
Adoptive T Cell Therapy for Solid Tumors: Pathway to Personalized Standard of Care



Abstract
1. Introduction
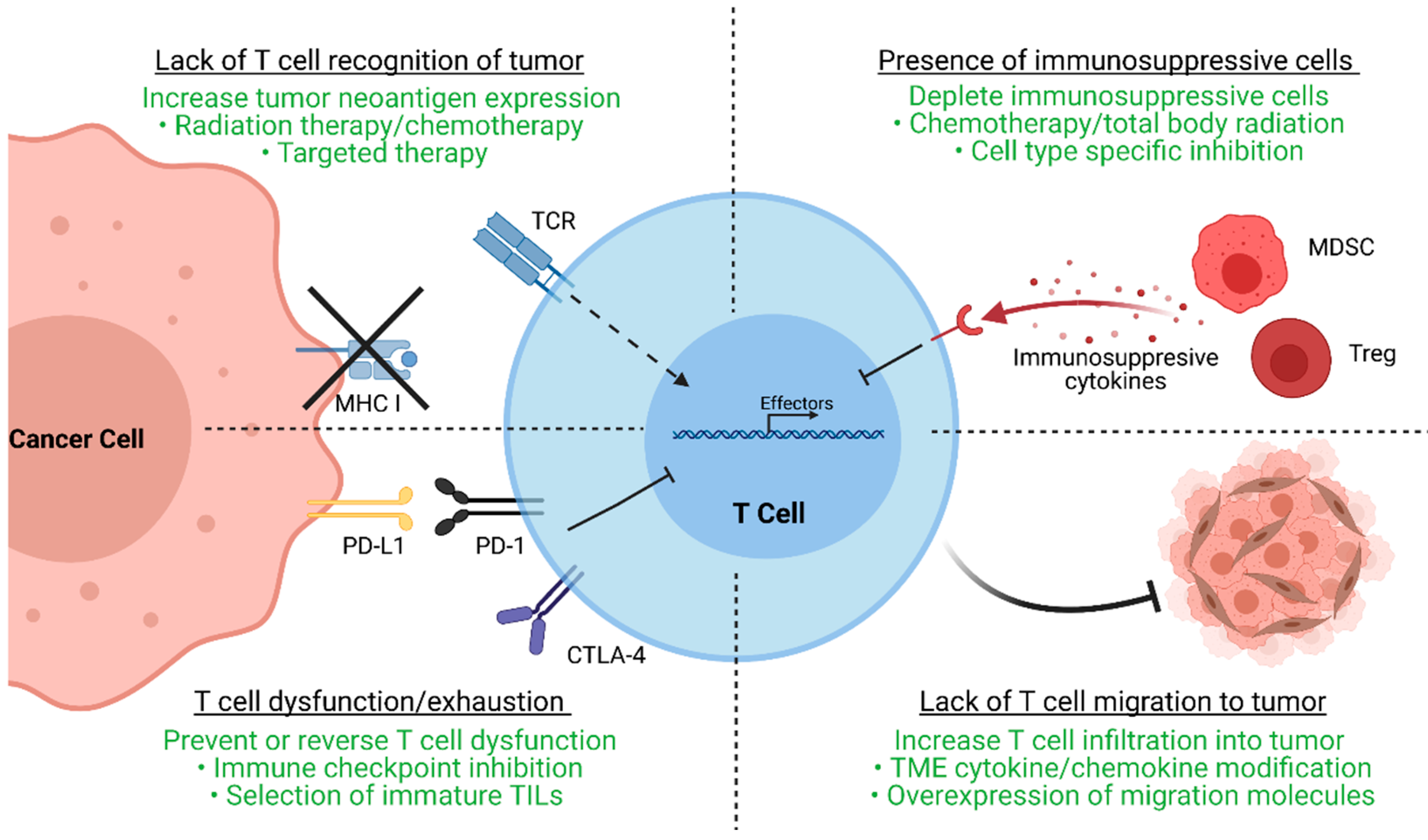
2. Mechanisms of ACT-TIL Resistance
3. TIL-ACT Protocol Refinement
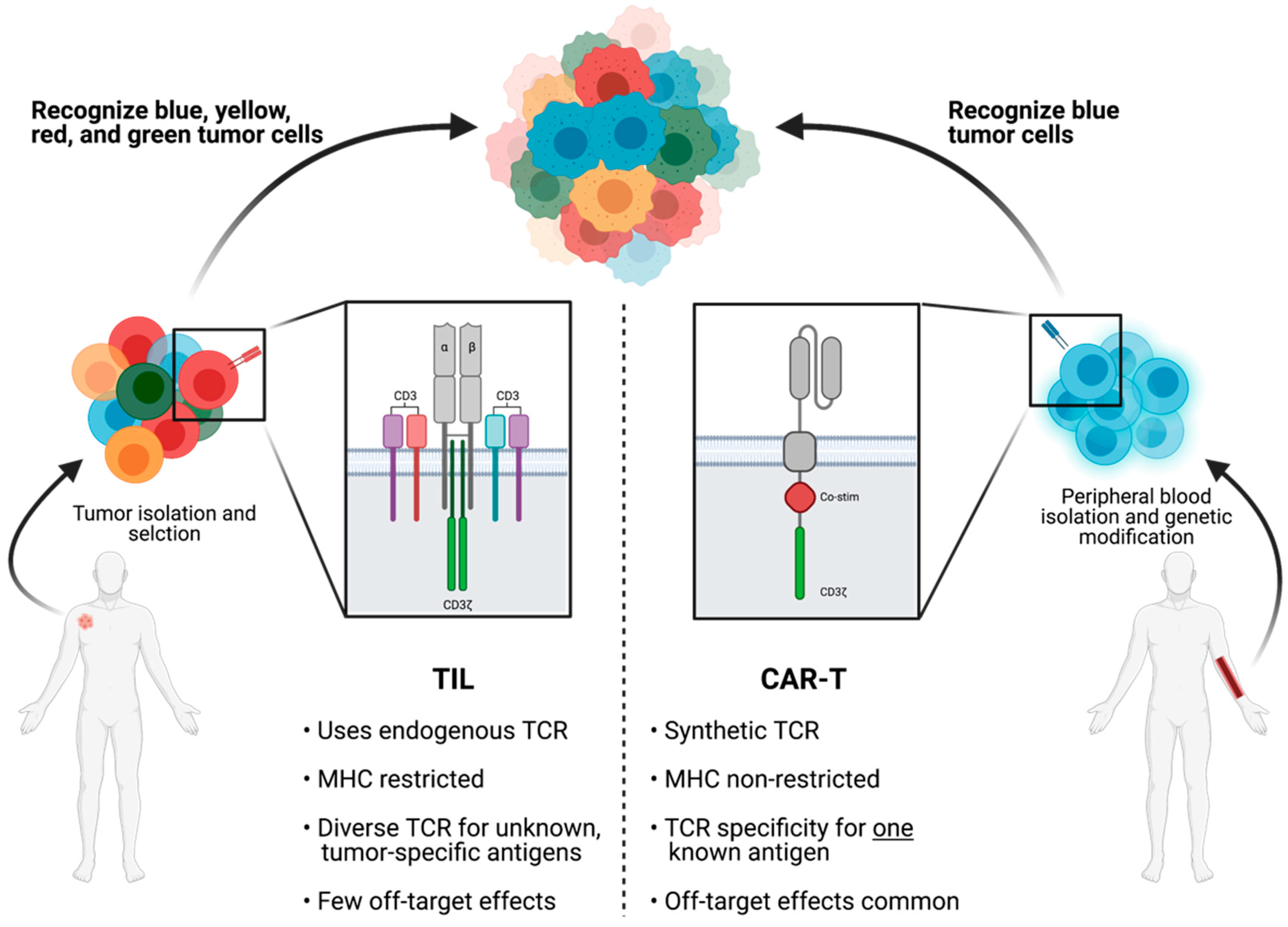
3.1. TIL Cell Type Selection
3.2. Role of Cytokine Support
3.3. Role of Pre-Conditioning Regimens
4. Combination Treatments
4.1. Immune Checkpoint Inhibitors
4.2. Targeted Therapy
4.3. Other Investigative Therapies
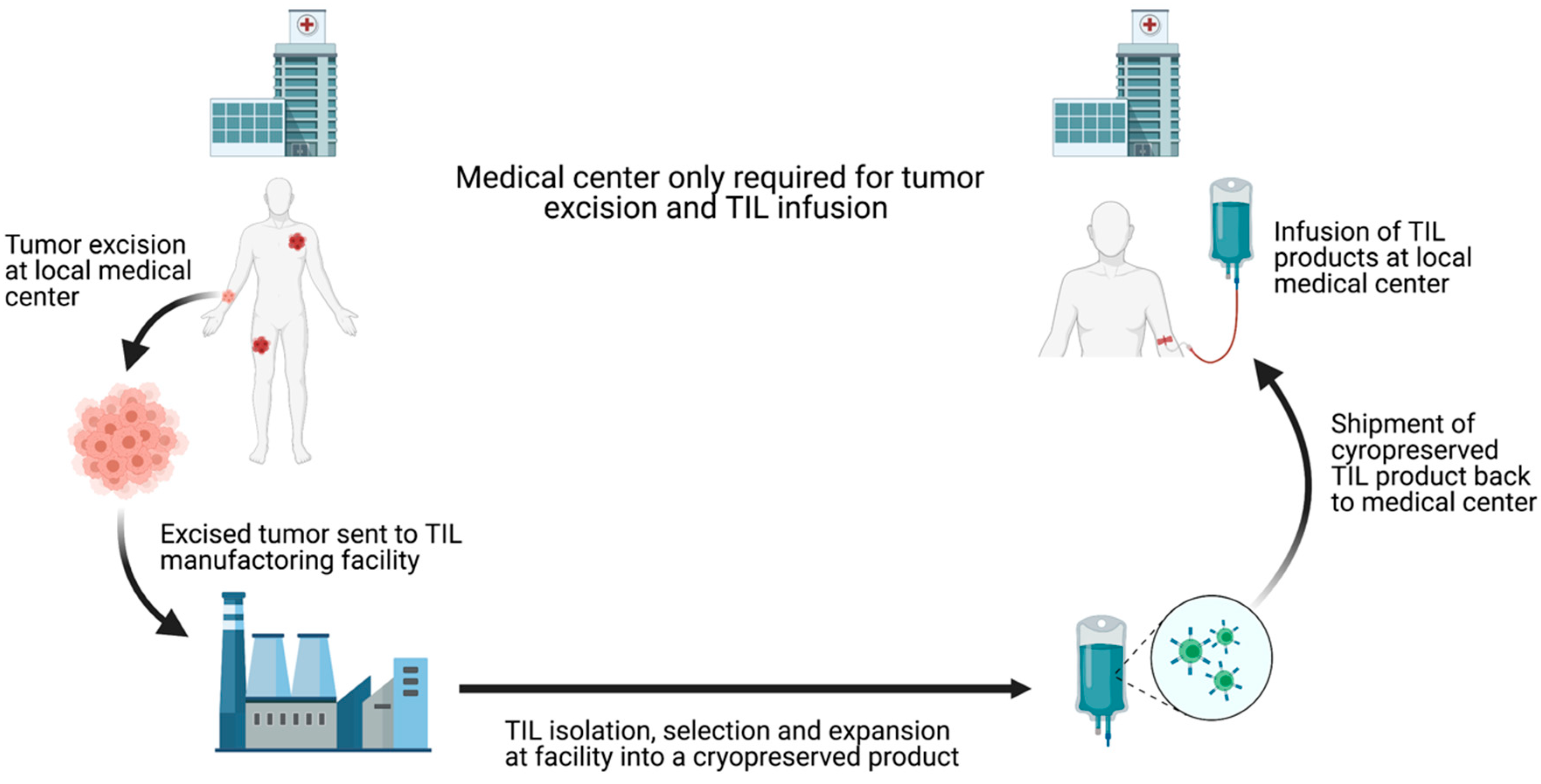
5. The Future of TIL-ACT
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rohaan, M.W.; van den Berg, J.H.; Kvistborg, P.; Haanen, J. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: A viable treatment option. J. Immunother. Cancer 2018, 6, 102. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson, B.A., 3rd; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 569. [Google Scholar] [CrossRef]
- Groscurth, P.; Filgueira, L. Killing Mechanisms of Cytotoxic T Lymphocytes. News Physiol. Sci 1998, 13, 17–21. [Google Scholar] [CrossRef]
- Halle, S.; Halle, O.; Forster, R. Mechanisms and Dynamics of T Cell-Mediated Cytotoxicity In Vivo. Trends Immunol. 2017, 38, 432–443. [Google Scholar] [CrossRef]
- Chandran, S.S.; Klebanoff, C.A. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol. Rev. 2019, 290, 127–147. [Google Scholar] [CrossRef] [PubMed]
- Rohaan, M.W.; Wilgenhof, S.; Haanen, J. Adoptive cellular therapies: The current landscape. Virchows Arch. 2019, 474, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Spiess, P.; Lafreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- van den Berg, J.H.; Heemskerk, B.; van Rooij, N.; Gomez-Eerland, R.; Michels, S.; van Zon, M.; de Boer, R.; Bakker, N.A.M.; Jorritsma-Smit, A.; van Buuren, M.M.; et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: Boosting of neoantigen-specific T cell reactivity and long-term follow-up. J. Immunother. Cancer 2020, 8, e000848. [Google Scholar] [CrossRef] [PubMed]
- Pockaj, B.A.; Sherry, R.M.; Wei, J.P.; Yannelli, J.R.; Carter, C.S.; Leitman, S.F.; Carasquillo, J.A.; Steinberg, S.M.; Rosenberg, S.A.; Yang, J.C. Localization of 111indium-labeled tumor infiltrating lymphocytes to tumor in patients receiving adoptive immunotherapy. Augmentation with cyclophosphamide and correlation with response. Cancer 1994, 73, 1731–1737. [Google Scholar] [CrossRef]
- Itzhaki, O.; Hovav, E.; Ziporen, Y.; Levy, D.; Kubi, A.; Zikich, D.; Hershkovitz, L.; Treves, A.J.; Shalmon, B.; Zippel, D.; et al. Establishment and large-scale expansion of minimally cultured “young” tumor infiltrating lymphocytes for adoptive transfer therapy. J. Immunother. 2011, 34, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Besser, M.J.; Shapira-Frommer, R.; Itzhaki, O.; Treves, A.J.; Zippel, D.B.; Levy, D.; Kubi, A.; Shoshani, N.; Zikich, D.; Ohayon, Y.; et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: Intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin. Cancer Res. 2013, 19, 4792–4800. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Stratton, M.R. Mutational signatures: The patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014, 24, 52–60. [Google Scholar] [CrossRef]
- Stevanovic, S.; Helman, S.R.; Wunderlich, J.R.; Langhan, M.M.; Doran, S.L.; Kwong, M.L.M.; Somerville, R.P.T.; Klebanoff, C.A.; Kammula, U.S.; Sherry, R.M.; et al. A Phase II Study of Tumor-infiltrating Lymphocyte Therapy for Human Papillomavirus-associated Epithelial Cancers. Clin. Cancer Res. 2019, 25, 1486–1493. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- van der Woude, L.L.; Gorris, M.A.J.; Halilovic, A.; Figdor, C.G.; de Vries, I.J.M. Migrating into the Tumor: A Roadmap for T Cells. Trends Cancer 2017, 3, 797–808. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8(+) T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Kuhns, M.S.; Davis, M.M.; Garcia, K.C. Deconstructing the form and function of the TCR/CD3 complex. Immunity 2006, 24, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Riviere, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Krause, A.; Guo, H.F.; Latouche, J.B.; Tan, C.; Cheung, N.K.; Sadelain, M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef]
- Eshhar, Z.; Bach, N.; Fitzer-Attas, C.J.; Gross, G.; Lustgarten, J.; Waks, T.; Schindler, D.G. The T-body approach: Potential for cancer immunotherapy. Springer Semin Immunopathol. 1996, 18, 199–209. [Google Scholar] [CrossRef]
- Mohanty, R.; Chowdhury, C.R.; Arega, S.; Sen, P.; Ganguly, P.; Ganguly, N. CAR T cell therapy: A new era for cancer treatment (Review). Oncol. Rep. 2019, 42, 2183–2195. [Google Scholar] [CrossRef]
- Wolf, B.; Zimmermann, S.; Arber, C.; Irving, M.; Trueb, L.; Coukos, G. Safety and Tolerability of Adoptive Cell Therapy in Cancer. Drug Saf. 2019, 42, 315–334. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Robbins, P.F.; Tran, E.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Ivey, G.; Li, Y.F.; El-Gamil, M.; Lalani, A.; et al. Unique Neoantigens Arise from Somatic Mutations in Patients with Gastrointestinal Cancers. Cancer Discov. 2019, 9, 1022–1035. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef]
- Zhou, J.; Shen, X.; Huang, J.; Hodes, R.J.; Rosenberg, S.A.; Robbins, P.F. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J. Immunol. 2005, 175, 7046–7052. [Google Scholar] [CrossRef]
- Dudley, M.E.; Gross, C.A.; Langhan, M.M.; Garcia, M.R.; Sherry, R.M.; Yang, J.C.; Phan, G.Q.; Kammula, U.S.; Hughes, M.S.; Citrin, D.E.; et al. CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin. Cancer Res. 2010, 16, 6122–6131. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Gross, C.A.; Somerville, R.P.; Hong, Y.; Schaub, N.P.; Rosati, S.F.; White, D.E.; Nathan, D.; Restifo, N.P.; Steinberg, S.M.; et al. Randomized selection design trial evaluating CD8+-enriched versus unselected tumor-infiltrating lymphocytes for adoptive cell therapy for patients with melanoma. J. Clin. Oncol. 2013, 31, 2152–2159. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 2019, 129, 1109–1114. [Google Scholar] [CrossRef]
- Lu, Y.C.; Zheng, Z.; Robbins, P.F.; Tran, E.; Prickett, T.D.; Gartner, J.J.; Li, Y.F.; Ray, S.; Franco, Z.; Bliskovsky, V.; et al. An Efficient Single-Cell RNA-Seq Approach to Identify Neoantigen-Specific T Cell Receptors. Mol. Ther. 2018, 26, 379–389. [Google Scholar] [CrossRef]
- Yossef, R.; Tran, E.; Deniger, D.C.; Gros, A.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Cafri, G.; Robbins, P.F.; et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 2018, 3, e122467. [Google Scholar] [CrossRef]
- Radvanyi, L.G.; Bernatchez, C.; Zhang, M.; Fox, P.S.; Miller, P.; Chacon, J.; Wu, R.; Lizee, G.; Mahoney, S.; Alvarado, G.; et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2012, 18, 6758–6770. [Google Scholar] [CrossRef]
- Goff, S.L.; Dudley, M.E.; Citrin, D.E.; Somerville, R.P.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. J. Clin. Oncol. 2016, 34, 2389–2397. [Google Scholar] [CrossRef]
- Yao, X.; Ahmadzadeh, M.; Lu, Y.C.; Liewehr, D.J.; Dudley, M.E.; Liu, F.; Schrump, D.S.; Steinberg, S.M.; Rosenberg, S.A.; Robbins, P.F. Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood 2012, 119, 5688–5696. [Google Scholar] [CrossRef]
- Antony, P.A.; Piccirillo, C.A.; Akpinarli, A.; Finkelstein, S.E.; Speiss, P.J.; Surman, D.R.; Palmer, D.C.; Chan, C.C.; Klebanoff, C.A.; Overwijk, W.W.; et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J. Immunol. 2005, 174, 2591–2601. [Google Scholar] [CrossRef]
- Li, K.; Donaldson, B.; Young, V.; Ward, V.; Jackson, C.; Baird, M.; Young, S. Adoptive cell therapy with CD4(+) T helper 1 cells and CD8(+) cytotoxic T cells enhances complete rejection of an established tumour, leading to generation of endogenous memory responses to non-targeted tumour epitopes. Clin. Transl. Immunol. 2017, 6, e160. [Google Scholar] [CrossRef]
- Imai, N.; Tawara, I.; Yamane, M.; Muraoka, D.; Shiku, H.; Ikeda, H. CD4(+) T cells support polyfunctionality of cytotoxic CD8(+) T cells with memory potential in immunological control of tumor. Cancer Sci. 2020, 111, 1958–1968. [Google Scholar] [CrossRef]
- Friedman, K.M.; Prieto, P.A.; Devillier, L.E.; Gross, C.A.; Yang, J.C.; Wunderlich, J.R.; Rosenberg, S.A.; Dudley, M.E. Tumor-specific CD4+ melanoma tumor-infiltrating lymphocytes. J. Immunother. 2012, 35, 400–408. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Prieto, P.A.; Durflinger, K.H.; Wunderlich, J.R.; Rosenberg, S.A.; Dudley, M.E. Enrichment of CD8+ cells from melanoma tumor-infiltrating lymphocyte cultures reveals tumor reactivity for use in adoptive cell therapy. J. Immunother. 2010, 33, 547–556. [Google Scholar] [CrossRef]
- Robbins, P.F.; Dudley, M.E.; Wunderlich, J.; El-Gamil, M.; Li, Y.F.; Zhou, J.; Huang, J.; Powell, D.J., Jr.; Rosenberg, S.A. Cutting edge: Persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J. Immunol. 2004, 173, 7125–7130. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Desmarais, C.; Emerson, R.; Schmitt, T.M.; Shibuya, K.; Lai, I.; Wagener, F.; Chou, J.; Roberts, I.M.; Coffey, D.G.; et al. Tracking the Fate and Origin of Clinically Relevant Adoptively Transferred CD8(+) T Cells In Vivo. Sci. Immunol. 2017, 2, eaal2568. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhang, W.; Shao, H.; Bo, H.; Shen, H.; Li, J.; Liu, Y.; Wang, T.; Ma, W.; Huang, S. Human effector T cells derived from central memory cells rather than CD8(+)T cells modified by tumor-specific TCR gene transfer possess superior traits for adoptive immunotherapy. Cancer Lett. 2013, 339, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Scott, C.D.; Leonardi, A.J.; Yamamoto, T.N.; Cruz, A.C.; Ouyang, C.; Ramaswamy, M.; Roychoudhuri, R.; Ji, Y.; Eil, R.L.; et al. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J. Clin. Investig. 2016, 126, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, C.S.; Borman, Z.A.; Cassard, L.; Gattinoni, L.; Spolski, R.; Yu, Z.; Sanchez-Perez, L.; Muranski, P.; Kern, S.J.; Logun, C.; et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 17469–17474. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Jia, L.; Zheng, Z.; Tran, E.; Robbins, P.F.; Rosenberg, S.A. Single-Cell Transcriptome Analysis Reveals Gene Signatures Associated with T-cell Persistence Following Adoptive Cell Therapy. Cancer Immunol. Res. 2019, 7, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef]
- Sprent, J.; Surh, C.D. Normal T cell homeostasis: The conversion of naive cells into memory-phenotype cells. Nat. Immunol. 2011, 12, 478–484. [Google Scholar] [CrossRef]
- Furlan, S.N.; Singh, K.; Lopez, C.; Tkachev, V.; Hunt, D.J.; Hibbard, J.; Betz, K.M.; Blazar, B.R.; Trapnell, C.; Kean, L.S. IL-2 enhances ex vivo-expanded regulatory T-cell persistence after adoptive transfer. Blood Adv. 2020, 4, 1594–1605. [Google Scholar] [CrossRef]
- Ross, S.H.; Cantrell, D.A. Signaling and Function of Interleukin-2 in T Lymphocytes. Annu. Rev. Immunol. 2018, 36, 411–433. [Google Scholar] [CrossRef]
- Liu, J.; Cao, S.; Kim, S.; Chung, E.Y.; Homma, Y.; Guan, X.; Jimenez, V.; Ma, X. Interleukin-12: An update on its immunological activities, signaling and regulation of gene expression. Curr. Immunol. Rev. 2005, 1, 119–137. [Google Scholar] [CrossRef]
- Tucker, C.G.; Mitchell, J.S.; Martinov, T.; Burbach, B.J.; Beura, L.K.; Wilson, J.C.; Dwyer, A.J.; Singh, L.M.; Mescher, M.F.; Fife, B.T. Adoptive T Cell Therapy with IL-12-Preconditioned Low-Avidity T Cells Prevents Exhaustion and Results in Enhanced T Cell Activation, Enhanced Tumor Clearance, and Decreased Risk for Autoimmunity. J. Immunol. 2020, 205, 1449–1460. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Naga, O.; Zidan, A.A.; Salem, M.L.; Pallin, M.; Parmigiani, A.; Walker, G.; Wieder, E.; Komanduri, K.; Cole, D.J.; et al. Synergy of brief activation of CD8 T-cells in the presence of IL-12 and adoptive transfer into lymphopenic hosts promotes tumor clearance and anti-tumor memory. Am. J. Cancer Res. 2011, 1, 882–896. [Google Scholar] [PubMed]
- Curtsinger, J.M.; Johnson, C.M.; Mescher, M.F. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J. Immunol. 2003, 171, 5165–5171. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.B.; Riesenberg, B.P.; May, B.R.; Gilreath, S.C.; Li, G.; Staveley-O’Carroll, K.F.; Garrett-Mayer, E.; Mehrotra, S.; Cole, D.J.; Rubinstein, M.P. Effector CD8+ T-cell Engraftment and Antitumor Immunity in Lymphodepleted Hosts Is IL7Ralpha Dependent. Cancer Immunol. Res. 2015, 3, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Drake, A.; Kaur, M.; Iliopoulou, B.P.; Phennicie, R.; Hanson, A.; Chen, J. Interleukins 7 and 15 Maintain Human T Cell Proliferative Capacity through STAT5 Signaling. PLoS ONE 2016, 11, e0166280. [Google Scholar] [CrossRef]
- Boyman, O.; Purton, J.F.; Surh, C.D.; Sprent, J. Cytokines and T-cell homeostasis. Curr. Opin. Immunol. 2007, 19, 320–326. [Google Scholar] [CrossRef]
- Gong, W.; Hoffmann, J.M.; Stock, S.; Wang, L.; Liu, Y.; Schubert, M.L.; Neuber, B.; Huckelhoven-Krauss, A.; Gern, U.; Schmitt, A.; et al. Comparison of IL-2 vs IL-7/IL-15 for the generation of NY-ESO-1-specific T cells. Cancer Immunol. Immunother. 2019, 68, 1195–1209. [Google Scholar] [CrossRef]
- Yee, C.; Thompson, J.A.; Byrd, D.; Riddell, S.R.; Roche, P.; Celis, E.; Greenberg, P.D. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: In vivo persistence, migration, and antitumor effect of transferred T cells. Proc. Natl. Acad. Sci. USA 2002, 99, 16168–16173. [Google Scholar] [CrossRef]
- Dudley, M.E.; Yang, J.C.; Sherry, R.; Hughes, M.S.; Royal, R.; Kammula, U.; Robbins, P.F.; Huang, J.; Citrin, D.E.; Leitman, S.F.; et al. Adoptive cell therapy for patients with metastatic melanoma: Evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. 2008, 26, 5233–5239. [Google Scholar] [CrossRef]
- Pilon-Thomas, S.; Kuhn, L.; Ellwanger, S.; Janssen, W.; Royster, E.; Marzban, S.; Kudchadkar, R.; Zager, J.; Gibney, G.; Sondak, V.K.; et al. Efficacy of adoptive cell transfer of tumor-infiltrating lymphocytes after lymphopenia induction for metastatic melanoma. J. Immunother. 2012, 35, 615–620. [Google Scholar] [CrossRef]
- Dafni, U.; Michielin, O.; Lluesma, S.M.; Tsourti, Z.; Polydoropoulou, V.; Karlis, D.; Besser, M.J.; Haanen, J.; Svane, I.M.; Ohashi, P.S.; et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: A systematic review and meta-analysis. Ann. Oncol. 2019, 30, 1902–1913. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, A.G.; Thompson, J.A.; Margolin, K.A.; Rodmyre, R.; Lai, I.P.; Dowdy, K.; Farrar, E.A.; Bhatia, S.; Sabath, D.E.; Cao, J.; et al. Transferred melanoma-specific CD8+ T cells persist, mediate tumor regression, and acquire central memory phenotype. Proc. Natl. Acad. Sci. USA 2012, 109, 4592–4597. [Google Scholar] [CrossRef] [PubMed]
- Ellebaek, E.; Iversen, T.Z.; Junker, N.; Donia, M.; Engell-Noerregaard, L.; Met, O.; Holmich, L.R.; Andersen, R.S.; Hadrup, S.R.; Andersen, M.H.; et al. Adoptive cell therapy with autologous tumor infiltrating lymphocytes and low-dose Interleukin-2 in metastatic melanoma patients. J. Transl. Med. 2012, 10, 169. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.; Donia, M.; Ellebaek, E.; Borch, T.H.; Kongsted, P.; Iversen, T.Z.; Holmich, L.R.; Hendel, H.W.; Met, O.; Andersen, M.H.; et al. Long-Lasting Complete Responses in Patients with Metastatic Melanoma after Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes and an Attenuated IL2 Regimen. Clin. Cancer Res. 2016, 22, 3734–3745. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Saibil, S.D.; Sotov, V.; Le, M.X.; Khoja, L.; Ghazarian, D.; Bonilla, L.; Majeed, H.; Hogg, D.; Joshua, A.M.; et al. Phase II clinical trial of adoptive cell therapy for patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and low-dose interleukin-2. Cancer Immunol. Immunother. 2019, 68, 773–785. [Google Scholar] [CrossRef]
- North, R.J. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J. Exp. Med. 1982, 155, 1063–1074. [Google Scholar] [CrossRef]
- Dye, E.S.; North, R.J. T cell-mediated immunosuppression as an obstacle to adoptive immunotherapy of the P815 mastocytoma and its metastases. J. Exp. Med. 1981, 154, 1033–1042. [Google Scholar] [CrossRef]
- Berendt, M.J.; North, R.J. T-cell-mediated suppression of anti-tumor immunity. An explanation for progressive growth of an immunogenic tumor. J. Exp. Med. 1980, 151, 69–80. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Khong, H.T.; Antony, P.A.; Palmer, D.C.; Restifo, N.P. Sinks, suppressors and antigen presenters: How lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005, 26, 111–117. [Google Scholar] [CrossRef]
- Wei, S.; Egenti, M.U.; Teitz-Tennenbaum, S.; Zou, W.; Chang, A.E. Effects of tumor irradiation on host T-regulatory cells and systemic immunity in the context of adoptive T-cell therapy in mice. J. Immunother. 2013, 36, 124–132. [Google Scholar] [CrossRef]
- Gattinoni, L.; Finkelstein, S.E.; Klebanoff, C.A.; Antony, P.A.; Palmer, D.C.; Spiess, P.J.; Hwang, L.N.; Yu, Z.; Wrzesinski, C.; Heimann, D.M.; et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005, 202, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Woo, E.Y.; Yeh, H.; Chu, C.S.; Schlienger, K.; Carroll, R.G.; Riley, J.L.; Kaiser, L.R.; June, C.H. Cutting edge: Regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. J. Immunol. 2002, 168, 4272–4276. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Wrzesinski, C.; Paulos, C.M.; Kaiser, A.; Muranski, P.; Palmer, D.C.; Gattinoni, L.; Yu, Z.; Rosenberg, S.A.; Restifo, N.P. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J. Immunother. 2010, 33, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.M.; Marincola, F.M.; Leitman, S.F.; Seipp, C.A.; et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J. Immunother. 2002, 25, 243–251. [Google Scholar] [CrossRef]
- Muranski, P.; Boni, A.; Wrzesinski, C.; Citrin, D.E.; Rosenberg, S.A.; Childs, R.; Restifo, N.P. Increased intensity lymphodepletion and adoptive immunotherapy--how far can we go? Nat. Clin. Pract. Oncol. 2006, 3, 668–681. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Poma, S.M.; Salas-Benito, D.; Lozano, T.; Casares, N.; Riezu-Boj, J.I.; Mancheno, U.; Elizalde, E.; Alignani, D.; Zubeldia, N.; Otano, I.; et al. Expansion of Tumor-Infiltrating CD8(+) T cells Expressing PD-1 Improves the Efficacy of Adoptive T-cell Therapy. Cancer Res. 2017, 77, 3672–3684. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.; Kjeldsen, J.W.; Andersen, R.; Westergaard, M.C.W.; Bianchi, V.; Legut, M.; Attaf, M.; Szomolay, B.; Ott, S.; Dolton, G.; et al. PD-1(+) Polyfunctional T Cells Dominate the Periphery after Tumor-Infiltrating Lymphocyte Therapy for Cancer. Clin. Cancer Res. 2017, 23, 5779–5788. [Google Scholar] [CrossRef]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef] [PubMed]
- Watson, H.A.; Durairaj, R.R.P.; Ohme, J.; Alatsatianos, M.; Almutairi, H.; Mohammed, R.N.; Vigar, M.; Reed, S.G.; Paisey, S.J.; Marshall, C.; et al. L-Selectin Enhanced T Cells Improve the Efficacy of Cancer Immunotherapy. Front. Immunol. 2019, 10, 1321. [Google Scholar] [CrossRef]
- Shi, L.Z.; Goswami, S.; Fu, T.; Guan, B.; Chen, J.; Xiong, L.; Zhang, J.; Ng Tang, D.; Zhang, X.; Vence, L.; et al. Blockade of CTLA-4 and PD-1 Enhances Adoptive T-cell Therapy Efficacy in an ICOS-Mediated Manner. Cancer Immunol. Res. 2019, 7, 1803–1812. [Google Scholar] [CrossRef]
- Blake, S.J.; Ching, A.L.; Kenna, T.J.; Galea, R.; Large, J.; Yagita, H.; Steptoe, R.J. Blockade of PD-1/PD-L1 promotes adoptive T-cell immunotherapy in a tolerogenic environment. PLoS ONE 2015, 10, e0119483. [Google Scholar] [CrossRef]
- Nie, W.; Wei, W.; Zuo, L.; Lv, C.; Zhang, F.; Lu, G.H.; Li, F.; Wu, G.; Huang, L.L.; Xi, X.; et al. Magnetic Nanoclusters Armed with Responsive PD-1 Antibody Synergistically Improved Adoptive T-Cell Therapy for Solid Tumors. ACS Nano 2019, 13, 1469–1478. [Google Scholar] [CrossRef]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Roberts, I.M.; Thompson, J.A.; Margolin, K.A.; Bhatia, S.; Lee, S.M.; Sloan, H.L.; Lai, I.P.; Farrar, E.A.; Wagener, F.; et al. T-Cell Therapy Using Interleukin-21-Primed Cytotoxic T-Cell Lymphocytes Combined With Cytotoxic T-Cell Lymphocyte Antigen-4 Blockade Results in Long-Term Cell Persistence and Durable Tumor Regression. J. Clin. Oncol. 2016, 34, 3787–3795. [Google Scholar] [CrossRef]
- Kverneland, A.H.; Pedersen, M.; Westergaard, M.C.W.; Nielsen, M.; Borch, T.H.; Olsen, L.R.; Aasbjerg, G.; Santegoets, S.J.; van der Burg, S.H.; Milne, K.; et al. Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget 2020, 11, 2092–2105. [Google Scholar] [CrossRef]
- Conde-Perez, A.; Larue, L. Human relevance of NRAS/BRAF mouse melanoma models. Eur. J. Cell Biol. 2014, 93, 82–86. [Google Scholar] [CrossRef]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef]
- Atay, C.; Kwak, T.; Lavilla-Alonso, S.; Donthireddy, L.; Richards, A.; Moberg, V.; Pilon-Thomas, S.; Schell, M.; Messina, J.L.; Rebecca, V.W.; et al. BRAF Targeting Sensitizes Resistant Melanoma to Cytotoxic T Cells. Clin. Cancer Res. 2019, 25, 2783–2794. [Google Scholar] [CrossRef]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.N.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef]
- Koya, R.C.; Mok, S.; Otte, N.; Blacketor, K.J.; Comin-Anduix, B.; Tumeh, P.C.; Minasyan, A.; Graham, N.A.; Graeber, T.G.; Chodon, T.; et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res. 2012, 72, 3928–3937. [Google Scholar] [CrossRef]
- Wilmott, J.S.; Long, G.V.; Howle, J.R.; Haydu, L.E.; Sharma, R.N.; Thompson, J.F.; Kefford, R.F.; Hersey, P.; Scolyer, R.A. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin. Cancer Res. 2012, 18, 1386–1394. [Google Scholar] [CrossRef]
- Deniger, D.C.; Kwong, M.L.; Pasetto, A.; Dudley, M.E.; Wunderlich, J.R.; Langhan, M.M.; Lee, C.R.; Rosenberg, S.A. A Pilot Trial of the Combination of Vemurafenib with Adoptive Cell Therapy in Patients with Metastatic Melanoma. Clin. Cancer Res. 2017, 23, 351–362. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Verma, V.; Jafarzadeh, N.; Boi, S.; Kundu, S.; Jiang, Z.; Fan, Y.; Lopez, J.; Nandre, R.; Zeng, P.; Alolaqi, F.; et al. MEK inhibition reprograms CD8(+) T lymphocytes into memory stem cells with potent antitumor effects. Nat. Immunol. 2021, 22, 53–66. [Google Scholar] [CrossRef]
- Peng, W.; Ye, Y.; Rabinovich, B.A.; Liu, C.; Lou, Y.; Zhang, M.; Whittington, M.; Yang, Y.; Overwijk, W.W.; Lizee, G.; et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin. Cancer Res. 2010, 16, 5458–5468. [Google Scholar] [CrossRef]
- Thanarajasingam, U.; Sanz, L.; Diaz, R.; Qiao, J.; Sanchez-Perez, L.; Kottke, T.; Thompson, J.; Chester, J.; Vile, R.G. Delivery of CCL21 to metastatic disease improves the efficacy of adoptive T-cell therapy. Cancer Res. 2007, 67, 300–308. [Google Scholar] [CrossRef]
- Walsh, S.R.; Simovic, B.; Chen, L.; Bastin, D.; Nguyen, A.; Stephenson, K.; Mandur, T.S.; Bramson, J.L.; Lichty, B.D.; Wan, Y. Endogenous T cells prevent tumor immune escape following adoptive T cell therapy. J. Clin. Investig. 2019, 129, 5400–5410. [Google Scholar] [CrossRef]
- Cho, H.I.; Reyes-Vargas, E.; Delgado, J.C.; Celis, E. A potent vaccination strategy that circumvents lymphodepletion for effective antitumor adoptive T-cell therapy. Cancer Res. 2012, 72, 1986–1995. [Google Scholar] [CrossRef]
- Hu, J.; Sun, C.; Bernatchez, C.; Xia, X.; Hwu, P.; Dotti, G.; Li, S. T-cell Homing Therapy for Reducing Regulatory T Cells and Preserving Effector T-cell Function in Large Solid Tumors. Clin. Cancer Res. 2018, 24, 2920–2934. [Google Scholar] [CrossRef] [PubMed]
- Grenier, J.M.; Yeung, S.T.; Qiu, Z.; Jellison, E.R.; Khanna, K.M. Combining Adoptive Cell Therapy with Cytomegalovirus-Based Vaccine Is Protective against Solid Skin Tumors. Front. Immunol. 2017, 8, 1993. [Google Scholar] [CrossRef]
- Nowicki, T.S.; Berent-Maoz, B.; Cheung-Lau, G.; Huang, R.R.; Wang, X.; Tsoi, J.; Kaplan-Lefko, P.; Cabrera, P.; Tran, J.; Pang, J.; et al. A Pilot Trial of the Combination of Transgenic NY-ESO-1-reactive Adoptive Cellular Therapy with Dendritic Cell Vaccination with or without Ipilimumab. Clin. Cancer Res. 2019, 25, 2096–2108. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lewis, C.M.; Lou, Y.; Xu, C.; Peng, W.; Yang, Y.; Gelbard, A.H.; Lizee, G.; Zhou, D.; Overwijk, W.W.; et al. Agonistic antibody to CD40 boosts the antitumor activity of adoptively transferred T cells in vivo. J. Immunother. 2012, 35, 276–282. [Google Scholar] [CrossRef]
- Sun, L.; Clavijo, P.E.; Robbins, Y.; Patel, P.; Friedman, J.; Greene, S.; Das, R.; Silvin, C.; Van Waes, C.; Horn, L.A.; et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight 2019, 4, e126853. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, D.; Trad, M.; Hanke, N.T.; Larmonier, C.B.; Janikashvili, N.; Bonnotte, B.; Katsanis, E.; Larmonier, N. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014, 74, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Kodumudi, K.N.; Weber, A.; Sarnaik, A.A.; Pilon-Thomas, S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J. Immunol. 2012, 189, 5147–5154. [Google Scholar] [CrossRef] [PubMed]
- Lindenberg, M.A.; Retel, V.P.; van den Berg, J.H.; Geukes Foppen, M.H.; Haanen, J.B.; van Harten, W.H. Treatment With Tumor-infiltrating Lymphocytes in Advanced Melanoma: Evaluation of Early Clinical Implementation of an Advanced Therapy Medicinal Product. J. Immunother. 2018, 41, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Lindenberg, M.; Retel, V.; Rohaan, M.; van den Berg, J.; Haanen, J.; van Harten, W. Evaluating different adoption scenarios for TIL-therapy and the influence on its (early) cost-effectiveness. BMC Cancer 2020, 20, 712. [Google Scholar] [CrossRef] [PubMed]
- Fardis, M.; DiTrapani, K.; Chartier, C.; Finckenstein, F.G. Current and future directions for tumor infiltrating lymphocyte therapy for the treatment of solid tumors. Cell Gene Ther. Insights 2020, 6, 855–863. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Number | Cell Type | Pre- and Post-Conditioning | Adjuvant Treatment | Tumor Types | Site Location | Status |
|---|---|---|---|---|---|---|
| TIL-ACT EFFICACY TRIALS | ||||||
| NCT03778814 | TIL enriched for tumor specificity | N/A | N/A | Solid tumors NSCLC | Second Affiliated Hospital of Guangzhou, Medical University, China | Recruiting 12/2021 |
| NCT04596033 | TIL | IL-2 +/− Cyclophosphamide Fludarabine | N/A | Multiple advanced solid tumors | Genocea Biosciences, Inc., Nashville, TN | Recruiting 5/2024 |
| NCT04625205 | TIL | N/A | N/A | Advanced and metastatic melanoma | BioNTech US Inc., Amsterdam, Netherlands | Recruiting 11/2023 |
| NCT03610490 | TIL | Cyclophosphamide Fludarabine IL-2 | N/A | Refractory and metastatic ovarian, PDAC, and CRC | M.D. Anderson Cancer Center Houston, TX | Active 09/2021 |
| NCT03449108 | Cryopreserved TIL | Cyclophosphamide Fludarabine IL-2 | N/A | Bone sarcoma Sarcoma Thyroid | M.D. Anderson Cancer Center Houston, TX | Recruiting 12/2022 |
| NCT03935893 | TIL | Cyclophosphamide Fludarabine IL-2 | N/A | Multiple solid advanced cancers | University of Pittsburgh Medical Center, Pittsburgh, PA | Recruiting 6/2030 |
| NCT03658785 | TIL | Cyclophosphamide Fludarabine IL-2 | N/A | Recurrent, metastatic, persistent carcinoma not amenable to currenttreatments | Tongji Hospital China | Not yet recruiting 12/2024 |
| NCT03991741 | T cell | IL-2 | N/A | Locally advanced and metastatic melanoma, Locally advanced and metastatic head and neck cancer | Immunotherapy Foundation, San Diego, CA | Recruiting 08/2023 |
| PRECONDITIONING | ||||||
| NCT03992326 | TIL | Cyclophosphamide Fludarabine +/− IL-2 LDI | Breast cancer NSCLC Colon cancer Ovarian cancer Other solid tumors (excluding brain, cutaneous, mucosal, ocular/uveal) | Centre Hospitalier, Universitaire Vaudois Lausanne, Switzerland | Recruiting 09/2025 | |
| NCT04643574 | TIL enriched for tumor specificity | Cyclophosphamide Fludarabine +/− IL-2 LDI | Solid tumors except CNS | Centre Hospitalier, Universitaire Vaudois Lausanne, Switzerland | Not yet recruiting 11/2027 | |
| IL-2 DOSAGE | ||||||
| NCT01462903 | TIL | Low dose IL-2 | N/A | Hepatocellular carcinoma Breast carcinoma Nasopharyngeal carcinoma | Sun Yat-sen University, China | Unknown |
| ADJUVANT THERAPIES | ||||||
| NCT00002733 | TIL | IL-2 | Cimetidine IFN-α | Metastatic melanoma Metastatic RCC | Hoag Memorial Hospital Presbyterian, Newport Beach, CA | Completed 01/2000 |
| NCT02876510 | TIL | Cyclophosphamide Fludarabine IL-2 | Atezolizumab | Advanced solid cancers | Immatics US, Inc., Houston, TX | Active not recruiting 12/2021 |
| NCT03645928 | Cryopreserved TIL | Cyclophosphamide Fludarabine IL-2 | Pembrolizumab Ipilimumab Nivolumab | Metastatic melanoma SCC of the head and neck NSCLC | Iovance Biotherapeutics, Inc., San Carlos, CA | Recruiting 12/2024 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, S.S.; Melucci, A.D.; Chacon, A.C.; Prieto, P.A. Adoptive T Cell Therapy for Solid Tumors: Pathway to Personalized Standard of Care. Cells 2021, 10, 808. https://doi.org/10.3390/cells10040808
Qin SS, Melucci AD, Chacon AC, Prieto PA. Adoptive T Cell Therapy for Solid Tumors: Pathway to Personalized Standard of Care. Cells. 2021; 10(4):808. https://doi.org/10.3390/cells10040808
Chicago/Turabian StyleQin, Shuyang S., Alexa D. Melucci, Alexander C. Chacon, and Peter A. Prieto. 2021. "Adoptive T Cell Therapy for Solid Tumors: Pathway to Personalized Standard of Care" Cells 10, no. 4: 808. https://doi.org/10.3390/cells10040808
APA StyleQin, S. S., Melucci, A. D., Chacon, A. C., & Prieto, P. A. (2021). Adoptive T Cell Therapy for Solid Tumors: Pathway to Personalized Standard of Care. Cells, 10(4), 808. https://doi.org/10.3390/cells10040808