
Aging and Interferons: Impacts on Inflammation and Viral Disease Outcomes


Abstract
1. Introduction
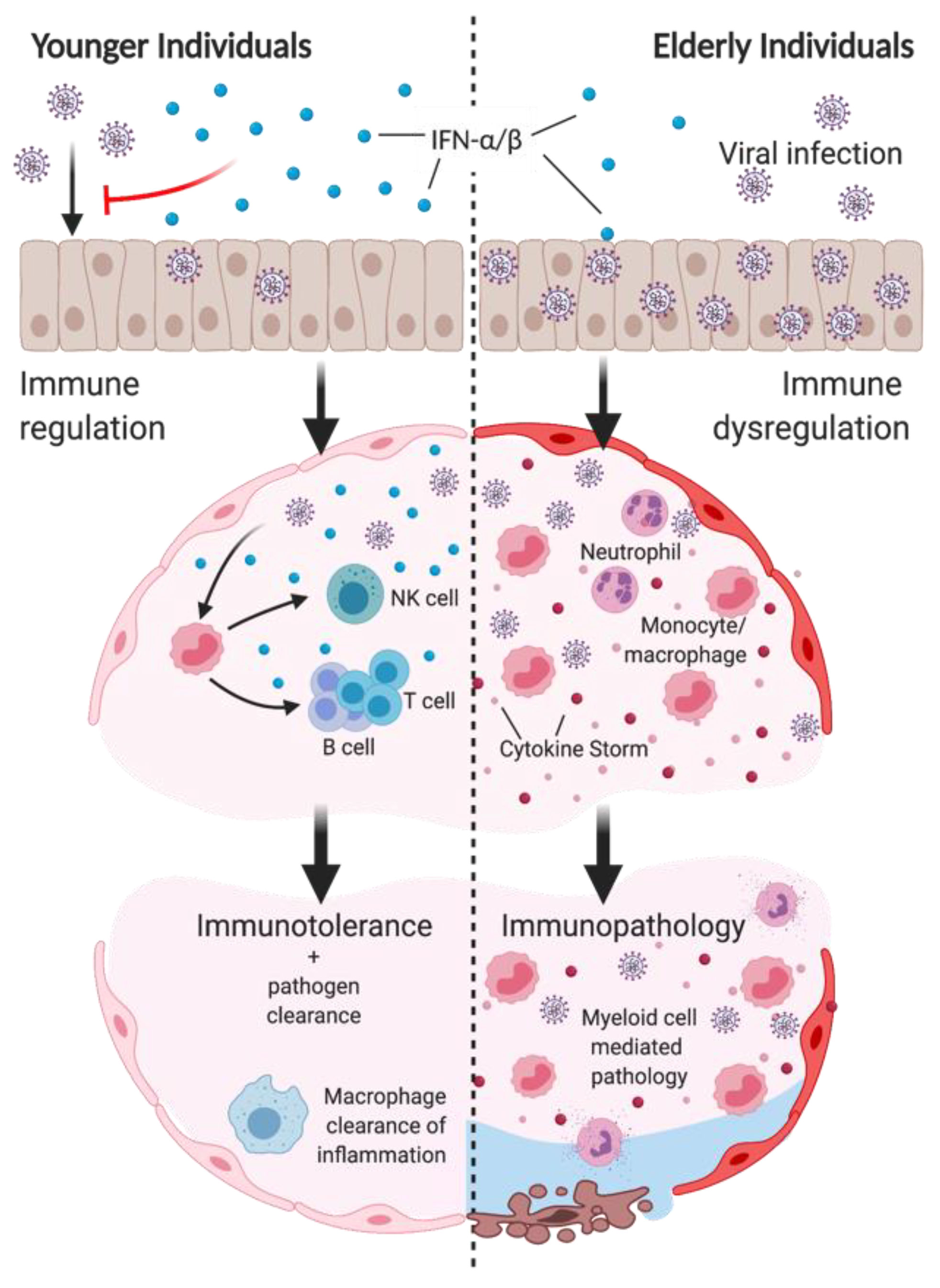
2. The Paradoxical Role of Type I IFNs in Inflammation and Disease Tolerance
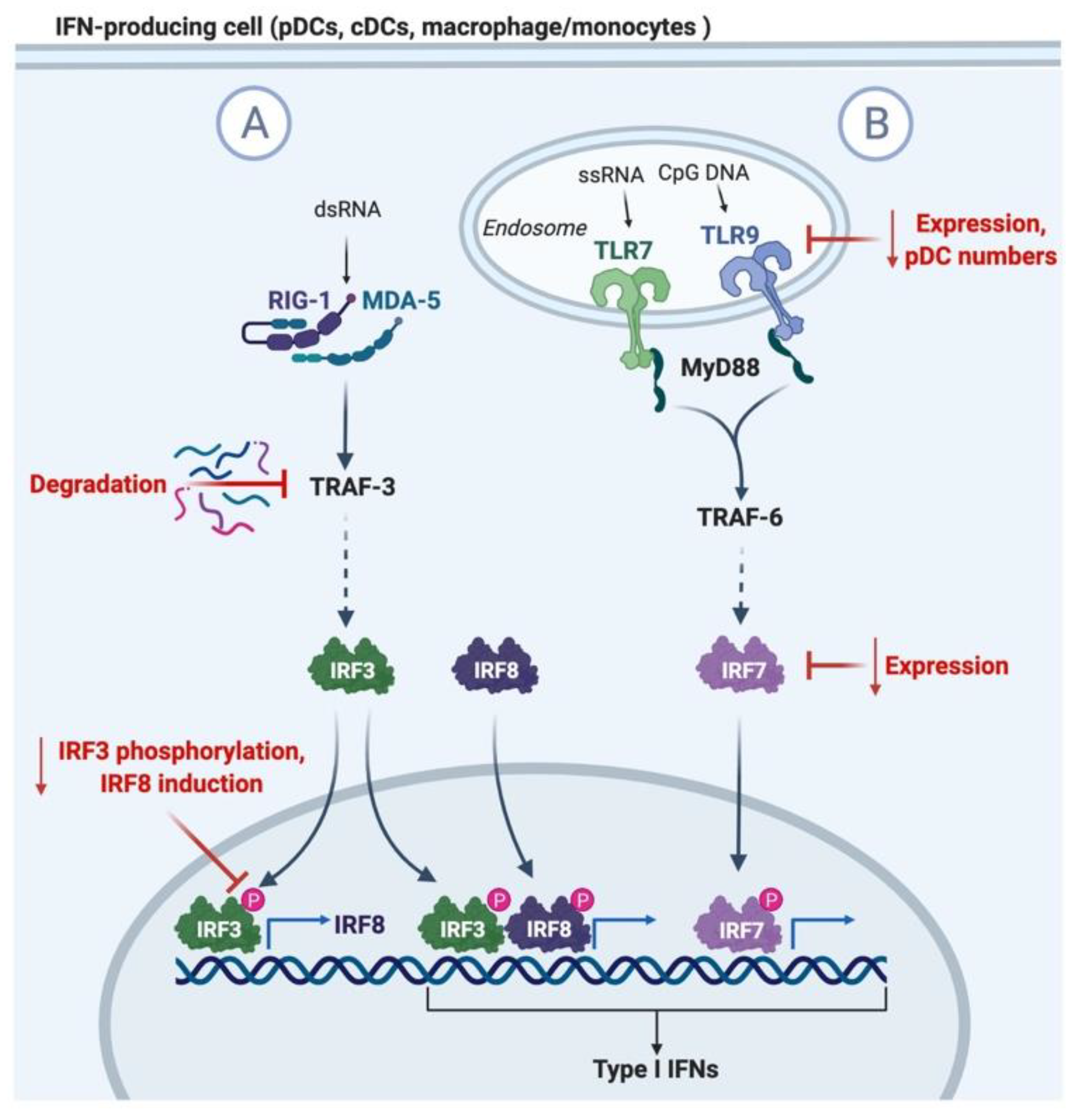
3. Induction and Regulation of Type I IFN Responses to Viral Infection
4. Mechanisms of Impaired Type I IFN Signaling in Aging
5. The Role of Interferon and Aging in Regulating the Innate Immune Response
5.1. Interferons Prevent Neutrophil-Mediated Inflammation
5.2. Macrophage Inflammatory and Anti-Inflammatory Functions Are Tightly Regulated by Type I IFNS
5.3. Type I IFNs Are Essential for NK Cell Antiviral Functions
6. Interferon, Aging, and the Adaptive Immune Response
6.1. Controlling the Balance between Protection and Destruction by T Cells
6.2. Promoting B Cell Responses and Memory to Infection
7. Type II Interferon Immunoregulatory Functions Are Dependent on Type I IFN Signaling
8. Type III Interferons Control Viral Replication at Mucosal Surfaces
9. The Therapeutic Potential of IFNs in the Treatment of Inflammation and Infection
10. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lieberman, N.A.P.; Peddu, V.; Xie, H.; Shrestha, L.; Huang, M.L.; Mears, M.C.; Cajimat, M.N.; Bente, D.A.; Shi, P.Y.; Bovier, F.; et al. In vivo antiviral host transcriptional response to SARS-CoV-2 by viral load, sex, and age. PLoS Biol. 2020, 18, e3000849. [Google Scholar] [CrossRef]
- Magleby, R.; Westblade, L.F.; Trzebucki, A.; Simon, M.S.; Rajan, M.; Park, J.; Goyal, P.; Safford, M.M.; Satlin, M.J. Impact of Severe Acute Respiratory Syndrome Coronavirus 2 Viral Load on Risk of Intubation and Mortality Among Hospitalized Patients With Coronavirus Disease 2019. Clin. Infect. Dis. 2020, 1–9. [Google Scholar] [CrossRef]
- To, K.K.W.K.W.; Hung, I.F.N.F.N.; Li, I.W.S.W.S.; Lee, K.L.; Koo, C.K.; Yan, W.W.; Liu, R.; Ho, K.Y.; Chu, K.H.; Watt, C.L.; et al. Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clin. Infect. Dis. 2010, 50, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Molony, R.D.; Nguyen, J.T.; Kong, Y.; Montgomery, R.R.; Shaw, A.C.; Iwasaki, A. Aging impairs both primary and secondary RIG-I signaling for interferon induction in human monocytes. Sci. Signal. 2017, 10, 1–12. [Google Scholar] [CrossRef]
- Pillai, P.S.; Molony, R.D.; Martinod, K.; Dong, H.; Pang, I.K.; Tal, M.C.; Solis, A.G.; Bielecki, P.; Mohanty, S.; Trentalange, M.; et al. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science 2017, 352, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cabezas, B.; Naranjo-Gómez, M.; Fernández, M.A.; Grífols, J.R.; Pujol-Borrell, R.; Borràs, F.E. Reduced numbers of plasmacytoid dendritic cells in aged blood donors. Exp. Gerontol. 2007, 42, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Shaheen, E.; Drake, R.R.; Chen, N.; Gravenstein, S.; Deng, Y. Aging is associated with a numerical and functional decline in plasmacytoid dendritic cells, whereas myeloid dendritic cells are relatively unaltered in human peripheral blood. Hum. Immunol. 2009, 70, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II- mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Takaoka, A.; Taniguchi, T. Regulation of the type I IFN induction: A current view. Int. Immunol. 2005, 17, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.U.; Kwon, H.J.; Ko, H.J.; Byun, Y.H.; Seong, B.L.; Uematsu, S.; Akira, S.; Kweon, M.N. Type I interferon signaling regulates Ly6Chi monocytes and neutrophils during acute viral pneumonia in mice. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Duerr, C.U.; Mccarthy, C.D.A.; Mindt, B.C.; Rubio, M.; Meli, A.P.; Pothlichet, J.; Eva, M.M.; Gauchat, J.; Qureshi, S.T.; Mazer, B.D.; et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat. Immunol. 2016, 17, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Cardenas-Rodriguez, M.; Osborn, D.P.; Irigoín, F.; Graña, M.; Romero, H.; Beales, P.L.; Badano, J.L. Characterization of CCDC28B reveals its role in ciliogenesis and provides insight to understand its modifier effect on Bardet-Biedl syndrome. Hum. Genet. 2013, 132, 91–105. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, Z.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Bigio, B.; Yang, R.; Arias, A.A.; Zhou, Q.; Han, J.E.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Wang, N.; Zhan, Y.; Zhu, L.; Hou, Z.; Liu, F.; Song, P.; Qiu, F.; Wang, X.; Zou, X.; Wan, D.; et al. Retrospective Multicenter Cohort Study Shows that Early Interferon Therapy is Associated with Favorable Clinical Responses in COVID-19 Patients. Cell Host Microbe 2020, 28, 455–464. [Google Scholar] [CrossRef]
- Price, A.; Okumura, A.; Haddock, E.; Feldmann, F.; Meade-White, K.; Sharma, P.; Artami, M.; Lipkin, W.I.; Threadgill, D.W.; Feldmann, H.; et al. Transcriptional Correlates of Tolerance and Lethality in Mice Predict Ebola Virus Disease Patient Outcomes. Cell Rep. 2020, 30, 1702–1713. [Google Scholar] [CrossRef]
- Leroy, E.M.; Baize, S.; Debre, P.; Lansoud-Soukate, J.; Mavoungou, E. Early immune responses accompanying human asymptomatic Ebola infections. Clin. Exp. Immunol. 2001, 124, 453–460. [Google Scholar] [CrossRef]
- Baize, S.; Leroy, E.M.; Georges, A.J.; Georges-Courbot, M.C.; Capron, M.; Bedjabaga, I.; Lansoud-Soukate, J.; Mavoungou, E. Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol. 2002, 128, 163–168. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B.; Meyerholz, D.K.; Perlman, S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Investig. 2019, 129, 3625–3639. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Chan, J.F.-W.; Wang, Y.; Yuen, T.T.-T.; Chai, Y.; Hou, Y.; Shuai, H.; Yang, D.; Hu, B.; Huang, X.; et al. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: An ex vivo study with implications for the pathogenesis of COVID-19. Clin. Infect. Dis. 2020, 29, 2341–2386. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; Crotta, S.; McCabe, T.M.; Wack, A. Pathogenic potential of interferon αβ in acute influenza infection. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Lee, A.J.; Chen, B.; Chew, M.V.; Barra, N.G.; Shenouda, M.M.; Nham, T.; van Rooijen, N.; Jordana, M.; Mossman, K.L.; Schreiber, R.D.; et al. Inflammatory monocytes require type I interferon receptor signaling to activate NK cells via IL-18 during a mucosal viral infection. J. Exp. Med. 2017, 214, 1153–1167. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Barbalat, R.; Lau, L.; Locksley, R.M.; Barton, G.M. Toll-like receptor 2 on inflammatory monocytes induces type i interferon in response to viral but not bacterial ligands. Nat. Immunol. 2009, 10, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Kong, K.-F.; Delroux, K.; Wang, X.; Qian, F.; Arjona, A.; Malawista, S.E.; Fikrig, E.; Montgomery, R.R. Dysregulation of TLR3 Impairs the Innate Immune Response to West Nile Virus in the Elderly. J. Virol. 2008, 82, 7613–7623. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Amakawa, R.; Inaba, M.; Hori, T.; Ota, M.; Nakamura, K.; Takebayashi, M.; Miyaji, M.; Yoshimura, T.; Inaba, K.; et al. Plasmacytoid Dendritic Cells Regulate Th Cell Responses through OX40 Ligand and Type I IFNs. J. Immunol. 2004, 172, 4253–4259. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, A.; Lutz, K.; Winheim, E.; Krug, A.B. What makes a PDC: Recent advances in understanding plasmacytoid DC development and heterogeneity. Front. Immunol. 2019, 10, 1222. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Wang, Y.; Vermi, W.; Gilfillan, S.; Schreiber, R.D.; Colonna, M. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J. Exp. Med. 2011, 208, 2367–2374. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Wang, Y.; Gilfillan, S.; Colonna, M. Plasmacytoid Dendritic Cells Contribute to Systemic but Not Local Antiviral Responses to HSV Infections. PLoS Pathog. 2013, 9, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Züst, R.; Weber, F.; Spiegel, M.; Lang, K.S.; Akira, S.; Thiel, V.; Ludewig, B. Control of coronavirus infection through plasmacytoid dendritic-cell- derived type I interferon. Blood 2007, 109, 1131–1137. [Google Scholar] [CrossRef]
- Fuertes Marraco, S.A.; Scott, C.L.; Bouillet, P.; Ives, A.; Masina, S.; Vremec, D.; Jansen, E.S.; O’Reilly, L.A.; Schneider, P.; Fasel, N.; et al. Type I interferon drives dendritic cell apoptosis via multiple BH3-only proteins following activation by polyic in vivo. PLoS ONE 2011, 6, e20189. [Google Scholar] [CrossRef] [PubMed]
- Nopora, A.; Brocker, T. Bcl-2 Controls Dendritic Cell Longevity In Vivo. J. Immunol. 2002, 169, 3006–3014. [Google Scholar] [CrossRef]
- Swiecki, M.; Gilfillan, S.; Vermi, W.; Wang, Y.; Colonna, M. Plasmacytoid Dendritic Cell Ablation Impacts Early Interferon Responses and Antiviral NK and CD8+ T Cell Accrual. Immunity 2010, 33, 955–966. [Google Scholar] [CrossRef]
- Cervantes-Barragan, L.; Lewis, K.L.; Firner, S.; Thiel, V.; Hugues, S.; Reith, W.; Ludewig, B.; Reizis, B. Plasmacytoid dendritic cells control T-cell response to chronic viral infection. Proc. Natl. Acad. Sci. USA 2012, 109, 3012–3017. [Google Scholar] [CrossRef]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef]
- Goritzka, M.; Makris, S.; Kausar, F.; Durant, L.R.; Pereira, C.; Kumagai, Y.; Culley, F.J.; Mack, M.; Akira, S.; Johansson, C. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J. Exp. Med. 2015, 212, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Montoya, M.; Unger, H.; Alexopoulou, L.; Roy, P.; Haswell, L.E.; Al-Shamkhani, A.; Flavell, R.; Borrow, P.; Reis e Sousa, C. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 2003, 424, 324–328. [Google Scholar] [CrossRef]
- Lee, P.Y.; Li, Y.; Kumagai, Y.; Xu, Y.; Weinstein, J.S.; Kellner, E.S.; Nacionales, D.C.; Butfiloski, E.J.; van Rooijen, N.; Akira, S.; et al. Type I Interferon Modulates Monocyte Recruitment and Maturation in Chronic Inflammation. Am. J. Pathol. 2009, 175, 2023–2033. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, Y.; Takeuchi, O.; Kato, H.; Kumar, H.; Matsui, K.; Morii, E.; Aozasa, K.; Kawai, T.; Akira, S. Alveolar Macrophages Are the Primary Interferon-α Producer in Pulmonary Infection with RNA Viruses. Immunity 2007, 27, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Canaday, D.H.; Amponsah, N.A.; Jones, L.; Tisch, D.J.; Hornick, T.R.; Ramachandra, L. Influenza-induced production of interferon-alpha is defective in geriatric individuals. J. Clin. Immunol. 2010, 30, 373–383. [Google Scholar] [CrossRef]
- Panda, A.; Qian, F.; Mohanty, S.; van Duin, D.; Newman, F.K.; Zhang, L.; Chen, S.; Towle, V.; Belshe, R.B.; Fikrig, E.; et al. Age-Associated Decrease in TLR Function in Primary Human Dendritic Cells Predicts Influenza Vaccine Response. J. Immunol. 2010, 184, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Wan, X.; Zhang, L.; Lin, A.; Zhao, H.; Fikrig, E.; Montgomery, R.R. Impaired interferon signaling in dendritic cells from older donors infected in vitro with west nile virus. J. Infect. Dis. 2011, 203, 1415–1424. [Google Scholar] [CrossRef]
- Van Splunter, M.; Perdijk, O.; Fick-Brinkhof, H.; Feitsma, A.L.; Floris-Vollenbroek, E.G.; Meijer, B.; Brugman, S.; Savelkoul, H.F.J.; Van Hoffen, E.; Van Neerven, R.J.J. Bovine lactoferrin enhances TLR7-mediated responses in plasmacytoid dendritic cells in elderly women: Results from a nutritional intervention study with bovine lactoferrin, GOS and Vitamin D. Front. Immunol. 2018, 9, 2677. [Google Scholar] [CrossRef]
- Stout-Delgado, H.W.; Yang, X.; Walker, W.E.; Tesar, B.M.; Goldstein, D.R. Aging Impairs IFN Regulatory Factor 7 Up-Regulation in Plasmacytoid Dendritic Cells during TLR9 Activation. J. Immunol. 2008, 181, 6747–6756. [Google Scholar] [CrossRef]
- Tailor, P.; Tamura, T.; Kong, H.J.; Kubota, T.; Kubota, M.; Borghi, P.; Gabriele, L.; Ozato, K. The Feedback Phase of Type I Interferon Induction in Dendritic Cells Requires Interferon Regulatory Factor 8. Immunity 2007, 27, 228–239. [Google Scholar] [CrossRef]
- Li, P.; Wong, J.J.Y.; Sum, C.; Sin, W.X.; Ng, K.Q.; Koh, M.B.C.; Chin, K.C. IRF8 and IRF3 cooperatively regulate rapid interferon-β induction in human blood monocytes. Blood 2011, 117, 2847–2854. [Google Scholar] [CrossRef]
- Macal, M.; Jo, Y.; Dallari, S.; Chang, A.Y.; Dai, J.; Swaminathan, S.; Wehrens, E.J.; Fitzgerald-Bocarsly, P.; Zúñiga, E.I. Self-Renewal and Toll-like Receptor Signaling Sustain Exhausted Plasmacytoid Dendritic Cells during Chronic Viral Infection. Immunity 2018, 48, 730–744.e5. [Google Scholar] [CrossRef]
- Zuniga, E.I.; Liou, L.Y.; Mack, L.; Mendoza, M.; Oldstone, M.B.A. Persistent Virus Infection Inhibits Type I Interferon Production by Plasmacytoid Dendritic Cells to Facilitate Opportunistic Infections. Cell Host Microbe 2008, 4, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, T.; Larbi, A.; Pawelec, G. Human T cell aging and the impact of persistent viral infections. Front. Immunol. 2013, 4, 1–9. [Google Scholar] [CrossRef]
- Derhovanessian, E.; Maier, A.B.; Hähnel, K.; McElhaney, J.E.; Slagboom, E.P.; Pawelec, G. Latent Infection with Cytomegalovirus Is Associated with Poor Memory CD4 Responses to Influenza A Core Proteins in the Elderly. J. Immunol. 2014, 193, 3624–3631. [Google Scholar] [CrossRef] [PubMed]
- Moro-García, M.A.; Alonso-Arias, R.; López-Vázquez, A.; Suárez-García, F.M.; Solano-Jaurrieta, J.J.; Baltar, J.; López-Larrea, C. Relationship between functional ability in older people, immune system status, and intensity of response to CMV. Age (Omaha). 2012, 34, 479–495. [Google Scholar] [CrossRef]
- Monteiro, R.; Azevedo, I. Chronic Inflammation in Obesity and the Metabolic Syndrome. Mediators Inflamm. 2010, 2010, 1–10. [Google Scholar] [CrossRef]
- Namkoong, H.; Ishii, M.; Fujii, H.; Asami, T.; Yagi, K.; Suzuki, S.; Azekawa, S.; Tasaka, S.; Hasegawa, N.; Betsuyaku, T. Obesity worsens the outcome of influenza virus infection associated with impaired type I interferon induction in mice. Biochem. Biophys. Res. Commun. 2019, 513, 405–411. [Google Scholar] [CrossRef]
- Teran-Cabanillas, E.; Montalvo-Corral, M.; Caire-Juvera, G.; Moya-Camarena, S.Y. Decreased interferon-a and interferon-b production in obesity and expression of suppressor of cytokine signaling. Nutrition 2013, 29, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, U.; Zemans, R.L.; Smith, C.A.; Wood, S.C.; Deng, J.C.; Goldstein, D.R. Excessive neutrophil levels in the lung underlie the age-associated increase in influenza mortality. Mucosal Immunol. 2019, 12, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Stout-Delgado, H.W.; Du, W.; Shirali, A.C.; Booth, C.J.; Goldstein, D.R. Aging Promotes Neutrophil-Induced Mortality by Augmenting IL-17 Production during Viral Infection. Cell Host Microbe 2009, 6, 446–456. [Google Scholar] [CrossRef]
- Stifter, S.A.; Bhattacharyya, N.; Pillay, R.; Flórido, M.; Triccas, J.A.; Britton, W.J.; Feng, C.G. Functional Interplay between Type I and II Interferons Is Essential to Limit Influenza A Virus-Induced Tissue Inflammation. PLoS Pathog. 2016, 12, e1005378. [Google Scholar] [CrossRef] [PubMed]
- Stock, A.T.; Smith, J.M.; Carbone, F.R. Type I IFN suppresses Cxcr2 driven neutrophil recruitment into the sensory ganglia during viral infection. J. Exp. Med. 2014, 211, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Hearps, A.C.; Martin, G.E.; Angelovich, T.A.; Cheng, W.-J.; Maisa, A.; Landay, A.L.; Jaworowski, A.; Crowe, S.M. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell 2012, 11, 867–875. [Google Scholar] [CrossRef]
- Wong, C.K.; Smith, C.A.; Sakamoto, K.; Kaminski, N.; Koff, J.L.; Goldstein, D.R. Aging Impairs Alveolar Macrophage Phagocytosis and Increases Influenza-Induced Mortality in Mice. J. Immunol. 2017, 199, 1060–1068. [Google Scholar] [CrossRef]
- Jia, T.; Leiner, I.; Dorothee, G.; Brandl, K.; Pamer, E.G. MyD88 and Type I Interferon Receptor-Mediated Chemokine Induction and Monocyte Recruitment during Listeria monocytogenes Infection. J. Immunol. 2009, 183, 1271–1278. [Google Scholar] [CrossRef]
- Crane, M.J.; Hokeness-Antonelli, K.L.; Salazar-Mather, T.P. Regulation of Inflammatory Monocyte/Macrophage Recruitment from the Bone Marrow during Murine Cytomegalovirus Infection: Role for Type I Interferons in Localized Induction of CCR2 Ligands. J. Immunol. 2009, 183, 2810–2817. [Google Scholar] [CrossRef]
- Pernet, E.; Downey, J.; Vinh, D.C.; Powell, W.S.; Divangahi, M. Leukotriene B4–type I interferon axis regulates macrophage-mediated disease tolerance to influenza infection. Nat. Microbiol. 2019, 4, 1389–1400. [Google Scholar] [CrossRef]
- Högner, K.; Wolff, T.; Pleschka, S.; Plog, S.; Gruber, A.D.; Kalinke, U.; Walmrath, H.D.; Bodner, J.; Gattenlöhner, S.; Lewe-Schlosser, P.; et al. Macrophage-expressed IFN-β Contributes to Apoptotic Alveolar Epithelial Cell Injury in Severe Influenza Virus Pneumonia. PLoS Pathog. 2013, 9, e1003188. [Google Scholar] [CrossRef]
- Herold, S.; Steinmueller, M.; Von Wulffen, W.; Cakarova, L.; Pinto, R.; Pleschka, S.; Mack, M.; Kuziel, W.A.; Corazza, N.; Brunner, T.; et al. Lung epithelial apoptosis in influenza virus pneumonia: The role of macrophage-expressed TNF-related apoptosis-inducing ligand. J. Exp. Med. 2008, 205, 3065–3077. [Google Scholar] [CrossRef]
- Satyanarayanan, S.K.; Kebir, D.; El Soboh, S.; Butenko, S.; Sekheri, M.; Saadi, J.; Peled, N.; Assi, S.; Othman, A.; Schif-zuck, S.; et al. IFN-β is a macrophage-derived effector cytokine facilitating the resolution of bacterial inflammation. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yángüez, E.; García-Culebras, A.; Frau, A.; Llompart, C.; Knobeloch, K.P.; Gutierrez-Erlandsson, S.; García-Sastre, A.; Esteban, M.; Nieto, A.; Guerra, S. ISG15 Regulates Peritoneal Macrophages Functionality against Viral Infection. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, F.; Jaworska, J.; Verway, M.; Tzelepis, F.; Massoud, A.; Gillard, J.; Wong, G.; Kobinger, G.; Xing, Z.; Couture, C.; et al. Targeted prostaglandin E2 inhibition enhances antiviral immunity through induction of type I interferon and apoptosis in macrophages. Immunity 2014, 40, 554–568. [Google Scholar] [CrossRef] [PubMed]
- Shirey, K.A.; Pletneva, L.M.; Puche, A.C.; Keegan, A.D.; Prince, G.A.; Blanco, J.C.G.; Vogel, S.N. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4Rα-, TLR4-, and IFN-Β-dependent. Mucosal Immunol. 2010, 3, 291–300. [Google Scholar] [CrossRef]
- Beli, E.; Clinthorne, J.F.; Duriancik, D.M.; Hwang, I.; Kim, S.; Gardner, E.M. Natural killer cell function is altered during the primary response of aged mice to influenza infection. Mech. Ageing Dev. 2011, 132, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Tilburgs, T.; Wong, B.; Strominger, J.L. Dysfunction of dendritic cells in aged C57BL/6 mice leads to failure of natural killer cell activation and of tumor eradication. Proc. Natl. Acad. Sci. USA 2014, 111, 14199–14204. [Google Scholar] [CrossRef]
- Plett, P.A.; Gardner, E.M.; Murasko, D.M. Age-related changes in interferon-α/β receptor expression, binding, and induction of apoptosis in natural killer cells from C57BL/6 mice. Mech. Ageing Dev. 2000, 118, 129–144. [Google Scholar] [CrossRef]
- Soudja, S.M.H.; Ruiz, A.L.; Marie, J.C.; Lauvau, G. Inflammatory Monocytes Activate Memory CD8+ T and Innate NK Lymphocytes Independent of Cognate Antigen during Microbial Pathogen Invasion. Immunity 2012, 37, 549–562. [Google Scholar] [CrossRef]
- Krug, A.; French, A.R.; Barchet, W.; Fischer, J.A.A.; Dzionek, A.; Pingel, J.T.; Orihuela, M.M.; Akira, S.; Yokoyama, W.M.; Colonna, M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 2004, 21, 107–119. [Google Scholar] [CrossRef]
- Romagnani, C.; Della Chiesa, M.; Kohler, S.; Moewes, B.; Radbruch, A.; Moretta, L.; Moretta, A.; Thiel, A. Activation of human NK cells by plasmacytoid dendritic cells and its modulation by CD4+ T helper cells and CD4+ CD25hi T regulatory cells. Eur. J. Immunol. 2005, 35, 2452–2458. [Google Scholar] [CrossRef]
- Madera, S.; Rapp, M.; Firth, M.A.; Beilke, J.N.; Lanier, L.L.; Sun, J.C. Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. J. Exp. Med. 2016, 213, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Geary, C.D.; Krishna, C.; Lau, C.M.; Adams, N.M.; Gearty, S.V.; Pritykin, Y.; Thomsen, A.R.; Leslie, C.S.; Sun, J.C. Non-redundant ISGF3 Components Promote NK Cell Survival in an Auto-regulatory Manner during Viral Infection. Cell Rep. 2018, 24, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Wong, C.H.Y.; Zemp, F.J.; McDonald, B.; Rahman, M.M.; Forsyth, P.A.; McFadden, G.; Kubes, P. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 2013, 13, 169–180. [Google Scholar] [CrossRef]
- Dienz, O.; Rud, J.G.; Eaton, S.M.; Lanthier, P.A.; Burg, E.; Drew, A.; Bunn, J.; Suratt, B.T.; Haynes, L.; Rincon, M. Essential role of IL-6 in protection against H1N1 influenza virus by promoting neutrophil survival in the lung. Mucosal Immunol. 2012, 5, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.A.; Phoon, M.C.; Van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, L.; Zhang, Y.; Pu, L.; Liu, J.; Li, X.; Chen, Z.; Hao, Y.; Wang, B.; Han, J.; et al. High Level of Neutrophil Extracellular Traps Correlates With Poor Prognosis of Severe Influenza A Infection. J. Infect. Dis. 2018, 217, 428–437. [Google Scholar] [CrossRef]
- Liu, K.; Chen, Y.; Lin, R.; Han, K. Clinical features of COVID-19 in elderly patients: A comparison with young and middle-aged patients. J. Infect. 2020, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. The Lancet Infectious Diseases Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Lancet 2020. [Google Scholar]
- Cook, L.E.; Locke, M.C.; Young, A.R.; Monte, K.; Hedberg, M.L.; Shimak, R.M.; Sheehan, K.C.F.; Veis, D.J.; Diamond, M.S.; Lenschow, D.J. Distinct Roles of Interferon Alpha and Beta in Controlling Chikungunya Virus Replication and Modulating Neutrophil-Mediated Inflammation. J. Virol. 2019, 94, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Neupane, B.; Acharya, D.; Nazneen, F.; Gonzalez-Fernandez, G.; Flynt, A.S.; Bai, F. Interleukin-17A Facilitates Chikungunya Virus Infection by Inhibiting IFN-α2 Expression. Front. Immunol. 2020, 11, 8382. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Iijima, N.; Mattei, L.M.; Iwasaki, A. Recruited inflammatory monocytes stimulate antiviral Th1 immunity in infected tissue. Proc. Natl. Acad. Sci. USA 2011, 108, 284–289. [Google Scholar] [CrossRef]
- Saura, M.; Zaragoza, C.; McMillan, A.; Quick, R.A.; Hohenadl, C.; Lowenstein, J.M.; Lowenstein, C.J. An antiviral mechanism of nitric oxide: Inhibition of a viral protease. Immunity 1999, 10, 21–28. [Google Scholar] [CrossRef]
- Croen, K.D. Evidence for an antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication. J. Clin. Investig. 1993, 91, 2446–2452. [Google Scholar] [CrossRef]
- Liu, C.Y.; Liu, Y.H.; Lin, S.M.; Yu, C.T.; Wang, C.H.; Lin, H.C.; Lin, C.H.; Kuo, H.P. Apoptotic neutrophils undergoing secondary necrosis induce human lung epithelial cell detachment. J. Biomed. Sci. 2003, 10, 746–756. [Google Scholar] [CrossRef]
- Rydell-Törmänen, K.; Uller, L.; Erjefält, J.S. Direct evidence of secondary necrosis of neutrophils during intense lung inflammation. Eur. Respir. J. 2006, 28, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, A.A.; Van den Bossche, J.; Mastroberardino, P.G.; de Winther, M.P.J.; Leenen, P.J.M. Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends Immunol. 2019, 40, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, T.; Grabiec, A.M.; Kaur, M.; Bell, T.J.; Fujino, N.; Cook, P.C.; Svedberg, F.R.; Macdonald, A.S.; Maciewicz, R.A.; Singh, D.; et al. The Axl receptor tyrosine kinase is a discriminator of macrophage function in the inflamed lung. Mucosal Immunol. 2015, 8, 1021–1030. [Google Scholar] [CrossRef]
- Takagi, H.; Fukaya, T.; Eizumi, K.; Sato, Y.; Sato, K.; Shibazaki, A.; Otsuka, H.; Hijikata, A.; Watanabe, T.; Ohara, O.; et al. Plasmacytoid Dendritic Cells Are Crucial for the Initiation of Inflammation and T Cell Immunity In Vivo. Immunity 2011, 35, 958–971. [Google Scholar] [CrossRef]
- Lee, A.J.; Mian, F.; Poznanski, S.M.; Stackaruk, M.; Chan, T.; Chew, M.V.; Ashkar, A.A. Type I Interferon Receptor on NK Cells Negatively Regulates Interferon- γ Production. Front. Immunol. 2019, 10, 1261. [Google Scholar] [CrossRef]
- Goodwin, K.; Viboud, C.; Simonsen, L. Antibody response to influenza vaccination in the elderly: A quantitative review. Vaccine 2006, 24, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Nichol, K.L.; Nordin, J.D.; Nelson, D.B.; Mullooly, J.P.; Hak, E. Effectiveness of Influenza Vaccine in the Community-Dwelling Elderly. N. Engl. J. Med. 2007, 357, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.H.; Kozlovsky, B.F.; Effros, R.B.; Grubeck-Loebenstein, B.; Edelman, R.; Sztein, M.B. Vaccination in the elderly: An immunological perspective. Trends Immunol. 2009, 30, 351–359. [Google Scholar] [CrossRef]
- Kang, I.; Hong, M.S.; Nolasco, H.; Park, S.H.; Dan, J.M.; Choi, J.-Y.; Craft, J. Age-Associated Change in the Frequency of Memory CD4 + T Cells Impairs Long Term CD4 + T Cell Responses to Influenza Vaccine. J. Immunol. 2004, 173, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Jego, G.; Palucka, A.K.; Blanck, J.P.; Chalouni, C.; Pascual, V.; Banchereau, J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 2003, 19, 225–234. [Google Scholar] [CrossRef]
- Fink, K.; Lang, K.S.; Manjarrez-Orduno, N.; Junt, T.; Senn, B.M.; Holdener, M.; Akira, S.; Zinkernagel, R.M.; Hengartner, H. Early type I interferon-mediated signals on B cells specifically enhance antiviral humoral responses. Eur. J. Immunol. 2006, 36, 2094–2105. [Google Scholar] [CrossRef]
- Le Bon, A.; Thompson, C.; Kamphuis, E.; Durand, V.; Rossmann, C.; Kalinke, U.; Tough, D.F. Cutting Edge: Enhancement of Antibody Responses Through Direct Stimulation of B and T Cells by Type I IFN. J. Immunol. 2006, 176, 2074–2078. [Google Scholar] [CrossRef] [PubMed]
- Zacca, E.R.; Crespo, M.I.; Acland, R.P.; Roselli, E.; Núñez, N.G.; Maccioni, M.; Maletto, B.A.; Pistoresi-Palencia, M.C.; Morón, G. Aging Impairs the ability of conventional dendritic cells to cross-prime CD8+ T cells upon stimulation with a TLR7 Ligand. PLoS ONE 2015, 10, e0140672. [Google Scholar] [CrossRef]
- Pereira, L.F.; Duarte de Souza, A.P.; Borges, T.J.; Bonorino, C. Impaired in vivo CD4+ T cell expansion and differentiation in aged mice is not solely due to T cell defects: Decreased stimulation by aged dendritic cells. Mech. Ageing Dev. 2011, 132, 187–194. [Google Scholar] [CrossRef]
- Gigley, J.P.; Khan, I.A. Plasmacytoid DC from aged mice down-regulate CD8 T cell responses by inhibiting CDC maturation after encephalitozoon cuniculi infection. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Moretto, M.M.; Lawlor, E.M.; Khan, I.A. Aging Mice Exhibit a Functional Defect in Mucosal Dendritic Cell Response against an Intracellular Pathogen. J. Immunol. 2008, 181, 7977–7984. [Google Scholar] [CrossRef] [PubMed]
- Lages, C.S.; Lewkowich, I.; Sproles, A.; Wills-Karp, M.; Chougnet, C. Partial restoration of T-cell function in aged mice by in vitro blockade of the PD-1/PD-L1 pathway. Aging Cell 2010, 9, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Luft, T.; Luetjens, P.; Hochrein, H.; Toy, T.; Masterman, K.A.; Rizkalla, M.; Maliszewski, C.; Shortman, K.; Cebon, J.; Maraskovsky, E. IFN-α enhances CD40 ligand-mediated activation of immature monocyte-derived dendritic cells. Int. Immunol. 2002, 14, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.M.; Lapenta, C.; Logozzi, M.; Parlato, S.; Spada, M.; Di Pucchio, T.; Belardelli, F. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J. Exp. Med. 2000, 191, 1777–1788. [Google Scholar] [CrossRef]
- Nakano, H.; Lin, K.L.; Yanagita, M.; Charbonneau, C.; Cook, D.N.; Kakiuchi, T.; Gunn, M.D. Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat. Immunol. 2009, 10, 394–402. [Google Scholar] [CrossRef]
- Le Bon, A.; Etchart, N.; Rossmann, C.; Ashton, M.; Hou, S.; Gewert, D.; Borrow, P.; Tough, D.F. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 2003, 4, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Shirley, J.L.; Keeler, G.D.; Sherman, A.; Zolotukhin, I.; Markusic, D.M.; Hoffman, B.E.; Morel, L.M.; Wallet, M.A.; Terhorst, C.; Herzog, R.W. Type I IFN Sensing by cDCs and CD4+ T Cell Help Are Both Requisite for Cross-Priming of AAV Capsid-Specific CD8+ T Cells. Mol. Ther. 2020, 28, 758–770. [Google Scholar] [CrossRef]
- Longhi, M.P.; Trumpfheller, C.; Idoyaga, J.; Caskey, M.; Matos, I.; Kluger, C.; Salazar, A.M.; Colonna, M.; Steinman, R.M. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J. Exp. Med. 2009, 206, 1589–1602. [Google Scholar] [CrossRef]
- Bosteels, C.; Neyt, K.; Vanheerswynghels, M.; van Helden, M.J.; Sichien, D.; Debeuf, N.; De Prijck, S.; Bosteels, V.; Vandamme, N.; Martens, L.; et al. Inflammatory Type 2 cDCs Acquire Features of cDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 2020, 52, 1039–1056. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Jing, Y.; Campbell, A.E.; Gravenstein, S. Age-Related Impaired Type 1 T Cell Responses to Influenza: Reduced Activation Ex Vivo, Decreased Expansion in CTL Culture In Vitro, and Blunted Response to Influenza Vaccination In Vivo in the Elderly. J. Immunol. 2004, 172, 3437–3446. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Gilmore, X.; Xu, K.; Chen, M.; Tebebi, P.; Mbawuike, I.N. Apoptosis and reduced influenza A virus specific CD8+ T cells in aging mice. Cell Death Differ. 2002, 9, 651–660. [Google Scholar] [CrossRef]
- Po, J.L.Z.; Gardner, E.M.; Anaraki, F.; Katsikis, P.D.; Murasko, D.M. Age-associated decrease in virus-specific CD8+ T lymphocytes during primary influenza infection. Mech. Ageing Dev. 2002, 123, 1167–1181. [Google Scholar] [CrossRef]
- Yager, E.J.; Ahmed, M.; Lanzer, K.; Randall, T.D.; Woodland, D.L.; Blackman, M.A. Age-associated decline in T cell repertoire diversity leads to holes in the repertoire and impaired immunity to influenza virus. J. Exp. Med. 2008, 205, 711–723. [Google Scholar] [CrossRef]
- Li, G.; Ju, J.; Weyand, C.M.; Goronzy, J.J. Age-Associated Failure To Adjust Type I IFN Receptor Signaling Thresholds after T Cell Activation. J. Immunol. 2015, 195, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Goplen, N.P.; Wu, Y.; Son, Y.M.; Li, C.; Wang, Z.; Cheon, I.S.; Jiang, L.; Zhu, B.; Ayasoufi, K.; Chini, E.N.; et al. Tissue-resident CD8+ T cells drive age-associated chronic lung sequelae after viral pneumonia. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Wang, Y.; Swiecki, M.; Cella, M.; Alber, G.; Schreiber, R.D.; Gilfillan, S.; Colonna, M. Timing and magnitude of type i interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe 2012, 11, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Kolumam, G.A.; Thomas, S.; Thompson, L.J.; Sprent, J.; Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 2005, 202, 637–650. [Google Scholar] [CrossRef]
- Krug, A.; Veeraswamy, R.; Pekosz, A.; Kanagawa, O.; Unanue, E.R.; Colonna, M.; Cella, M. Interferon-producing cells fail to induce proliferation of naive T cells but can promote expansion and T helper 1 differentiation of antigen-experienced unpolarized T cells. J. Exp. Med. 2003, 197, 899–906. [Google Scholar] [CrossRef]
- Jung, A.; Kato, H.; Kumagai, Y.; Kumar, H.; Kawai, T.; Takeuchi, O.; Akira, S. Lymphocytoid Choriomeningitis Virus Activates Plasmacytoid Dendritic Cells and Induces a Cytotoxic T-Cell Response via MyD88. J. Virol. 2008, 82, 196–206. [Google Scholar] [CrossRef]
- Pinto, A.K.; Daffis, S.; Brien, J.D.; Gainey, M.D.; Yokoyama, W.M.; Sheehan, K.C.F.; Murphy, K.M.; Schreiber, R.D.; Diamond, M.S. A temporal role of type I interferon signaling in CD8 + T cell maturation during acute West Nile virus infection. PLoS Pathog. 2011, 7, e1002407. [Google Scholar] [CrossRef] [PubMed]
- Kohlmeier, J.E.; Cookenham, T.; Roberts, A.D.; Miller, S.C.; Woodland, D.L. Type I interferons regulate cytolytic activity of memory CD8+ T cells in the lung airways during respiratory virus challenge. Immunity 2010, 33, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Bahl, K.; Kim, S.-K.; Calcagno, C.; Ghersi, D.; Puzone, R.; Celada, F.; Selin, L.K.; Welsh, R.M. IFN-Induced Attrition of CD8 T Cells in the Presence or Absence of Cognate Antigen during the Early Stages of Viral Infections. J. Immunol. 2006, 176, 4284–4295. [Google Scholar] [CrossRef]
- McNally, J.M.; Zarozinski, C.C.; Lin, M.-Y.; Brehm, M.A.; Chen, H.D.; Welsh, R.M. Attrition of Bystander CD8 T Cells during Virus-Induced T-Cell and Interferon Responses. J. Virol. 2001, 75, 5965–5976. [Google Scholar] [CrossRef]
- Sasaki, S.; Sullivan, M.; Narvaez, C.F.; Holmes, T.H.; Furman, D.; Zheng, N.Y.; Nishtala, M.; Wrammert, J.; Smith, K.; James, J.A.; et al. Limited efficacy of inactivated influenza vaccine in elderly individuals is associated with decreased production of vaccine-specific antibodies. J. Clin. Investig. 2011, 121, 3109–3119. [Google Scholar] [CrossRef]
- Frasca, D.; Blomberg, B.B. B cell function and influenza vaccine responses in healthy aging and disease. Curr. Opin. Immunol. 2014, 29, 112–118. [Google Scholar] [CrossRef][Green Version]
- Frasca, D.; Diaz, A.; Romero, M.; D’Eramo, F.; Blomberg, B.B. Aging effects on T-bet expression in human B cell subsets. Cell. Immunol. 2017, 321, 68–73. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Landin, A.M.; Phillips, M.; Lechner, S.C.; Ryan, J.G.; Blomberg, B.B. Intrinsic defects in B cell response to seasonal influenza vaccination in elderly humans. Vaccine 2010, 28, 8077–8084. [Google Scholar] [CrossRef] [PubMed]
- Deal, E.M.; Lahl, K.; Narváez, C.F.; Butcher, E.C.; Greenberg, H.B. Plasmacytoid dendritic cells promote rotavirus-induced human and murine B cell responses. J. Clin. Investig. 2013, 123, 2464–2474. [Google Scholar] [CrossRef] [PubMed]
- Bekeredjian-Ding, I.B.; Wagner, M.; Hornung, V.; Giese, T.; Schnurr, M.; Endres, S.; Hartmann, G. Plasmacytoid Dendritic Cells Control TLR7 Sensitivity of Naive B Cells via Type I IFN. J. Immunol. 2005, 174, 4043–4050. [Google Scholar] [CrossRef] [PubMed]
- Douagi, I.; Gujer, C.; Sundling, C.; Adams, W.C.; Smed-Sörensen, A.; Seder, R.A.; Karlsson Hedestam, G.B.; Loré, K. Human B Cell Responses to TLR Ligands Are Differentially Modulated by Myeloid and Plasmacytoid Dendritic Cells. J. Immunol. 2009, 182, 1991–2001. [Google Scholar] [CrossRef]
- Le Bon, A.; Schiavoni, G.; D’Agostino, G.; Gresser, I.; Belardelli, F.; Tough, D.F. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 2001, 14, 461–470. [Google Scholar] [CrossRef]
- Green, N.M.; Laws, A.; Kiefer, K.; Busconi, L.; Kim, Y.-M.; Brinkmann, M.M.; Trail, E.H.; Yasuda, K.; Christensen, S.R.; Shlomchik, M.J.; et al. Murine B Cell Response to TLR7 Ligands Depends on an IFN-β Feedback Loop. J. Immunol. 2009, 183, 1569–1576. [Google Scholar] [CrossRef]
- Oganesyan, G.; Saha, S.K.; Pietras, E.M.; Guo, B.; Miyahira, A.K.; Zarnegar, B.; Cheng, G. IRF3-dependent type I interferon response in B cells regulates CpG-mediated antibody production. J. Biol. Chem. 2008, 283, 802–808. [Google Scholar] [CrossRef]
- Ng, C.T.; Sullivan, B.M.; Teijaro, J.R.; Lee, A.M.; Welch, M.; Rice, S.; Sheehan, K.C.F.; Schreiber, R.D.; Oldstone, M.B.A. Blockade of interferon beta, but not interferon alpha, signaling controls persistent viral infection. Cell Host Microbe 2015, 17, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Osokine, I.; Snell, L.M.; Cunningham, C.R.; Yamada, D.H.; Wilson, E.B.; Elsaesser, H.J.; De La Torre, J.C.; Brooks, D. Type i interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc. Natl. Acad. Sci. USA 2014, 111, 7409–7414. [Google Scholar] [CrossRef]
- Shaabani, N.; Duhan, V.; Khairnar, V.; Gassa, A.; Ferrer-Tur, R.; Häussinger, D.; Recher, M.; Zelinskyy, G.; Liu, J.; Dittmer, U.; et al. CD169+ macrophages regulate PD-L1 expression via type I interferon and thereby prevent severe immunopathology after LCMV infection. Cell Death Dis. 2016, 7, e2446. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of Chronic Type I Interferon Signaling to Control Persistent LCMV Infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.F.; Welch, M.; Schreiber, R.D.; Carlos de la Torre, J.; Oldstone, M.B.A. Persistent LCMV Infection Is Controlled by Blockade of Type I Interferon Signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.L.G.; Pérez-Girón, J.V.; Lüdtke, A.; Gómez-Medina, S.; Ruibal, P.; Idoyaga, J.; Muñoz-Fontela, C. Monocyte-derived dendritic cells enhance protection against secondary influenza challenge by controlling the switch in CD8+ T-cell immunodominance. Eur. J. Immunol. 2017, 47, 345–352. [Google Scholar] [CrossRef]
- Haring, J.S.; Badovinac, V.P.; Harty, J.T. Inflaming the CD8+ T Cell Response. Immunity 2006, 25, 19–29. [Google Scholar] [CrossRef]
- Starbeck-Miller, G.R.; Xue, H.H.; Harty, J.T. IL-12 and type I interferon prolong the division of activated CD8 T cells by maintaining high-affinity IL-2 signaling in vivo. J. Exp. Med. 2014, 211, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.S.; Jeon, I.; Kim, B.S.; Kim, I.K.; Park, Y.J.; Koh, C.H.; Song, B.; Lee, J.M.; Lim, J.; Bae, E.A.; et al. Monocyte-derived dendritic cells dictate the memory differentiation of CD8+ T cells during acute infection. Front. Immunol. 2019, 10, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.D.; Fraire, A.E.; Joris, I.; Brehm, M.A.; Welsh, R.M.; Selin, L.K. Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung. Nat. Immunol. 2001, 2, 1067–1076. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Y.; Xu, L.T.; Jackson, K.J.L.; Roskin, K.M.; Pham, T.D.; Laserson, J.; Marshall, E.L.; Seo, K.; Lee, J.-Y.; et al. Effects of Aging, Cytomegalovirus Infection, and EBV Infection on Human B Cell Repertoires. J. Immunol. 2014, 192, 603–611. [Google Scholar] [CrossRef]
- Frasca, D.; Nguyen, D.; Riley, R.L.; Blomberg, B.B. Effects of aging on proliferation and E47 transcription factor activity induced by different stimuli in murine splenic B cells. Mech. Ageing Dev. 2003, 124, 361–369. [Google Scholar] [CrossRef]
- Hart, G.; Avin-Wittenberg, T.; Shachar, I. IL-15 regulates immature B-cell homing in an Ly49D-, IL-12-, and IL-18-dependent manner. Blood 2008, 111, 50–59. [Google Scholar] [CrossRef]
- Dubois, B.; Massacrier, C.; Vanbervliet, B.; Fayette, J.; Brière, F.; Banchereau, J.; Caux, C. Critical role of IL-12 in dendritic cell-induced differentiation of naive B lymphocytes. J. Immunol. 1998, 161, 2223–2231. [Google Scholar] [PubMed]
- Farsakoglu, Y.; Palomino-Segura, M.; Latino, I.; Zanaga, S.; Chatziandreou, N.; Pizzagalli, D.U.; Rinaldi, A.; Bolis, M.; Sallusto, F.; Stein, J.V.; et al. Influenza Vaccination Induces NK-Cell-Mediated Type-II IFN Response that Regulates Humoral Immunity in an IL-6-Dependent Manner. Cell Rep. 2019, 26, 2307–2315.e5. [Google Scholar] [CrossRef]
- Liu, N.; Ohnishi, N.; Ni, L.; Akira, S.; Bacon, K.B. CpG directly induces T-bet expression and inhibits IgG1 and IgE switching in B cells. Nat. Immunol. 2003, 4, 687–693. [Google Scholar] [CrossRef]
- Rubtsova, K.; Rubtsov, A.V.; Van Dyk, L.F.; Kappler, J.W.; Marrack, P. T-box transcription factor T-bet, a key player in a unique type of B-cell activation essential for effective viral clearance. Proc. Natl. Acad. Sci. USA 2013, 110. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-γ: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Herrero, C.; Marqués, L.; Lloberas, J.; Celada, A. IFN-γ-dependent transcription of MHC class II IA is impaired in macrophages from aged mice. J. Clin. Investig. 2001, 107, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Ostler, T.; Davidson, W.; Ehl, S. Virus clearance and immunopathology by CD8+ T cells during infection with respiratory syncytial virus are mediated by IFN-γ. Eur. J. Immunol. 2002, 32, 2117–2123. [Google Scholar] [CrossRef]
- Gill, N.; Chenoweth, M.J.; Verdu, E.F.; Ashkar, A.A. NK cells require type I IFN receptor for antiviral responses during genital HSV-2 infection. Cell. Immunol. 2011, 269, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Huang, X.; Yang, Y.; Alerts, E. Direct Action of Type I IFN on NK Cells Is Required for Their Activation in Response to Vaccinia Viral Infection In Vivo. J. Immunol. 2008, 180, 1592–1597. [Google Scholar] [CrossRef]
- Moro, K.; Kabata, H.; Tanabe, M.; Koga, S.; Takeno, N.; Mochizuki, M.; Fukunaga, K.; Asano, K.; Betsuyaku, T.; Koyasu, S. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat. Immunol. 2016, 17, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Goldszmid, R.S.; Caspar, P.; Rivollier, A.; White, S.; Dzutsev, A.; Hieny, S.; Kelsall, B.; Trinchieri, G.; Sher, A. NK Cell-Derived Interferon-γ Orchestrates Cellular Dynamics and the Differentiation of Monocytes into Dendritic Cells at the Site of Infection. Immunity 2012, 36, 1047–1059. [Google Scholar] [CrossRef]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Hemann, E.A.; Gale, M.; Savan, R. Interferon lambda genetics and biology in regulation of viral control. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Prakash, S.; Agrawal, S.; Cao, J.N.; Gupta, S.; Agrawal, A. Impaired secretion of interferons by dendritic cells from aged subjects to influenza. Age (Omaha) 2013, 35, 1785–1797. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, A.; Esposo, M.; Kaushal, K.; Tay, J.; Osann, K.; Agrawal, S.; Gupta, S.; Agrawal, A. Age-associated impaired plasmacytoid dendritic cell functions lead to decreased CD4 and CD8 T cell immunity. Age (Omaha) 2011, 33, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Mordstein, M.; Neugebauer, E.; Ditt, V.; Jessen, B.; Rieger, T.; Falcone, V.; Sorgeloos, F.; Ehl, S.; Mayer, D.; Kochs, G.; et al. Lambda Interferon Renders Epithelial Cells of the Respiratory and Gastrointestinal Tracts Resistant to Viral Infections. J. Virol. 2010, 84, 5670–5677. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-D.; Feng, N.; Sen, A.; Balan, M.; Tseng, H.C.; McElrath, C.; Smirnov, S.V.; Peng, J.; Yasukawa, L.L.; Durbin, R.K.; et al. Distinct Roles of Type I and Type III Interferons in Intestinal Immunity to Homologous and Heterologous Rotavirus Infections. PLoS Pathog. 2016, 12, e1005600. [Google Scholar] [CrossRef]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-lambda (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008, 4, 1–12. [Google Scholar] [CrossRef]
- Lazear, H.M.; Nice, T.J.; Diamond, M.S. Interferon-λ: Immune Functions at Barrier Surfaces and Beyond. Immunity 2015, 43, 15–28. [Google Scholar] [CrossRef]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-λ orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef]
- Okabayashi, T.; Kojima, T.; Masaki, T.; Yokota, S.I.; Imaizumi, T.; Tsutsumi, H.; Himi, T.; Fujii, N.; Sawada, N. Type-III interferon, not type-I, is the predominant interferon induced by respiratory viruses in nasal epithelial cells. Virus Res. 2011, 160, 360–366. [Google Scholar] [CrossRef]
- Jewell, N.A.; Cline, T.; Mertz, S.E.; Smirnov, S.V.; Flaño, E.; Schindler, C.; Grieves, J.L.; Durbin, R.K.; Kotenko, S.V.; Durbin, J.E. Lambda Interferon Is the Predominant Interferon Induced by Influenza A Virus Infection In Vivo. J. Virol. 2010, 84, 11515–11522. [Google Scholar] [CrossRef]
- Ank, N.; West, H.; Bartholdy, C.; Eriksson, K.; Thomsen, A.R.; Paludan, S.R. Lambda Interferon (IFN- ), a Type III IFN, Is Induced by Viruses and IFNs and Displays Potent Antiviral Activity against Select Virus Infections In Vivo. J. Virol. 2006, 80, 4501–4509. [Google Scholar] [CrossRef]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-λ Mediates Non-redundant Front-Line Antiviral Protection against Influenza Virus Infection without Compromising Host Fitness. Immunity 2017, 46, 875–890. [Google Scholar] [CrossRef]
- Davidson, S.; McCabe, T.M.; Crotta, S.; Gad, H.H.; Hessel, E.M.; Beinke, S.; Hartmann, R.; Wack, A. IFN λ is a potent anti-influenza therapeutic without the inflammatory side effects of IFN α treatment. EMBO Mol. Med. 2016, 8, 1099–1112. [Google Scholar] [CrossRef]
- Feld, J.J.; Kandel, C.; Biondi, M.J.; Kozak, R.A.; Zahoor, M.A.; Lemieux, C.; Borgia, S.M.; Boggild, A.K.; Powis, J.; McCready, J.; et al. Peginterferon lambda for the treatment of outpatients with COVID-19: A phase 2, placebo-controlled randomised trial. Lancet Respir. Med. 2021, 2600, 1–13. [Google Scholar] [CrossRef]
- Weiss, I.D.; Wald, O.; Wald, H.; Beider, K.; Abraham, M.; Galun, E.; Nagler, A.; Peled, A. IFN-γ treatment at early stages of influenza virus infection protects mice from death in a NK cell-dependent manner. J. Interf. Cytokine Res. 2010, 30, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Rhein, B.A.; Powers, L.S.; Rogers, K.; Anantpadma, M.; Singh, B.K.; Sakurai, Y.; Bair, T.; Miller-Hunt, C.; Sinn, P.; Davey, R.A.; et al. Interferon-γ Inhibits Ebola Virus Infection. PLoS Pathog. 2015, 11, 1–28. [Google Scholar] [CrossRef]
- US National Library of Medicine. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04578210 (accessed on 15 February 2021).
- US National Library of Medicine. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04365101 (accessed on 15 February 2021).
- US National Library of Medicine. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04280224 (accessed on 15 February 2021).
- Liu, B.; Bao, L.L.; Wang, L.; Li, F.; Wen, M.; Li, H.; Deng, W.; Zhang, X.; Cao, B. Anti-IFN-γ therapy alleviates acute lung injury induced by severe influenza A (H1N1) pdm09 infection in mice. J. Microbiol. Immunol. Infect. 2019. [Google Scholar] [CrossRef]
- Zhou, G.; Juang, S.W.W.; Kane, K.P. NK cells exacerbate the pathology of influenza virus infection in mice. Eur. J. Immunol. 2013, 43, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Sheridan, P.A.; Harp, J.B.; Beck, M.A. Diet-induced obese mice have increased mortality and altered immune responses when infected with influenza virus. J. Nutr. 2007, 137, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Cormier, S.A.; Shrestha, B.; Saravia, J.; Lee, G.I.; Shen, L.; DeVincenzo, J.P.; Kim, Y.-I.; You, D. Limited Type I Interferons and Plasmacytoid Dendritic Cells during Neonatal Respiratory Syncytial Virus Infection Permit Immunopathogenesis upon Reinfection. J. Virol. 2014, 88, 9350–9360. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cell Type | Aging | Absence/Reduction in Type I IFN |
|---|---|---|
| Neutrophils |
|
|
| Monocyte/Macrophages |
| |
| NK cells |
|
|
| Cell Type | Aging | Absence/Reduction in Type I IFN |
|---|---|---|
| DCs |
|
|
| T cells | ||
| B cells |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, E.; Balint, E.; Poznanski, S.M.; Ashkar, A.A.; Loeb, M. Aging and Interferons: Impacts on Inflammation and Viral Disease Outcomes. Cells 2021, 10, 708. https://doi.org/10.3390/cells10030708
Feng E, Balint E, Poznanski SM, Ashkar AA, Loeb M. Aging and Interferons: Impacts on Inflammation and Viral Disease Outcomes. Cells. 2021; 10(3):708. https://doi.org/10.3390/cells10030708
Chicago/Turabian StyleFeng, Emily, Elizabeth Balint, Sophie M. Poznanski, Ali A. Ashkar, and Mark Loeb. 2021. "Aging and Interferons: Impacts on Inflammation and Viral Disease Outcomes" Cells 10, no. 3: 708. https://doi.org/10.3390/cells10030708
APA StyleFeng, E., Balint, E., Poznanski, S. M., Ashkar, A. A., & Loeb, M. (2021). Aging and Interferons: Impacts on Inflammation and Viral Disease Outcomes. Cells, 10(3), 708. https://doi.org/10.3390/cells10030708