pathways for AMD after treatment with anti-VEGF drugs and TSA were measured.
4.1. Angiogenesis Genes
HIF-1α is the oxygen-sensitive subunit of HIF-1, a transcription factor upregulated in cells in response to hypoxia. Activation of HIF-1α results in overexpression of downstream genes, including pro-angiogenic genes, mainly VEGF-A [47]. Both class I and II HDACs are upregulated by hypoxia and induce angiogenesis, particularly HDAC1 [48,49]. HIF-1α is a non-histone protein target for acetylation. It is acetylated under normoxic conditions, which decreases its stability by facilitating its proteasomal degradation. Under hypoxic conditions, HIF-1α becomes deacetylated by HDACs, which makes it more stable and prolongs its half-life [50].
We found that all three anti-VEGF drugs downregulated the expression of HIF1-α gene at 2×, but not 1×, concentration. This suggests that these drugs might protect cells against oxidative damage. Treatment with TSA had the same inhibitory effect. This observation comes in agreement with previous studies demonstrating that TSA could downregulate both gene and protein expression of HIF-1α in vitro and in animal models [51,52]. Other HDAC inhibitors demonstrated a protective effect against oxidative damage in vitro and animal models. These include valproic acid (VPA), which is another broad-spectrum HDACi, and tubastatin A (TST); a specific inhibitor of HDAC6 [53,54]. The inhibitory effect displayed by anti-VEGF drugs and TSA in our study was reversed by the combination of bevacizumab and TSA, which significantly upregulated HIF1-α expression.
VEGF-A is the main pro-angiogenic factor implicated in the pathogenesis of wet AMD [55]. The expression levels of VEGF, as well as tissue response to its secretion, are modulated by both genetic and epigenetic factors. Epigenetic regulation of angiogenesis has been extensively studied in cancer cells. Both HDAC7 and SIRT1 promote angiogenesis during development and disease by inducing pro-angiogenic factors and suppressing anti-angiogenic ones [56,57,58]. HDAC6 has also been associated with angiogenesis due to its ability to bind several cytoskeletal proteins in the cytoplasm and stimulate vascular proliferation and sprouting. The anti-angiogenic properties of HDAC inhibitors are the basis for their use as anti-cancer agents and extend beyond gene silencing to directly acetylating angiogenic factors in the cytoplasm. Chan and colleagues demonstrated that TSA could downregulate VEGF-A by epigenetically silencing its expression in the presence of CoCl2, a hypoxia-inducing agent, in human RPE cells in vitro. Furthermore, they showed that TSA could attenuate a laser induced CNV in a mouse model. However, we found that TSA did not alter VEGF-A gene expression in ARPE-19 cells. The ability of TSA to silence gene expression might be more pronounced under conditions of hypoxic stress that induce aberrant upregulation of VEGF-A, which was not the case in our experiment.
Two studies explored the effect of either ranibizumab or bevacizumab on VEGF-A gene expression in primary RPE cells. In one experiment, bevacizumab had no effect on the baseline expression levels of VEGF-A [59]. Similarly, in another study ranibizumab treatment did not significantly alter VEGF-A mRNA overexpression induced by white light illumination of RPE cells [60]. In ARPE-19 cells, both aflibercept and ranibizumab induced VEGF-A mRNA expression after 24 h of treatment. The authors postulated that the cells upregulated the transcription of VEGF-A to compensate for blocking the VEGF-A protein in the culture media [43]. Similarly, another study demonstrated that ranibizumab and bevacizumab induced overexpression of VEGF-A inARPE-19 cells that were subjected to oxidative stress by preincubation in a hypoxic chamber [61]. Another study showed a similar compensatory upregulation in response to bevacizumab treatment in murine RPE cells in vivo, but not in ARPE-19 cells in vitro. These investigators suggested that the compensatory upregulation of VEGF-A expression in complex in vivo systems might not be captured by the simpler APRE-19 cell model. It could also be that the RPE of healthy young animals was able to compensate for VEGF-A neutralization, unlike ARPE-19 cells, which carry more similarities to aged RPE [62]. Other studies showed that anti-VEGF drugs could bring down VEGF-A gene expression to control levels in human retinal pericytes [63] and in ARPE-19 cells [19].
VEGF-A expression levels were not affected by any of the used drugs in our experiment, although its expression was significantly lower in cells treated with a combination of bevacizumab plus TSA compared to treatment with TSA alone. It seems that the effect of anti-angiogenic drugs on VEGF-A gene expression varies according to cell type, the nature of biologic stress that the cells are exposed to as well as the in vivo versus in vitro environment.
4.2. Oxidative Stress Genes
Oxidative stress occurs when ROS accumulation overwhelms the capacity of the cell to detoxify them. Neural and RPE cells in the retina have a high metabolic demand and are most prone to oxidative damage with aging. Aging is associated with differential gene expression and chromatin reorganization mediated by epigenetic mechanisms, ultimately leading to impaired ability of the cells to adapt to environmental stress [64].
The main antioxidant in the retina is the superoxide dismutase (SOD) family [65]. SOD2 gene encodes for a mitochondrial enzyme, which deactivates superoxide free radicals. Polymorphisms of SOD2 have been associated with exudative AMD [66]. SOD2 expression depends on acetylation. Being a mitochondrial enzyme, SOD2 is directly deacetylated by SIRT3, a mitochondrial Class III HDAC, resulting in its activation [67]. Forkhead box O3a (FoxO3a) activates the promoter of SOD2 gene inducing its expression. TSA was shown to increase the acetylation of FoxO3a promoter region, and upregulating its expression, as well as its target protein, SOD2, expression in vitro [68]. Treatment with bevacizumab 1× in our study significantly upregulated SOD2 expression, as did combined treatment with bevacizumab and TSA. Conversely, aflibercept significantly reduced SOD2 expression.
Glutathione peroxidase (GPX) is another antioxidant enzyme found in the RPE and photoreceptors that protects the retina from oxidative damage. GPX expression is upregulated in AMD patients, most likely due to oxidative stress [69]. VPA induced the expression of SOD2 and GPX genes in ARPE-19 cells in normal conditions and maintained their expression hypoxic conditions. A similar effect was demonstrated in rat retina [70]. In our study, only the combination of bevacizumab plus TSA was able to significantly increase GPX3A expression.
4.3. Inflammation Genes
Inflammation has been recently recognized as a key player in the pathogenesis of AMD. Both drusen components and intracellular lipofuscin can incite inflammasome activation and the release of the pro-inflammatory cytokines IL-18 and IL-1β in retinal tissues [71,72]. It is thought that epigenetic mechanisms are involved in initiating the immune response by altering the gene expression of immune cells to allow for cytokine production and chemotaxis [24]. HDAC6 inhibition by TST suppressed mRNA expression of IL-1β in an inflammation model of mammary epithelial cells in vitro [73]. We found that TSA treatment had the same effect on IL-1β gene expression in ARPE-19 cells. Cultures treated with ranibizumab 2× and the TSA+bevacizumab combination also displayed downregulation of IL-1β.
Although inflammation significantly contributes to tissue damage in AMD, a certain degree of inflammation might be needed to protect against neovascularization. IL-18 has exhibited anti-angiogenic properties in tumors and post-ischemic injury and is being investigated as a potential anti-angiogenic therapy in wet AMD [74]. Shen et al. demonstrated reciprocal suppression between VEGF and IL-18. They were able to detect increased levels of IL-18 in the aqueous of patients receiving intravitreal ranibizumab injections for macular edema secondary to retinal vein occlusion. Furthermore, they found that intravitreal injection of anti-VEGF antibody in a mouse model of ischemic retinopathy upregulated mRNA expression of IL-18. IL-18 was able to block VEGF-induced vascular leakage and neovascularization in mice. Thus, each agent can suppress both the production and function of the other [75]. This observation could explain our findings that IL-18 was significantly upregulated by both concentrations of ranibizumab and bevacizumab. TSA also upregulated IL-18 expression, possibly owing to its anti-angiogenic properties. Aflibercept 2×, however, suppressed its expression. Aflibercept has a different molecular structure than both ranibizumab and bevacizumab and could have triggered another signaling mechanism that reduced IL-18 transcription.
4.4. Apoptosis Genes
RPE cell loss characteristic of advanced AMD is thought to represent cell death by apoptosis. The BCL-2 family regulates the intrinsic mitochondrial pathway of apoptosis. Accumulation of oxidized low-density lipoproteins (LDL) in drusen and basal linear deposits, both hallmark lesions of AMD, results in upregulation of BAX, a pro-apoptotic BCL-2 family member, and downregulation of BCL-2, an anti-apoptotic member. The increased BAX/BCL-2 ratio tips the balance in favor of apoptosis [76]. Activation of pro-apoptotic BCL-2 members results in opening of mitochondrial membrane pores with subsequent release of pro-apoptotic factors, such as cytochrome c, into the cytoplasm. Cytochrome c can recruit and activate caspase-9, an initiator caspase, which activates effector caspases, such as caspase-3, that eventually cause degradation of genomic DNA and cell death [77].
Anti-VEGF drug treatment of ARPE-19 cells resulted in upregulation of the 4 pro-apoptotic genes in this study. This effect was seen with the three drugs tested at both 1× and 2× concentrations. We previously demonstrated some degree of reduced mitochondrial membrane potential (MMP), which is an early sign of apoptosis, in ARPE-19 cells after 24 h of treatment with higher-than-clinical concentrations of ranibizumab and aflibercept. Only bevacizumab decreased MMP at 1× concentration [42]. Another study found that bevacizumab significantly increased apoptosis in an ARPE-19 cell model of oxidative stress and as stress levels increased, the dose of bevacizumab capable of inducing apoptosis decreased. The authors postulated that bevacizumab blocked the protective effects of VEGF under high oxidative stress conditions and downregulated BCL-2 gene expression [78]. Another study showed that ranibizumab could enhance the anti-proliferation effects of oxidative stress on ARPE-19 cells [42].
These results warrant further in vivo investigations since the net effect on retinal cells in vivo is subject to a complex interplay of many protective and detrimental factors. Anti-angiogenic therapy has demonstrated protective effects in our in vitro study, as evidenced by suppressing oxidative stress and inflammatory cytokine gene transcription. However, further research is warranted as concerns have been raised about the development of geographic atrophy in 98% of wet AMD patients receiving chronic anti-VEGF injections over prolonged periods of time [79].
5. Conclusions
In conclusion, we demonstrated that anti-VEGF drugs can (1) alter expression profiles for genes involved in oxidative stress, inflammation, and apoptosis pathways and (2) modulate intracellular signal transduction in addition to blocking VEGF-A. This could have implications in management of resistance or nonresponse to anti-VEGF therapy in some AMD patients. The phenomenon of individual variation in response to anti-VEGF treatment has also been observed in different cancers treated with bevacizumab [80,81,82]. Genetic variations may render vascular tissue more responsive or resistant to drug effects. Epigenetic mechanisms may render tissues less sensitive to the anti-VEGF treatment and influence pharmacogenetic interactions, as evidenced by miRNA regulation of enzymes involves in drug uptake and metabolism [83,84]. The fact that these drugs could influence the epigenome might guide precision medicine in the future by obtaining an “epigenetic” profile for wet AMD patients to predict resistance and direct the choice of therapy.
Another avenue we believe is worth exploring is the possibility of adding HDAC inhibitors to the therapeutic armamentarium of AMD. Epigenetic drugs have shown a great promise in immunomodulation, neuroprotection, and angiogenesis suppression [85]. To date, 6 epigenetic drugs have been approved by the FDA for cancer therapy [51]. A variety of epigenetic drugs, including DNMT and HDAC inhibitors are currently under investigation as potential therapeutic agents in AMD, owing to their ability to reverse inflammation and angiogenesis [55,86]. Specific HDAC inhibitors might be preferable to pan-inhibitors, such as TSA, as the latter can cause undesirable alterations in gene expression by inducing histone hyperacetylation [53].
Further studies including in vivo tests are required. Other retinal cell types are involved in the evolution of AMD pathogenesis, and the impact of the studied drugs on these cells needs to be explored as well. Human pluripotent stem cell (hPSC)-derived retinal organoids could provide an alternative platform to study drug interactions and intracellular signaling mechanisms that more closely approximates the retinal environment in vivo [87].
Author Contributions
Conceptualization: M.C.K. and B.D.K.; methodology: M.A.H., M.T.M., S.N., R.D.C., K.S. and S.R.A.; investigation: M.A.H., M.T.M., S.N., R.D.C., K.S. and S.R.A.; validation: M.A.H., M.T.M. and S.N.; data curation: M.A.H., M.T.M., S.N. and R.D.C.; formal analysis: M.A.H. and M.T.M.; resources: M.C.K. and B.D.K.; writing—original draft preparation: M.A.H., M.T.M. and S.N.; writing—review and editing: S.N., M.C.K. and B.D.K.; supervision: B.D.K. and M.C.K.; project administration: B.D.K. and M.C.K.; funding acquisition: B.D.K. and M.C.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Discovery Eye Foundation, Polly and Michael Smith Foundation, Iris and B. Gerald Cantor Foundation, Beckman Initiative for Macular Research, Max Factor Family Foundation, and Guenther Foundation. This study was also supported in part by an unrestricted grant from Research to Prevent Blindness, Inc. The authors acknowledge the support of the Institute for Clinical and Translational Science grant number: ICTS (ULI TR001414/TR/NCATS) at University of California, Irvine.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data are presented within the manuscript.
Acknowledgments
NEI grant: NEI R01 EY027363 (MCK).
Conflicts of Interest
B.D.K. is consultant to Alcon, Alimera, Allegro, Allergan, Genentech, Glaukos, GSK, Neurotech, Novagali, Novartis, Ophthotech, Pfizer, Regeneron, Santen, SecondSight, Teva, ThromboGenics. The rest of the authors declare that they have no competing interests. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Gehrs, K.M.; Anderson, D.H.; Johnson, L.V.; Hageman, G.S. Age-related macular degeneration--emerging pathogenetic and therapeutic concepts. Ann. Med. 2006, 38, 450–471. [Google Scholar] [CrossRef]
- Wong, T.Y.; Chakravarthy, U.; Klein, R.; Mitchell, P.; Zlateva, G.; Buggage, R.; Fahrbach, K.; Probst, C.; Sledge, I. The natural history and prognosis of neovascular age-related macular degeneration: A systematic review of the literature and meta-analysis. Ophthalmology 2008, 115, 116–126. [Google Scholar] [CrossRef]
- Amadio, M.; Govoni, S.; Pascale, A. Targeting VEGF in eye neovascularization: What’s new? A comprehensive review on current therapies and oligonucleotide-based interventions under development. Pharmacol. Res. 2016, 103, 253–269. [Google Scholar] [CrossRef]
- Fogli, S.; Del Re, M.; Rofi, E.; Posarelli, C.; Figus, M.; Danesi, R. Clinical pharmacology of intravitreal anti-VEGF drugs. Eye 2018, 32, 1010–1020. [Google Scholar] [CrossRef]
- Grisanti, S.; Tatar, O. The role of vascular endothelial growth factor and other endogenous interplayers in age-related macular degeneration. Prog. Retin. Eye Res. 2008, 27, 372–390. [Google Scholar] [CrossRef]
- Brown, D.M.; Kaiser, P.K.; Michels, M.; Soubrane, G.; Heier, J.S.; Kim, R.Y.; Sy, J.P.; Schneider, S. Ranibizumab versus Verteporfin for Neovascular Age-Related Macular Degeneration. N. Engl. J. Med. 2006, 355, 1432–1444. [Google Scholar] [CrossRef]
- Chakravarthy, U.; Harding, S.P.; Rogers, C.A.; Downes, S.M.; Lotery, A.J.; Wordsworth, S.; Reeves, B.C. Ranibizumab versus Bevacizumab to Treat Neovascular Age-related Macular Degeneration: One-Year Findings from the IVAN Randomized Trial. Ophthalmology 2012, 119, 1399–1411. [Google Scholar] [CrossRef]
- Group, T.C.R. Ranibizumab and Bevacizumab for Neovascular Age-Related Macular Degeneration. N. Engl. J. Med. 2011, 364, 1897–1908. [Google Scholar]
- Heier, J.S.; Brown, D.M.; Chong, V.; Korobelnik, J.-F.; Kaiser, P.K.; Nguyen, Q.D.; Kirchhof, B.; Ho, A.; Ogura, Y.; Yancopoulos, G.D.; et al. Intravitreal Aflibercept (VEGF Trap-Eye) in Wet Age-related Macular Degeneration. Ophthalmology 2012, 119, 2537–2548. [Google Scholar] [CrossRef]
- Rosenfeld, P.J.; Brown, D.M.; Heier, J.S.; Boyer, D.S.; Kaiser, P.K.; Chung, C.Y.; Kim, R.Y. Ranibizumab for Neovascular Age-Related Macular Degeneration. N. Engl. J. Med. 2006, 355, 1419–1431. [Google Scholar] [CrossRef]
- Shah, A.R.; Williams, S.; Baumal, C.R.; Rosner, B.; Duker, J.S.; Seddon, J.M. Predictors of Response to Intravitreal Anti-Vascular Endothelial Growth Factor Treatment of Age-Related Macular Degeneration. Am. J. Ophthalmol. 2016, 163, 154–166.e158. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Strafella, C.; Caputo, V.; Errichiello, V.; Zampatti, S.; Milano, F.; Potenza, S.; Mauriello, S.; Novelli, G.; Ricci, F.; et al. Towards the application of precision medicine in Age-Related Macular Degeneration. Prog. Retin. Eye Res. 2018, 63, 132–146. [Google Scholar] [CrossRef]
- Hagstrom, S.A.; Ying, G.-S.; Pauer, G.J.T.; Sturgill-Short, G.M.; Huang, J.; Callanan, D.G.; Kim, I.K.; Klein, M.L.; Maguire, M.G.; Martin, D.F. Pharmacogenetics for Genes Associated with Age-related Macular Degeneration in the Comparison of AMD Treatments Trials (CATT). Ophthalmology 2013, 120, 593–599. [Google Scholar] [CrossRef]
- Hagstrom, S.A.; Ying, G.; Pauer, G.T.; Sturgill-Short, G.M.; Huang, J.; Maguire, M.G.; Martin, D.F. Vegfa and vegfr2 gene polymorphisms and response to anti–vascular endothelial growth factor therapy: Comparison of age-related macular degeneration treatments trials (catt). JAMA Ophthalmol. 2014, 132, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Hagstrom, S.A.; Ying, G.S.; Maguire, M.G.; Martin, D.F.; Gibson, J.; Lotery, A.; Chakravarthy, U. VEGFR2 Gene Polymorphisms and Response to Anti-Vascular Endothelial Growth Factor Therapy in Age-Related Macular Degeneration. Ophthalmology 2015, 122, 1563–1568. [Google Scholar] [CrossRef]
- Hagstrom, S.A.; Ying, G.S.; Pauer, G.J.; Huang, J.; Maguire, M.G.; Martin, D.F. Endothelial PAS domain-containing protein 1 (EPAS1) gene polymorphisms and response to anti-VEGF therapy in the comparison of AMD treatments trials (CATT). Ophthalmology 2014, 121, 1663–1664.e1661. [Google Scholar] [CrossRef][Green Version]
- Lotery, A.J.; Gibson, J.; Cree, A.J.; Downes, S.M.; Harding, S.P.; Rogers, C.A.; Reeves, B.C.; Ennis, S.; Chakravarthy, U. Pharmacogenetic Associations with Vascular Endothelial Growth Factor Inhibition in Participants with Neovascular Age-related Macular Degeneration in the IVAN Study. Ophthalmology 2013, 120, 2637–2643. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Tzekov, R.; Li, W.; Jiang, F.; Mao, S.; Tong, Y. Pharmacogenetics of Complement Factor H Y402H Polymorphism and Treatment of Neovascular AMD with Anti-VEGF Agents: A Meta-Analysis. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef]
- Park, U.C.; Shin, J.Y.; McCarthy, L.C.; Kim, S.J.; Park, J.H.; Chung, H.; Yu, H.G. Pharmacogenetic associations with long-term response to anti-vascular endothelial growth factor treatment in neovascular AMD patients. Mol. Vis. 2014, 20, 1680–1694. [Google Scholar]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef]
- Hjelmeland, L.M. Dark matters in AMD genetics: Epigenetics and stochasticity. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1622–1631. [Google Scholar] [CrossRef]
- Gemenetzi, M.; Lotery, A.J. The role of epigenetics in age-related macular degeneration. Eye 2014, 28, 1407–1417. [Google Scholar] [CrossRef]
- Oliver, V.F.; Jaffe, A.E.; Song, J.; Wang, G.; Zhang, P.; Branham, K.E.; Swaroop, A.; Eberhart, C.G.; Zack, D.J.; Qian, J.; et al. Differential DNA methylation identified in the blood and retina of AMD patients. Epigenetics 2015, 10, 698–707. [Google Scholar] [CrossRef]
- Wen, X.; Hu, X.; Miao, L.; Ge, X.; Deng, Y.; Bible, P.W.; Wei, L. Epigenetics, microbiota, and intraocular inflammation: New paradigms of immune regulation in the eye. Prog. Retin. Eye Res. 2018, 64, 84–95. [Google Scholar] [CrossRef]
- Wu, C.; Morris, J.R. Genes, Genetics, and Epigenetics: A Correspondence. Science 2001, 293, 1103–1105. [Google Scholar] [CrossRef] [PubMed]
- Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Resnikoff, S.; Pascolini, D.; Etya’ale, D.; Kocur, I.; Pararajasegaram, R.; Pokharel, G.P.; Mariotti, S.P. Global data on visual impairment in the year 2002. Bull. World Health Organ. 2004, 82, 844–851. [Google Scholar]
- Klein, R.; Klein, B.E.K.; Knudtson, M.D.; Meuer, S.M.; Swift, M.; Gangnon, R.E. Fifteen-Year Cumulative Incidence of Age-Related Macular Degeneration: The Beaver Dam Eye Study. Ophthalmology 2007, 114, 253–262. [Google Scholar] [CrossRef]
- Ranjbar, M.; Brinkmann, M.P.; Tura, A.; Rudolf, M.; Miura, Y.; Grisanti, S. Ranibizumab interacts with the VEGF-A/VEGFR-2 signaling pathway in human RPE cells at different levels. Cytokine 2016, 83, 210–216. [Google Scholar] [CrossRef]
- Mahendra, C.K.; Tan, L.T.H.; Pusparajah, P.; Htar, T.T.; Chuah, L.H.; Lee, V.S.; Low, L.E.; Tang, S.Y.; Chan, K.G.; Goh, B.H. Detrimental Effects of UVB on Retinal Pigment Epithelial Cells and Its Role in Age-Related Macular Degeneration. Oxid Med. Cell Longev. 2020, 2020, 1904178. [Google Scholar] [CrossRef]
- Moustafa, M.T.; Ramirez, C.; Schneider, K.; Atilano, S.R.; Limb, G.A.; Kuppermann, B.D.; Kenney, M.C. Protective Effects of Memantine on Hydroquinone-Treated Human Retinal Pigment Epithelium Cells and Human Retinal Müller Cells. J. Ocul. Pharmacol. Ther. 2017, 33, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Dunn, K.; Aotaki-Keen, A.; Putkey, F.; Hjelmeland, L. ARPE-19, A Human Retinal Pigment Epithelial Cell Line with Differentiated Properties. Exp. Eye Res. 1996, 62, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.L.; DeAngelis, M.M. Epigenetic Mechanisms of the Aging Human Retina. J. Exp. Neurosci. 2015, 9, 51–79. [Google Scholar] [CrossRef] [PubMed]
- Sobrin, L.; Seddon, J.M. Nature and nurture- genes and environment- predict onset and progression of macular degeneration. Prog. Retin. Eye Res. 2014, 40, 1–15. [Google Scholar] [CrossRef]
- Stewart, M.W. Extended Duration Vascular Endothelial Growth Factor Inhibition in the Eye: Failures, Successes, and Future Possibilities. Pharmaceutics 2018, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Damico, L.; Shams, N.; Lowman, H.; Kim, R. Development of ranibizumab, an anti-vascular endothelial growth factor antigen binding fragment, as therapy for neovascular age-related macular degeneration. Retina 2006, 26, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Kramer, I.; Lipp, H.P. Bevacizumab, a humanized anti-angiogenic monoclonal antibody for the treatment of colorectal cancer. J. Clin. Pharm. Ther. 2007, 32, 1–14. [Google Scholar] [CrossRef]
- Ohr, M.; Kaiser, P.K. Aflibercept in wet age-related macular degeneration: A perspective review. Ther. Adv. Chronic Dis. 2012, 3, 153–161. [Google Scholar] [CrossRef]
- Avery, R.L.; Castellarin, A.A.; Steinle, N.C.; Dhoot, D.S.; Pieramici, D.J.; See, R.; Couvillion, S.; Nasir, M.A.; Rabena, M.D.; Le, K.; et al. Systemic pharmacokinetics following intravitreal injections of ranibizumab, bevacizumab or aflibercept in patients with neovascular AMD. Br. J. Ophthalmol. 2014, 98, 1636–1641. [Google Scholar] [CrossRef]
- Puddu, A.; Sanguineti, R.; Traverso, C.E.; Viviani, G.L.; Nicolo, M. Response to anti-VEGF-A treatment of retinal pigment epithelial cells in vitro. Eur. J. Ophthalmol. 2016, 26, 425–430. [Google Scholar] [CrossRef]
- Luthra, S.; Sharma, A.; Dong, J.; Neekhra, A.; Gramajo, A.L.; Seigel, G.M.; Kenney, M.C.; Kuppermann, B.D. Effect of bevacizumab (Avastin (TM)) on mitochondrial function of in vitro retinal pigment epithelial, neurosensory retinal and microvascular endothelial cells. Indian J. Ophthalmol. 2013, 61, 705–710. [Google Scholar]
- Malik, D.; Tarek, M.; Caceres del Carpio, J.; Ramirez, C.; Boyer, D.; Kenney, M.C.; Kuppermann, B.D. Safety profiles of anti-VEGF drugs: Bevacizumab, ranibizumab, aflibercept and ziv-aflibercept on human retinal pigment epithelium cells in culture. Br. J. Ophthalmol. 2014, 98 (Suppl. S1), i11–i16. [Google Scholar] [CrossRef]
- Ammar, D.A.; Mandava, N.; Kahook, M.Y. The effects of aflibercept on the viability and metabolism of ocular cells in vitro. Retina 2013, 33, 1056–1061. [Google Scholar] [CrossRef]
- Schnichels, S.; Hagemann, U.; Januschowski, K.; Hofmann, J.; Bartz-Schmidt, K.U.; Szurman, P.; Spitzer, M.S.; Aisenbrey, S. Comparative toxicity and proliferation testing of aflibercept, bevacizumab and ranibizumab on different ocular cells. Br. J. Ophthalmol. 2013, 97, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Dinc, E.; Ayaz, L.; Kurt, A.H. Effects of Bevacizumab, Ranibizumab, and Aflibercept on MicroRNA Expression in a Retinal Pigment Epithelium Cell Culture Model of Oxidative Stress. J. Ocul. Pharmacol. Ther. 2018, 34, 346–353. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Dai, Y.; Dong, F.; Zhang, X.; Yang, Z. Internal limiting membrane transplantation for unclosed and large macular holes. Graefes Arch. Clin. Ex. P Ophthalmol. 2016, 254, 2095–2099. [Google Scholar] [CrossRef] [PubMed]
- Elmasry, K.; Mohamed, R.; Sharma, I.; Elsherbiny, N.M.; Liu, Y.; Al-Shabrawey, M.; Tawfik, A. Epigenetic modifications in hyperhomocysteinemia: Potential role in diabetic retinopathy and age-related macular degeneration. Oncotarget 2018, 9, 12562–12590. [Google Scholar] [CrossRef]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Berndsen, R.H.; Abdul, U.K.; Weiss, A.; Zoetemelk, M.; Te Winkel, M.T.; Dyson, P.J.; Griffioen, A.W.; Nowak-Sliwinska, P. Epigenetic approach for angiostatic therapy: Promising combinations for cancer treatment. Angiogenesis 2017, 20, 245–267. [Google Scholar] [CrossRef] [PubMed]
- Turtoi, A.; Peixoto, P.; Castronovo, V.; Bellahcene, A. Histone deacetylases and cancer-associated angiogenesis: Current understanding of the biology and clinical perspectives. Crit. Rev. Oncog. 2015, 20, 119–137. [Google Scholar]
- Kothary, P.C.; Rossi, B.; Del Monte, M.A. Valproic Acid Induced Human Retinal Pigment Epithelial Cell Death as Well as its Survival after Hydrogen Peroxide Damage is Mediated by P38 Kinase. Adv. Exp. Med. Biol. 2016, 854, 765–772. [Google Scholar] [PubMed]
- Leyk, J.; Daly, C.; Janssen-Bienhold, U.; Kennedy, B.N.; Richter-Landsberg, C. HDAC6 inhibition by tubastatin A is protective against oxidative stress in a photoreceptor cell line and restores visual function in a zebrafish model of inherited blindness. Cell Death Dis. 2017, 8, e3028. [Google Scholar] [CrossRef]
- Chan, N.; He, S.; Spee, C.K.; Ishikawa, K.; Hinton, D.R. Attenuation of choroidal neovascularization by histone deacetylase inhibitor. PLoS ONE 2015, 10, e0120587. [Google Scholar] [CrossRef]
- Kwa, F.A.; Thrimawithana, T.R. Epigenetic modifications as potential therapeutic targets in age-related macular degeneration and diabetic retinopathy. Drug. Discov. Today 2014, 19, 1387–1393. [Google Scholar] [CrossRef]
- Mottet, D.; Bellahcene, A.; Pirotte, S.; Waltregny, D.; Deroanne, C.; Lamour, V.; Lidereau, R.; Castronovo, V. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ. Res. 2007, 101, 1237–1246. [Google Scholar] [CrossRef]
- Potente, M.; Ghaeni, L.; Baldessari, D.; Mostoslavsky, R.; Rossig, L.; Dequiedt, F.; Haendeler, J.; Mione, M.; Dejana, E.; Alt, F.W.; et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007, 21, 2644–2658. [Google Scholar] [CrossRef]
- Subramani, M.; Murugeswari, P.; Dhamodaran, K.; Chevour, P.; Gunasekaran, S.; Kumar, R.S.; Jayadev, C.; Shetty, R.; Begum, N.; Das, D. Short Pulse of Clinical Concentration of Bevacizumab Modulates Human Retinal Pigment Epithelial Functionality. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1140–1152. [Google Scholar] [CrossRef]
- Kernt, M.; Thiele, S.; Neubauer, A.S.; Koenig, S.; Hirneiss, C.; Haritoglou, C.; Ulbig, M.W.; Kampik, A. Inhibitory activity of ranibizumab, sorafenib, and pazopanib on light-induced overexpression of platelet-derived growth factor and vascular endothelial growth factor A and the vascular endothelial growth factor A receptors 1 and 2 and neuropilin 1 and 2. Retina 2012, 32, 1652–1663. [Google Scholar] [CrossRef]
- Golan, S.; Entin-Meer, M.; Semo, Y.; Maysel-Auslender, S.; Mezad-Koursh, D.; Keren, G.; Loewenstein, A.; Barak, A. Gene profiling of human VEGF signaling pathways in human endothelial and retinal pigment epithelial cells after anti VEGF treatment. BMC Res. Notes 2014, 7, 617. [Google Scholar] [CrossRef][Green Version]
- Ford, K.M.; Saint-Geniez, M.; Walshe, T.; Zahr, A.; D’Amore, P.A. Expression and role of VEGF in the adult retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9478–9487. [Google Scholar] [CrossRef]
- Giurdanella, G.; Anfuso, C.D.; Olivieri, M.; Lupo, G.; Caporarello, N.; Eandi, C.M.; Drago, F.; Bucolo, C.; Salomone, S. Aflibercept, bevacizumab and ranibizumab prevent glucose-induced damage in human retinal pericytes in vitro, through a PLA2/COX-2/VEGF-A pathway. Biochem. Pharmacol. 2015, 96, 278–287. [Google Scholar] [CrossRef]
- Corso-Diaz, X.; Jaeger, C.; Chaitankar, V.; Swaroop, A. Epigenetic control of gene regulation during development and disease: A view from the retina. Prog. Retin Eye Res. 2018, 65, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Pujol-Lereis, L.M.; Schäfer, N.; Kuhn, L.B.; Rohrer, B.; Pauly, D. Interrelation Between Oxidative Stress and Complement Activation in Models of Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2016, 854, 87–93. [Google Scholar] [PubMed]
- Kan, M.; Liu, F.; Weng, X.; Ye, J.; Wang, T.; Xu, M.; He, L.; Liu, Y. Association study of newly identified age-related macular degeneration susceptible loci SOD2, MBP, and C8orf42 in Han Chinese population. Diagn. Pathol. 2014, 9, 1–4. [Google Scholar] [CrossRef][Green Version]
- Gao, J.; Zheng, Z.; Gu, Q.; Chen, X.; Liu, X.; Xu, X. Deacetylation of MnSOD by PARP-regulated SIRT3 protects retinal capillary endothelial cells from hyperglycemia-induced damage. Biochem. Biophys. Res. Commun. 2016, 472, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, Z.; Shi, C.; Li, J.; Yao, M.; Chen, X. Trichostatin A attenuates oxidative stress-mediated myocardial injury through the FoxO3a signaling pathway. Int. J. Mol. Med. 2017, 40, 999–1008. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, M.; Xu, J.; Li, M.; Yu, Y. Identification of pathogenic genes and upstream regulators in age-related macular degeneration. BMC Ophthalmol. 2017, 17, 102. [Google Scholar] [CrossRef]
- Tokarz, P.; Kaarniranta, K.; Blasiak, J. Inhibition of DNA methyltransferase or histone deacetylase protects retinal pigment epithelial cells from DNA damage induced by oxidative stress by the stimulation of antioxidant enzymes. Eur. J. Pharm. 2016, 776, 167–175. [Google Scholar] [CrossRef]
- Brandstetter, C.; Mohr, L.K.M.; Latz, E.; Holz, F.G.; Krohne, T.U. Light induces NLRP3 inflammasome activation in retinal pigment epithelial cells via lipofuscin-mediated photooxidative damage. J. Mol. Med. 2015, 93, 905–916. [Google Scholar] [CrossRef]
- Doyle, S.L.; Campbell, M.; Ozaki, E.; Salomon, R.G.; Mori, A.; Kenna, P.F.; Farrar, G.J.; Kiang, A.-S.; Humphries, M.M.; Lavelle, E.C.; et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat. Med. 2012, 18, 791–798. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, L.; Wei, Z.; Zhang, X.; Wang, Y.; Li, F.; Fu, Y.; Liu, B. Inhibition of histone deacetylase reduces lipopolysaccharide-induced-inflammation in primary mammary epithelial cells by regulating ROS-NF-кB signaling pathways. Int. Immunopharmacol. 2018, 56, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Doyle, S.; Humphries, P. IL-18: A new player in immunotherapy for age-related macular degeneration? Expert Rev. Clin. Immunol. 2014, 10, 1273–1275. [Google Scholar] [CrossRef][Green Version]
- Shen, J.; Choy, D.F.; Yoshida, T.; Iwase, T.; Hafiz, G.; Xie, B.; Hackett, S.F.; Arron, J.R.; Campochiaro, P.A. Interleukin-18 has antipermeablity and antiangiogenic activities in the eye: Reciprocal suppression with VEGF. J. Cell Physiol. 2014, 229, 974–983. [Google Scholar] [CrossRef]
- Yating, Q.; Yuan, Y.; Wei, Z.; Qing, G.; Xingwei, W.; Qiu, Q.; Lili, Y. Oxidized LDL induces apoptosis of human retinal pigment epithelium through activation of ERK-Bax/Bcl-2 signaling pathways. Curr. Eye Res. 2015, 40, 415–422. [Google Scholar] [CrossRef]
- Hanus, J.; Anderson, C.; Wang, S. RPE necroptosis in response to oxidative stress and in AMD. Ageing Res. Rev. 2015, 24Pt. B, 286–298. [Google Scholar] [CrossRef]
- Kim, S.; Kim, Y.J.; Kim, N.R.; Chin, H.S. Effects of Bevacizumab on Bcl-2 Expression and Apoptosis in Retinal Pigment Epithelial Cells under Oxidative Stress. Korean J. Ophthalmol. 2015, 29, 424–432. [Google Scholar] [CrossRef]
- Rofagha, S.; Bhisitkul, R.B.; Boyer, D.S.; Sadda, S.R.; Zhang, K. SEVEN-UP Study Group. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: A multicenter cohort study (SEVEN-UP). Ophthalmology 2013, 120, 2292–2299. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Miller, K.D.; Chap, L.I.; Holmes, F.A.; Cobleigh, M.A.; Marcom, P.K.; Fehrenbacher, L.; Dickler, M.; Overmoyer, B.A.; Reimann, J.D.; Sing, A.P.; et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J. Clin. Oncol. 2005, 23, 792–799. [Google Scholar] [CrossRef]
- Sandler, A.; Gray, R.; Perry, M.C.; Brahmer, J.; Schiller, J.H.; Dowlati, A.; Lilenbaum, R.; Johnson, D.H. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 2006, 355, 2542–2550, Erratum in: N. Engl. J. Med. 2007, 356, 318. [Google Scholar] [CrossRef]
- Buysschaert, I.; Schmidt, T.; Roncal, C.; Carmeliet, P.; Lambrechts, D. Genetics, epigenetics and pharmaco-(epi)genomics in angiogenesis. J. Cell Mol. Med. 2008, 12, 2533–2551. [Google Scholar] [CrossRef]
- Yu, A.-M.; Tian, Y.; Tu, M.-J.; Ho, P.Y.; Jilek, J.L. MicroRNA Pharmacoepigenetics: Posttranscriptional Regulation Mechanisms behind Variable Drug Disposition and Strategy to Develop More Effective Therapy. Drug. Metab. Dispos. 2016, 44, 308–319. [Google Scholar] [CrossRef]
- Ahmadi, M.; Gharibi, T.; Dolati, S.; Rostamzadeh, D.; Aslani, S.; Baradaran, B.; Younesi, V.; Yousefi, M. Epigenetic modifications and epigenetic based medication implementations of autoimmune diseases. Biomed. Pharmacother. 2017, 87, 596–608. [Google Scholar] [CrossRef]
- Christen, W.G.; Glynn, R.J.; Chew, E.Y.; Albert, C.M.; Manson, J.E. Folic Acid, Vitamin B6, and Vitamin B12 in Combination and Age-Related Cataract in a Randomized Trial of Women. Ophthalmic. Epidemiol. 2016, 23, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Browne, A.W.; Arnesano, C.; Harutyunyan, N.; Khuu, T.; Martinez, J.C.; Pollack, H.A.; Koos, D.S.; Lee, T.C.; Fraser, S.E.; Moats, R.A.; et al. Structural and Functional Characterization of Human Stem-Cell-Derived Retinal Organoids by Live Imaging. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3311–3318. [Google Scholar] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).