
Macrophages and Autoantibodies in Demyelinating Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Role of Macrophages and Autoantibodies in Demyelinating Diseases
2.1. GBS
2.2. CIDP
2.3. MS and Related Diseases
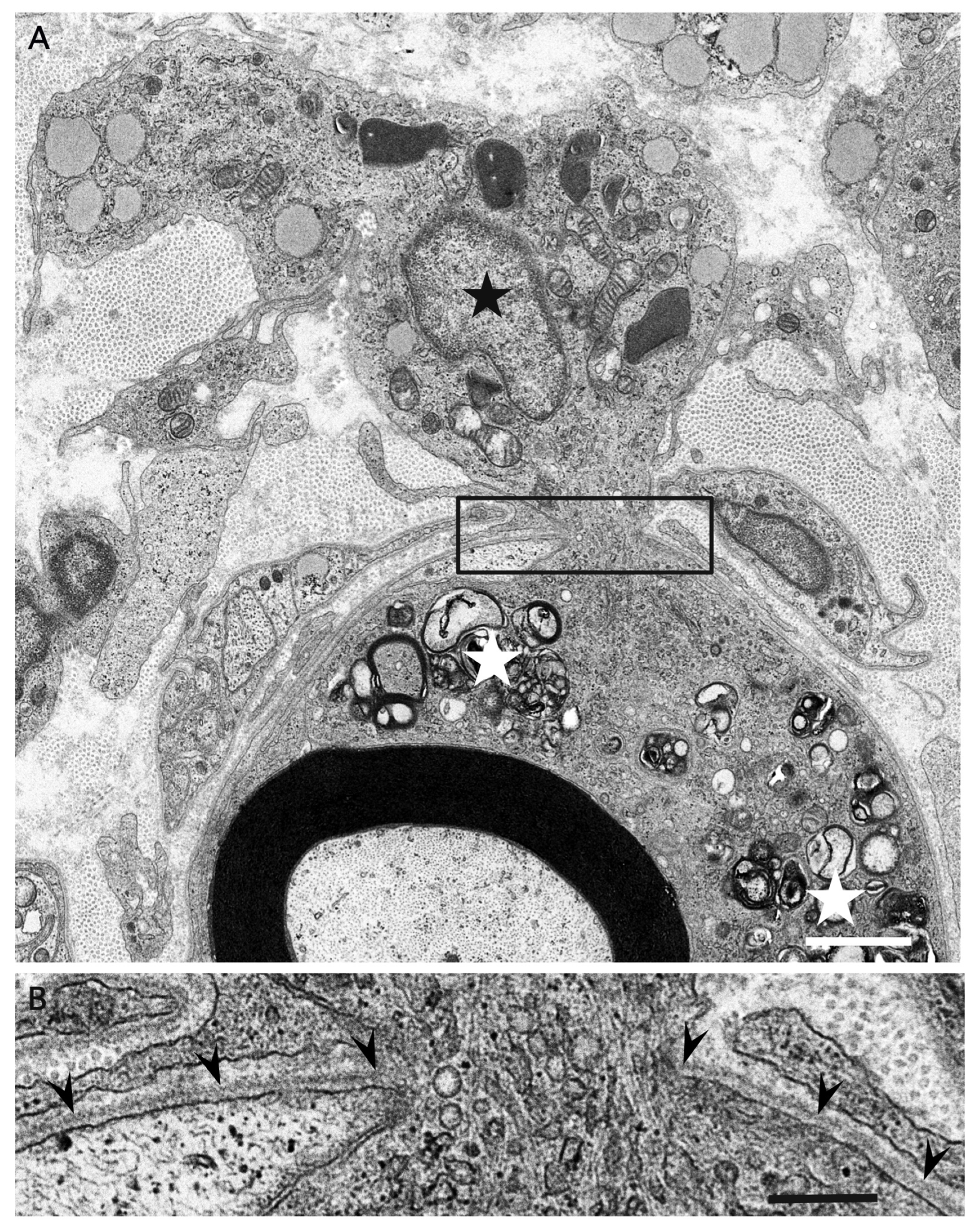
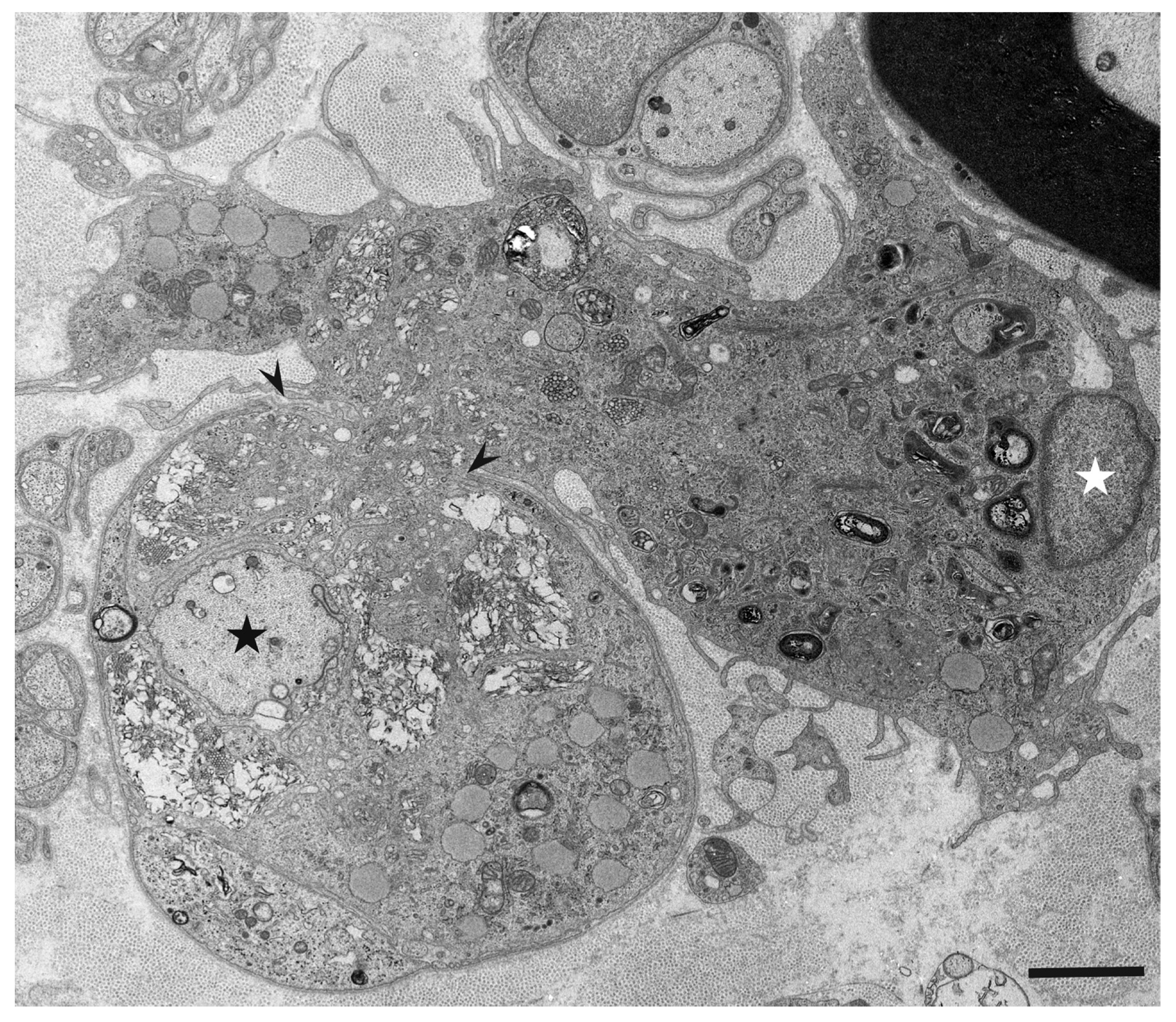
3. Morphology of Macrophages in Demyelination
4. What Attracts Macrophages to Myelin?
5. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Prineas, J.W. Acute idiopathic polyneuritis. An electron microscope study. Lab. Investig. 1972, 26, 133–147. [Google Scholar] [PubMed]
- Dyck, P.J.; Lais, A.C.; Ohta, M.; Bastron, J.A.; Okazaki, H.; Groover, R.V. Chronic inflammatory polyradiculoneuropathy. Mayo Clin Proc. 1975, 50, 621–637. [Google Scholar] [PubMed]
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis—The plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Nishi, R.; Ikeda, S.; Kawagashira, Y.; Iijima, M.; Katsuno, M.; Sobue, G. Ultrastructural mechanisms of macrophage-induced demyelination in CIDP. Neurology 2018, 91, 1051–1060. [Google Scholar] [CrossRef]
- Koike, H.; Fukami, Y.; Nishi, R.; Kawagashira, Y.; Iijima, M.; Katsuno, M.; Sobue, G. Ultrastructural mechanisms of macrophage-induced demyelination in Guillain-Barré syndrome. J. Neurol. Neurosurg. Psychiatry 2020, 91, 650–659. [Google Scholar] [CrossRef]
- Prineas, J.; McLeod, J. Chronic relapsing polyneuritis. J. Neurol. Sci. 1976, 27, 427–458. [Google Scholar] [CrossRef]
- Kuwabara, S.; Yuki, N. Axonal Guillain-Barré syndrome: Concepts and controversies. Lancet Neurol. 2013, 12, 1180–1188. [Google Scholar] [CrossRef]
- Koike, H.; Kadoya, M.; Kaida, K.-I.; Ikeda, S.; Kawagashira, Y.; Iijima, M.; Kato, D.; Ogata, H.; Yamasaki, R.; Matsukawa, N.; et al. Paranodal dissection in chronic inflammatory demyelinating polyneuropathy with anti-neurofascin-155 and anti-contactin-1 antibodies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 465–473. [Google Scholar] [CrossRef]
- Koike, H.; Katsuno, M. Pathophysiology of Chronic Inflammatory Demyelinating Polyneuropathy: Insights into Classification and Therapeutic Strategy. Neurol. Ther. 2020, 9, 213–227. [Google Scholar] [CrossRef]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef]
- Griffin, J.W.; Li, C.Y.; Ho, T.W.; Xue, P.; Macko, C.; Gao, C.Y.; Yang, C.; Tian, M.; Mishu, B.; Cornblath, D.R.; et al. Guillain-Barré syndrome in northern China: The spectrum of neuropathological changes in clinically defined cases. Brain 1995, 118, 577–595. [Google Scholar] [CrossRef]
- Misu, T.; Fujihara, K.; Kakita, A.; Konno, H.; Nakamura, M.; Watanabe, S.; Takahashi, T.; Nakashima, I.; Itoyama, Y. Loss of aquaporin 4 in lesions of Neuromyelitis optica: Distinction from multiple sclerosis. Brain 2007, 130, 1224–1234. [Google Scholar] [CrossRef]
- Yuki, N.; Hartung, H.P. Guillain-Barré syndrome. N. Engl. J. Med. 2012, 366, 2294–2304. [Google Scholar] [CrossRef] [PubMed]
- Feasby, T.E.; Gilbert, J.J.; Brown, W.F.; Bolton, C.F.; Hahn, A.F.; Koopman, W.F.; Zochodne, D.W. An acute axonal form of Guillain-Barré polyneuropathy. Brain 1986, 109, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Sobue, G.; Li, M.; Terao, S.; Aoki, S.; Ichimura, M.; Ieda, T.; Doyu, M.; Yasuda, T.; Hashizume, Y.; Mitsuma, T. Axonal pathology in Japanese Guillain-Barré syndrome: A study of 15 autopsied cases. Neurology 1997, 48, 1694–1700. [Google Scholar] [CrossRef]
- Hafer-Macko, C.; Hsieh, S.T.; Li, C.Y.; Ho, T.W.; Sheikh, K.; Cornblath, D.R.; McKhann, G.M.; Asbury, A.K.; Griffin, J.W. Acute motor axonal neuropathy: An antibody-mediated attack on axolemma. Ann. Neurol. 1996, 40, 635–644. [Google Scholar] [CrossRef]
- Susuki, K.; Rasband, M.N.; Tohyama, K.; Koibuchi, K.; Okamoto, S.; Funakoshi, K.; Hirata, K.; Baba, H.; Yuki, N. Anti-GM1 Antibodies Cause Complement-Mediated Disruption of Sodium Channel Clusters in Peripheral Motor Nerve Fibers. J. Neurosci. 2007, 27, 3956–3967. [Google Scholar] [CrossRef] [PubMed]
- Matsui, N.; Nodera, H.; Kuzume, D.; Iwasa, N.; Unai, Y.; Sakai, W.; Miyazaki, Y.; Yamazaki, H.; Osaki, Y.; Mori, A.; et al. Guillain-Barré syndrome in a local area in Japan, 2006–2015: An epidemiological and clinical study of 108 patients. Eur. J. Neurol. 2018, 25, 718–724. [Google Scholar] [CrossRef]
- Sawai, S.; Satoh, M.; Mori, M.; Misawa, S.; Sogawa, K.; Kazami, T.; Ishibashi, M.; Beppu, M.; Shibuya, K.; Ishige, T.; et al. Moesin is a possible target molecule for cytomegalovirus-related Guillain-Barre syndrome. Neurology 2014, 83, 113–117. [Google Scholar] [CrossRef]
- Uncini, A.; Shahrizaila, N.; Kuwabara, S. Zika virus infection and Guillain-Barré syndrome: A review focused on clinical and electrophysiological subtypes. J. Neurol. Neurosurg. Psychiatry 2017, 88, 266–271. [Google Scholar] [CrossRef]
- Leonhard, S.E.; Halstead, S.; Lant, S.B.; Albuquerque, M.D.F.P.M.D.; De Brito, C.A.A.; De Albuquerque, L.B.B.; Ellul, M.A.; França, R.F.D.O.; Gourlay, D.; Griffiths, M.J.; et al. Guillain-Barré syndrome during the Zika virus outbreak in Northeast Brazil: An observational cohort study. J. Neurol. Sci. 2021, 420, 117272. [Google Scholar] [CrossRef]
- Lucchese, G.; Kanduc, D. Zika virus and autoimmunity: From microcephaly to Guillain-Barré syndrome, and beyond. Autoimmun. Rev. 2016, 15, 801–808. [Google Scholar] [CrossRef]
- Lucchese, G.; Kanduc, D. Minimal immune determinants connect Zika virus, human Cytomegalovirus, and Toxoplasma gondiito microcephaly-related human proteins. Am. J. Reprod. Immunol. 2017, 77, e12608. [Google Scholar] [CrossRef]
- Fragiel, M.; Miró, Ò.; Llorens, P.; Jiménez, S.; Piñera, P.; Burillo, G.; Martín, A.; Martín-Sánchez, F.J.; García-Lamberechts, E.J.; Jacob, J.; et al. SIESTA (Spanish Investigators in Emergency Situations Team) network. Incidence, clinical, risk factors and outcomes of Guillain-Barré in Covid-19. Ann. Neurol. 2021, 89, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Keddie, S.; Pakpoor, J.; Mousele, C.; Pipis, M.; Machado, P.M.; Foster, M.; Record, C.J.; Keh, R.Y.S.; Fehmi, J.; Paterson, R.W.; et al. Epidemiological and cohort study finds no association between COVID-19 and Guillain-Barré syndrome. Brain 2021, 144, 682–693. (in press). [CrossRef]
- Sriwastava, S.; Kataria, S.; Tandon, M.; Patel, J.; Patel, R.; Jowkar, A.; Daimee, M.; Bernitsas, E.; Jaiswal, P.; Lisak, R.P. Guillain Barré Syndrome and its variants as a manifestation of COVID-19: A systematic review of case reports and case series. J. Neurol. Sci. 2021, 420, 117263. [Google Scholar] [CrossRef] [PubMed]
- Cornblath, D.R.; Asbury, A.K.; Albers, J.W.; Feasby, T.E. Research criteria for diagnosis of chronic inflammatory demyelinating polyneuropathy (CIDP). Neurology 1991, 41, 617–618. [Google Scholar] [CrossRef]
- Van den Bergh, P.Y.; Hadden, R.D.; Bouche, P.; Cornblath, D.R.; Hahn, A.; Illa, I.; Koski, C.L.; Léger, J.M.; Nobile-Orazio, E.; Pollard, J.; et al. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society—First revision. Eur. J. Neurol. 2010, 17, 356–363. [Google Scholar] [PubMed]
- Ikeda, S.; Koike, H.; Nishi, R.; Kawagashira, Y.; Iijima, M.; Katsuno, M.; Sobue, G. Clinicopathological characteristics of subtypes of chronic inflammatory demyelinating polyradiculoneuropathy. J. Neurol. Neurosurg. Psychiatry 2019, 90, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Querol, L.; Nogales-Gadea, G.; Rojas-Garcia, R.; Martinez-Hernandez, E.; Diaz-Manera, J.; Suárez-Calvet, X.; Navas, M.; Araque, J.; Gallardo, E.; Illa, I. Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann. Neurol. 2013, 73, 370–380. [Google Scholar] [CrossRef]
- Doppler, K.; Appeltshauser, L.; Wilhelmi, K.; Villmann, C.; Dib-Hajj, S.D.; Waxman, S.G.; Mäurer, M.; Weishaupt, A.; Sommer, C. Destruction of paranodal architecture in inflammatory neuropathy with anti-contactin-1 autoantibodies. J. Neurol. Neurosurg. Psychiatry 2015, 86, 720–728. [Google Scholar] [CrossRef]
- Ogata, H.; Yamasaki, R.; Hiwatashi, A.; Oka, N.; Kawamura, N.; Matsuse, D.; Kuwahara, M.; Suzuki, H.; Kusunoki, S.; Fujimoto, Y.; et al. Characterization of IgG4 anti-neurofascin 155 antibody-positive polyneuropathy. Ann. Clin. Transl. Neurol. 2015, 2, 960–971. [Google Scholar] [CrossRef]
- Kadoya, M.; Kaida, K.; Koike, H.; Takazaki, H.; Ogata, H.; Moriguchi, K.; Shimizu, J.; Nagata, E.; Takizawa, S.; Chiba, A.; et al. IgG4 anti-neurofascin155 antibodies in chronic inflammatory demyelinating polyradiculoneuropathy: Clinical significance and diagnostic utility of a conventional assay. J. Neuroimmunol. 2016, 301, 16–22. [Google Scholar] [CrossRef]
- Vallat, J.M.; Magy, L.; Corcia, P.; Boulesteix, J.M.; Uncini, A.; Mathis, S. Ultrastructural lesions of nodo-paranodopathies in peripheral neuropathies. J. Neuropathol. Exp. Neurol. 2020, 79, 247–255. [Google Scholar] [CrossRef]
- Vallat, J.-M.; Yuki, N.; Sekiguchi, K.; Kokubun, N.; Oka, N.; Mathis, S.; Magy, L.; Sherman, D.L.; Brophy, P.J.; Devaux, J.J. Paranodal lesions in chronic inflammatory demyelinating polyneuropathy associated with anti-Neurofascin 155 antibodies. Neuromuscul. Disord. 2017, 27, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Nishi, R.; Ikeda, S.; Kawagashira, Y.; Iijima, M.; Atsuta, N.; Nakamura, T.; Hirayama, M.; Ogata, H.; Yamasaki, R.; et al. Restoration of a conduction block after the long-term treatment of CIDP with anti-neurofascin 155 antibodies: Follow-up of a case over 23 years. Intern. Med. 2018, 57, 2061–2066. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Ann. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-Cell Depletion with Rituximab in Relapsing–Remitting Multiple Sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.K.; Yong, V.W. Myeloid cells—Targets of medication in multiple sclerosis. Nat. Rev. Neurol. 2016, 12, 539–551. [Google Scholar] [CrossRef]
- Grajchen, E.; Hendriks, J.J.A.; Bogie, J.F.J. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol. Commun. 2018, 6, 124. [Google Scholar] [CrossRef] [PubMed]
- Prineas, J.W.; Graham, J.S. Multiple sclerosis: Capping of surface immunoglobulin G on macrophages engaged in myelin breakdown. Ann. Neurol. 1981, 10, 149–158. [Google Scholar] [CrossRef]
- Kira, J. Multiple sclerosis in the Japanese population. Lancet Neurol. 2003, 2, 117–127. [Google Scholar] [CrossRef]
- Jung, J.S.; Bhat, R.V.; Preston, G.M.; Guggino, W.B.; Baraban, J.M.; Agre, P. Molecular characterization of an aquaporin cDNA from brain: Candidate osmoreceptor and regulator of water balance. Proc. Natl. Acad. Sci. USA 1994, 91, 13052–13056. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; De Seze, J.; Fujihara, K.; Greenberg, B.M.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.W.; Stoll, G.; Li, C.Y.; Tyor, W.; Cornblath, D.R. Macrophage responses in inflammatory demyelinating neuropathies. Ann. Neurol. 1990, 27, S64–S68. [Google Scholar] [CrossRef]
- Hartung, H.-P.; Reiners, K.; Michels, M.; Hughes, R.; Heidenreich, F.; Zielasek, J.; Enders, U.; Toyka, K.V. Serum levels of soluble E-selectin (ELAM-1) in immune-mediated neuropathies. Neurology 1994, 44, 1153. [Google Scholar] [CrossRef]
- Enders, U.; Lobb, R.; Pepinsky, R.B.; Hartung, H.P.; Toyka, K.V.; Gold, R. The role of the very late antigen-4 and its counterligand vascular cell adhesion molecule-1 in the pathogenesis of experimental autoimmune neuritis of the Lewis rat. Brain 1998, 121, 1257–1266. [Google Scholar] [CrossRef]
- Trojano, M.; Avolio, C.; Ruggieri, M.; De Robertis, F.; Giuliani, F.; Paolicelli, D.; Livrea, P. Soluble intercellular adhesion molecule-I (sICAM-I) in serum and cerebrospinal fluid of demyelinating diseases of the central and peripheral nervous system. Mult. Scler. 1998, 4, 39–44. [Google Scholar] [CrossRef]
- Lund, H.; Pieber, M.; Parsa, R.; Han, J.; Grommisch, D.; Ewing, E.; Kular, L.; Needhamsen, M.; Espinosa, A.; Nilsson, E.; et al. Competitive repopulation of an empty microglial niche yields functionally distinct subsets of microglia-like cells. Nat. Commun. 2018, 9, 4845. [Google Scholar] [CrossRef] [PubMed]
- Park, H.T.; Kim, Y.H.; Lee, K.E.; Kim, J.K. Behind the pathology of macrophage-associated demyelination in inflammatory neuropathies: Demyelinating Schwann cells. Cell. Mol. Life Sci. 2020, 77, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Katsuno, M.; Sobue, G. Deciphering the mechanism and spectrum of chronic inflammatory demyelinating polyneuropathy using morphology. Clin. Exp. Neuroimmunol. 2018, 9, 35–46. [Google Scholar] [CrossRef]
- Wiśniewski, H.; Terry, R.D.; Whitaker, J.N.; Cook, S.D.; Dowling, P.C. Landry-Guillain-Barré syndrome. A primary demyelinating disease. Arch. Neurol. 1969, 21, 269–276. [Google Scholar] [CrossRef]
- Carpenter, S. An ultrastructural study of an acute fatal case of the Guillain-Barré syndrome. J. Neurol. Sci. 1972, 15, 125–140. [Google Scholar] [CrossRef]
- Hafer-Macko, C.E.; Sheikh, K.A.; Li, C.Y.; Ho, T.W.; Cornblath, D.R.; McKhann, G.M.; Asbury, A.K.; Griffin, J.W. Immune attack on the schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann. Neurol. 1996, 39, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Berciano, J.; Figols, J.; García, A.; Calle, E.; Illa, I.; Lafarga, M.; Berciano, M.T. Fulminant Guillain-Barré syndrome with universal inexcitability of peripheral nerves: A clinicopathological study. Muscle Nerve 1997, 20, 846–857. [Google Scholar] [CrossRef]
- Raine, C.S.; Cannella, B.; Hauser, S.L.; Genain, C.P. Demyelination in primate autoimmune encephalomyelitis and acute multiple sclerosis lesions: A case for antigen-specific antibody mediation. Ann. Neurol. 1999, 46, 144–160. [Google Scholar] [CrossRef]
- Koike, H.; Katsuno, M. The role of macrophages in Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy. Neurol. Clin. Neurosci 2021, in press. [Google Scholar] [CrossRef]
- Lampert, P.W. Mechanism of demyelination in experimental allergic neuritis. Electron microscopic studies. Lab. Investig. 1969, 20, 127–138. [Google Scholar]
- Schröder, J.M.; Krücke, W. Zur Feinstruktur der experimentell-allergischen Neuritis beim Kaninchen [Ultrastructure of experimental allergic neuritis in the rabbit]. Acta Neuropathol. 1970, 14, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Lampert, P. Electron microscopic studies on ordinary and hyperacute experimental allergic encephalomyelitis. Acta Neuropathol. 1967, 9, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Lampert, P.W.; Kies, M.W. Mechanism of demyelination in allergic encephalomyelitis of guinea pigs. An electron microscopic study. Exp. Neurol. 1967, 18, 210–223. [Google Scholar] [CrossRef]
- Lassmann, H.; Wisniewski, H. Chronic relapsing experimental allergic encephalomyelitis: Morphological sequence of myelin degradation. Brain Res. 1979, 169, 357–368. [Google Scholar] [CrossRef]
- Ilyas, A.A.; Mithen, F.A.; Dalakas, M.C.; Chen, Z.W.; Cook, S.D. Antibodies to acidic glycolipids in Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy. J. Neurol. Sci. 1992, 107, 111–121. [Google Scholar] [CrossRef]
- Kusunoki, S.; Chiba, A.; Hitoshi, S.; Takizawa, H.; Kanazawa, I. Anti-gal-C antibody in autoimmune neuropathies subsequent to mycoplasma infection. Muscle Nerve 1995, 18, 409–413. [Google Scholar] [CrossRef]
- Kuwahara, M.; Suzuki, S.; Takada, K.; Kusunoki, S. Antibodies to LM1 and LM1-containing ganglioside complexes in Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy. J. Neuroimmunol. 2011, 239, 87–90. [Google Scholar] [CrossRef]
- Devaux, J.J.; Odaka, M.; Yuki, N. Nodal proteins are target antigens in Guillain-Barré syndrome. J. Peripher. Nerv. Syst. 2012, 17, 62–71. [Google Scholar] [CrossRef]
- Koike, H.; Ikeda, S.; Fukami, Y.; Nishi, R.; Kawagashira, Y.; Iijima, M.; Nakamura, T.; Kuwahara, M.; Kusunoki, S.; Katsuno, M.; et al. Complement deposition and macrophage-induced demyelination in CIDP with anti-LM1 antibodies. J. Neurol. Sci. 2020, 408, 116509. [Google Scholar] [CrossRef]
- Kuwahara, M.; Suzuki, H.; Samukawa, M.; Hamada, Y.; Takada, K.; Kusunoki, S. Clinical features of CIDP with LM1-associated antibodies. J. Neurol. Neurosurg. Psychiatry 2013, 84, 573–575. [Google Scholar] [CrossRef]
- Sato, D.K.; Callegaro, D.; Lana-Peixoto, M.A.; Waters, P.J.; Jorge, F.M.D.H.; Takahashi, T.; Nakashima, I.; Apostolos-Pereira, S.L.; Talim, N.; Simm, R.F.; et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 2014, 82, 474–481. [Google Scholar] [CrossRef]
- Cobo-Calvo, Á.; d’Indy, H.; Ruiz, A.; Collongues, N.; Kremer, L.; Durand-Dubief, F.; Rollot, F.; Casey, R.; Vukusic, S.; De Seze, J.; et al. Frequency of myelin oligodendrocyte glycoprotein antibody in multiple sclerosis: A multicenter cross-sectional study. Neurol. Neuroimmunol. Neuroinflamm. 2019, 7, e649. [Google Scholar] [CrossRef] [PubMed]
- Misawa, S.; Kuwabara, S.; Sato, Y.; Yamaguchi, N.; Nagashima, K.; Katayama, K.; Sekiguchi, Y.; Iwai, Y.; Amino, H.; Suichi, T.; et al. Safety and efficacy of eculizumab in Guillain-Barré syndrome: A multicentre, double-blind, randomised phase 2 trial. Lancet. Neurol. 2018, 17, 519–529. [Google Scholar] [CrossRef]
- Shen, D.; Chu, F.; Lang, Y.; Geng, Y.; Zheng, X.; Zhu, J.; Liu, K. Beneficial or Harmful Role of Macrophages in Guillain-Barré Syndrome and Experimental Autoimmune Neuritis. Mediat. Inflamm. 2018, 2018, 4286364. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Boyd, A.; Zhao, J.-W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; Van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koike, H.; Katsuno, M. Macrophages and Autoantibodies in Demyelinating Diseases. Cells 2021, 10, 844. https://doi.org/10.3390/cells10040844
Koike H, Katsuno M. Macrophages and Autoantibodies in Demyelinating Diseases. Cells. 2021; 10(4):844. https://doi.org/10.3390/cells10040844
Chicago/Turabian StyleKoike, Haruki, and Masahisa Katsuno. 2021. "Macrophages and Autoantibodies in Demyelinating Diseases" Cells 10, no. 4: 844. https://doi.org/10.3390/cells10040844
APA StyleKoike, H., & Katsuno, M. (2021). Macrophages and Autoantibodies in Demyelinating Diseases. Cells, 10(4), 844. https://doi.org/10.3390/cells10040844