
Rapid and Progressive Loss of Multiple Retinal Cell Types in Cathepsin D-Deficient Mice—An Animal Model of CLN10 Disease †

Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Immunohistochemistry
2.3. Retina Thickness Measurements and Cell Counting
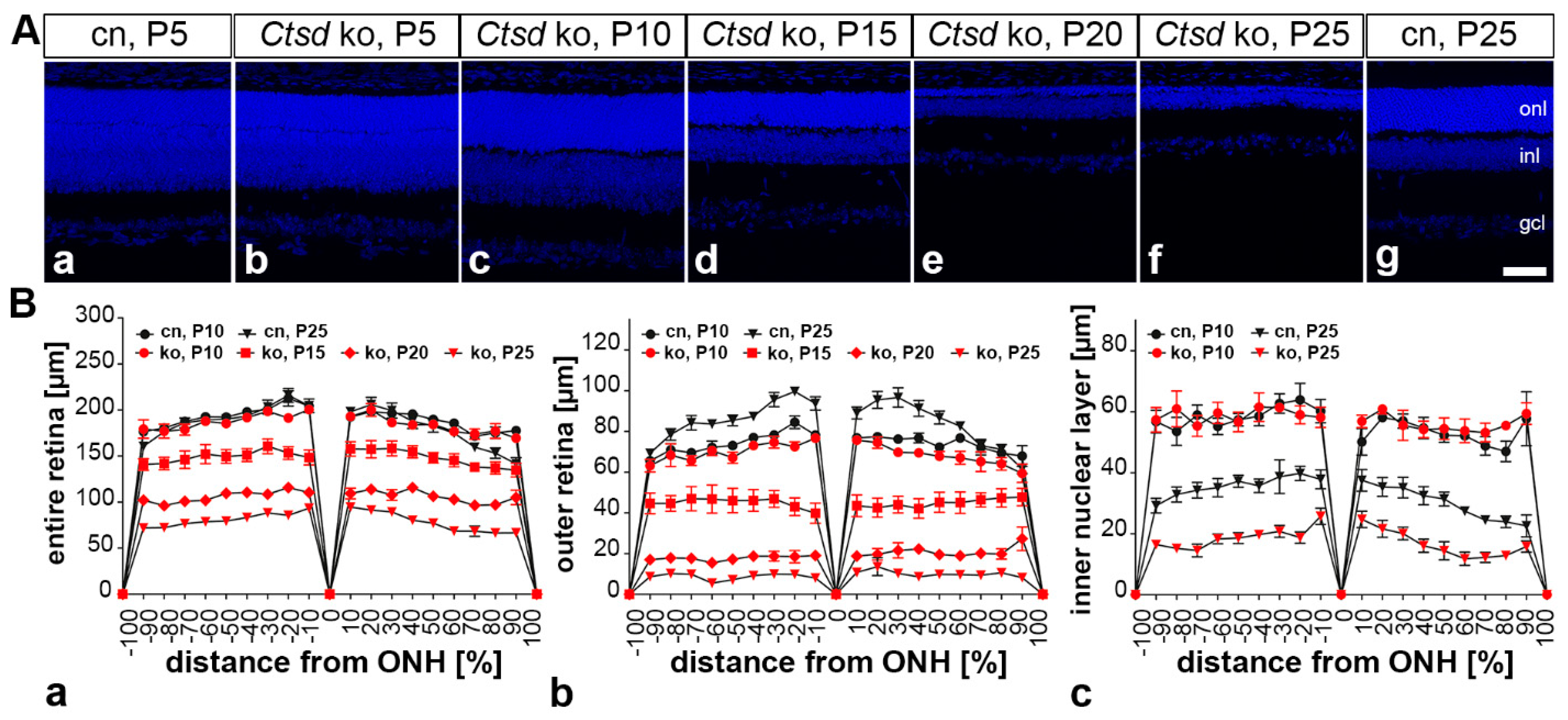
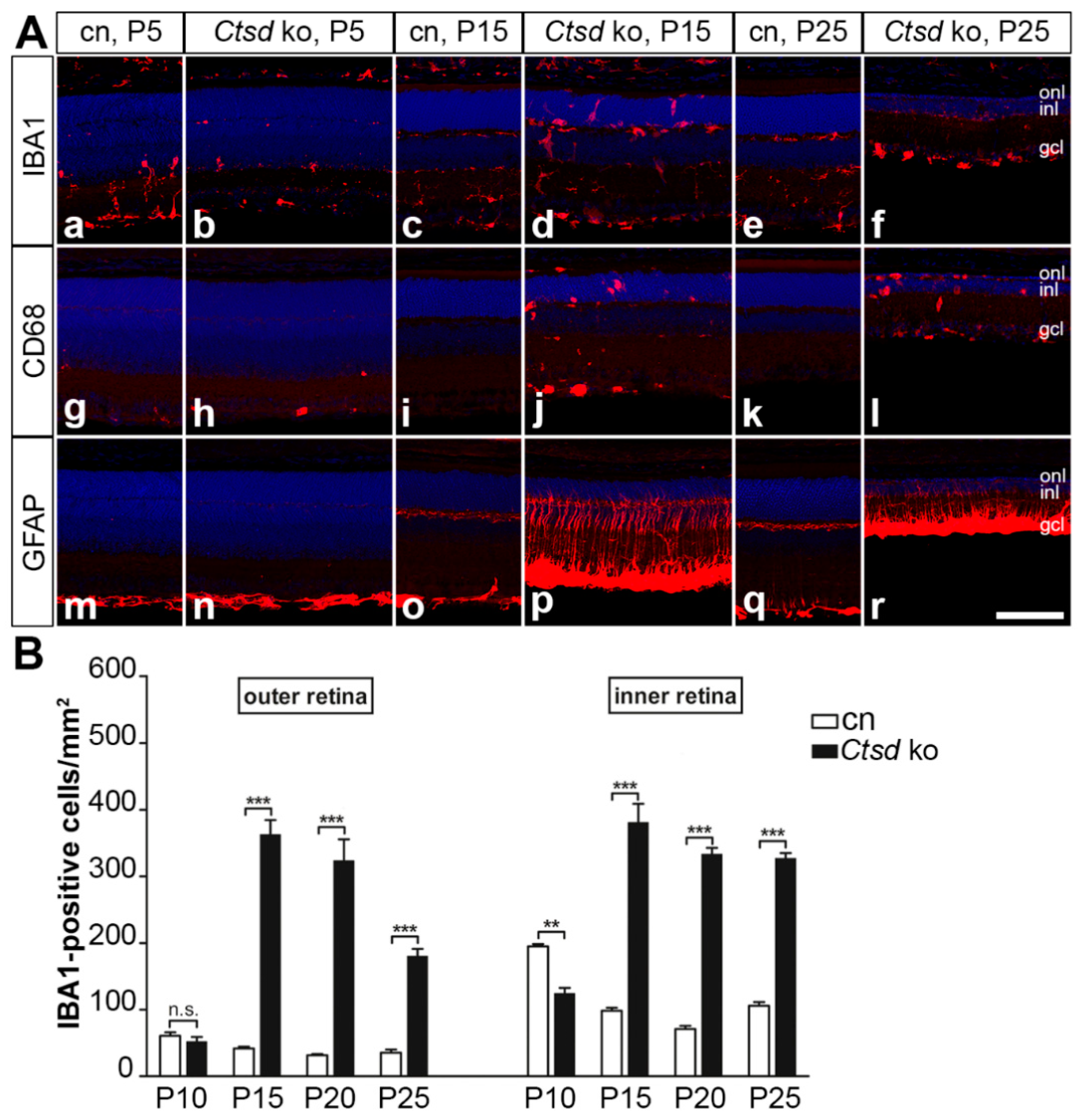
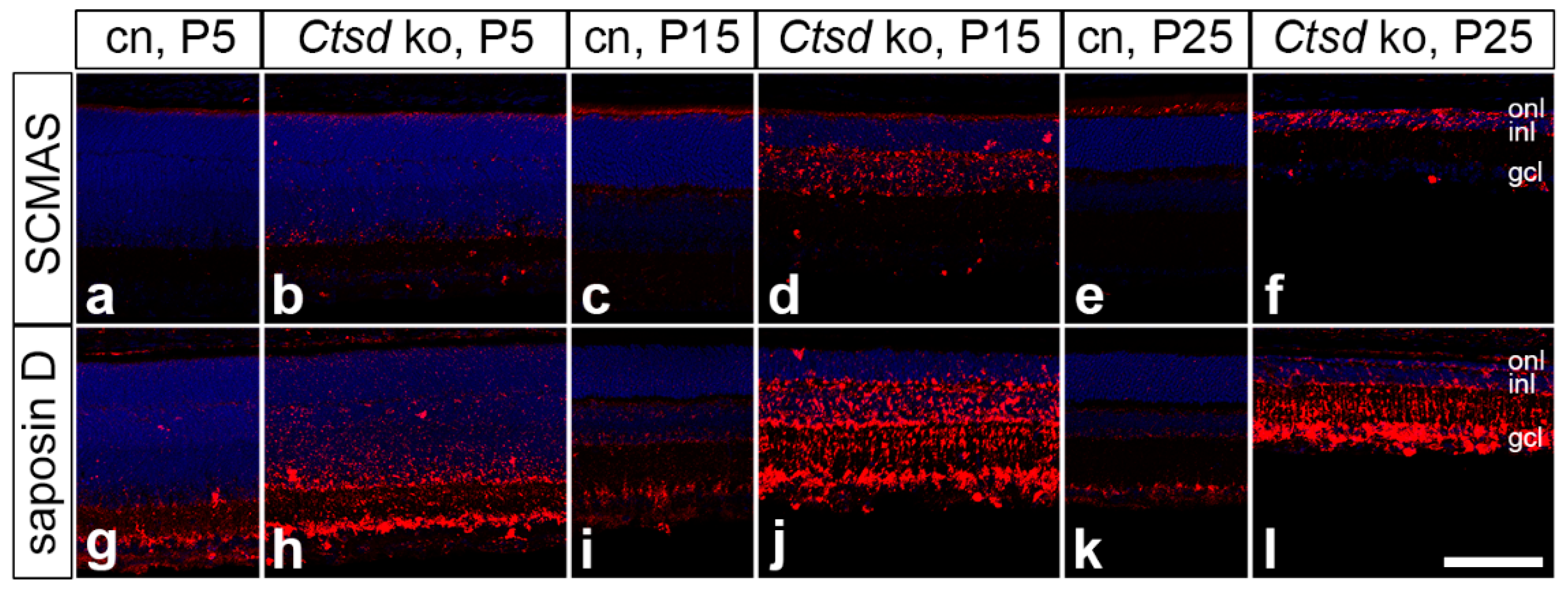
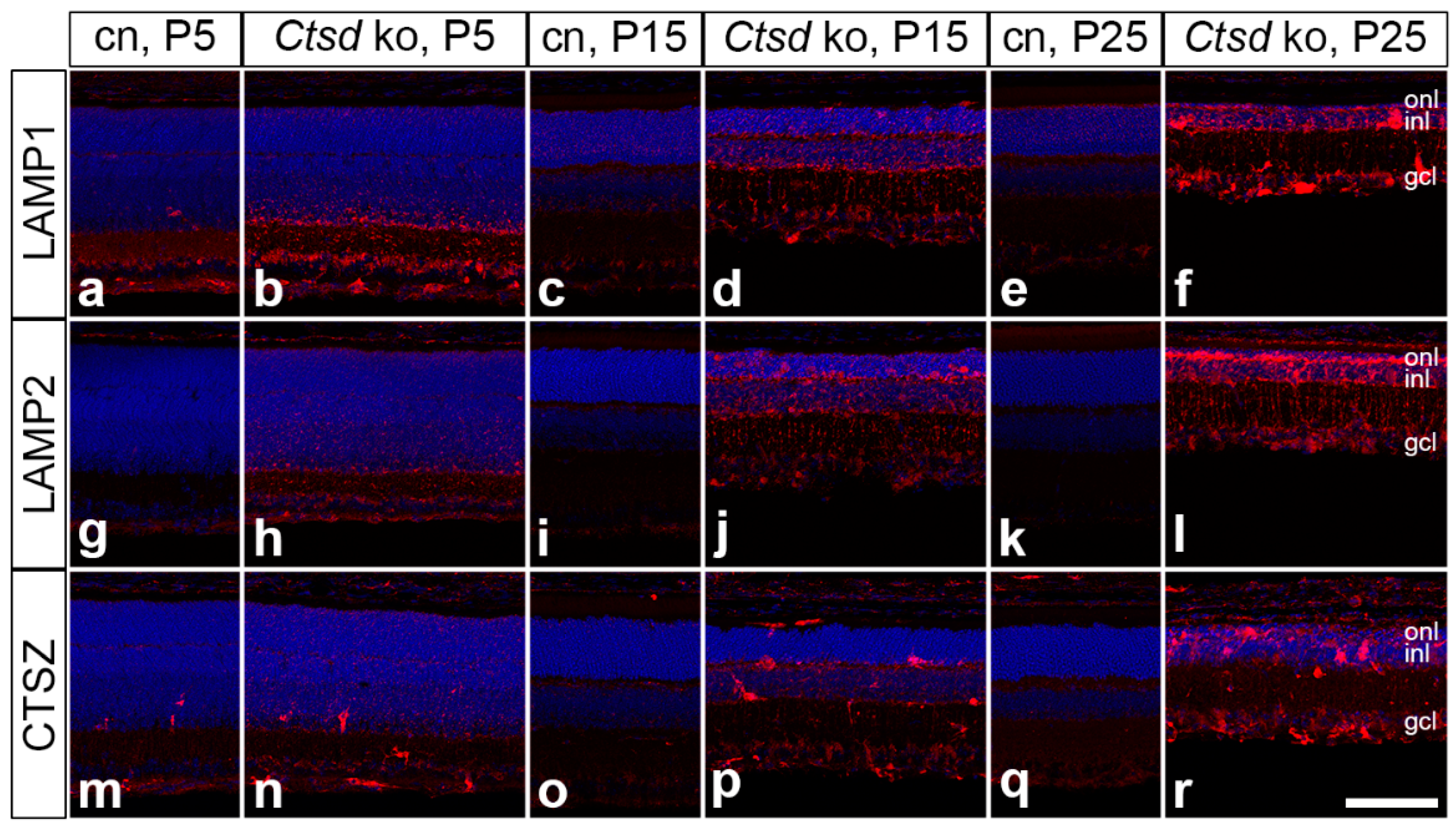
3. Results
3.1. Progressive Thinning of Ctsd ko Retinas
3.2. Reactive Microgliosis and Astrogliosis
3.3. Accumulation of Storage Material and Dysregulation of Lysosomal Proteins
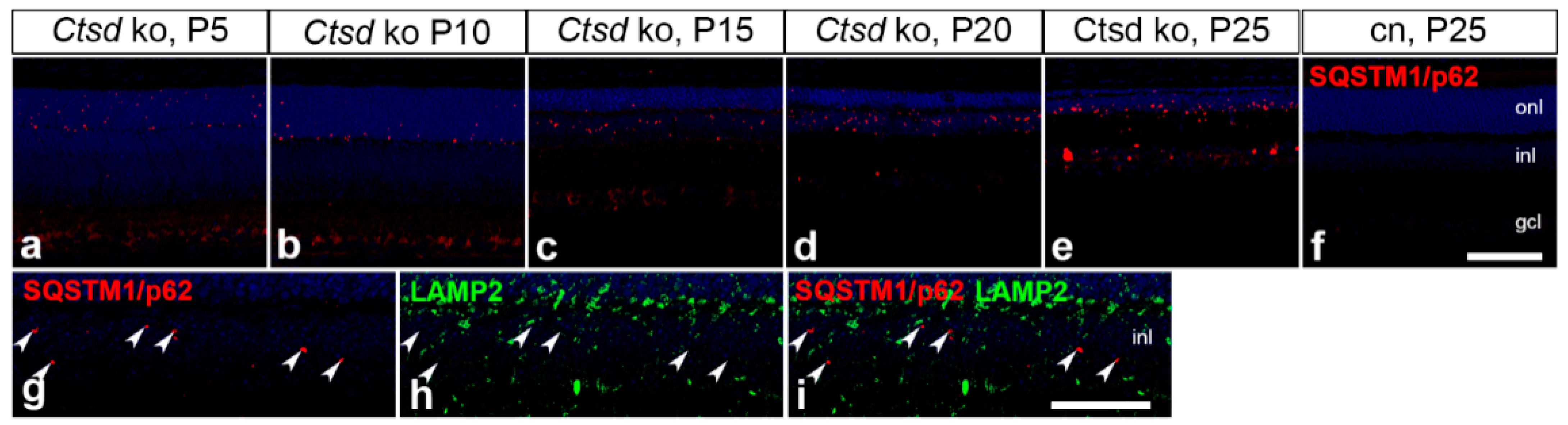
3.4. Expression of the Autophagy Marker SQSTM1/p62
3.5. Progressive Loss of Multiple Retinal Cell Types
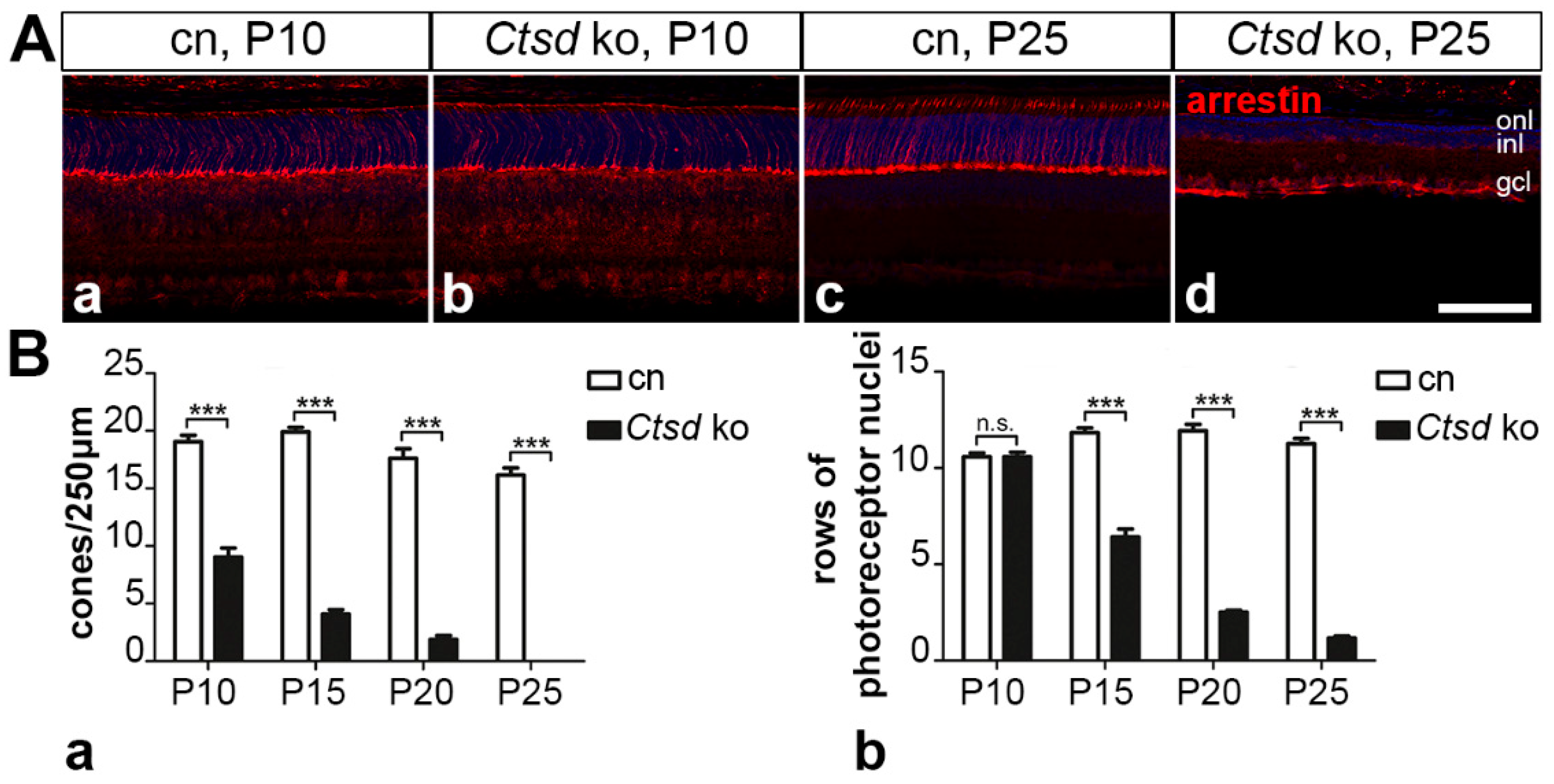
3.5.1. Rod and Cone Photoreceptor Cells
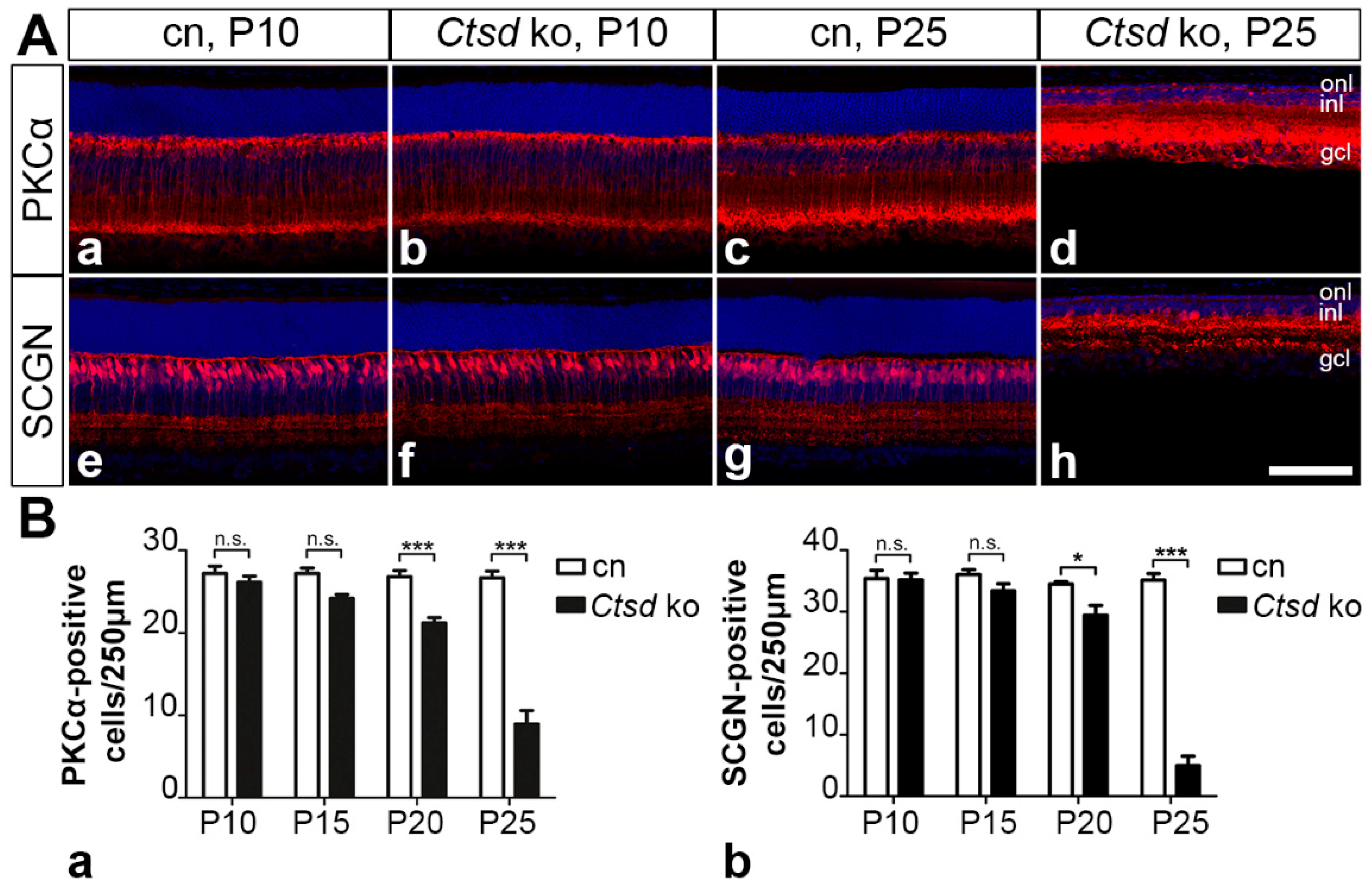
3.5.2. Rod and Cone Bipolar Cells
3.5.3. Retinal Ganglion Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cárcel-Trullols, J.; Kovács, A.D.; Pearce, D.A. Cell biology of the NCL proteins: What they do and don’t do. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2242–2255. [Google Scholar] [CrossRef] [PubMed]
- Mole, S.E.; Cotman, S.L. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2237–2241. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.B.; Appu, A.P.; Sadhukhan, T.; Casey, S.; Mondal, A.; Zhang, Z.; Bagh, M.B. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol. Neurodegener. 2019, 14, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Seehafer, S.S.; Pearce, D.A. You say lipofuscin, we say ceroid: Defining autofluorescent storage material. Neurobiol. Aging 2006, 27, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.N. The relevance of the storage of subunit c of ATP synthase in different forms and models of Batten disease (NCLs). Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2287–2291. [Google Scholar] [CrossRef] [PubMed]
- Haltia, M.; Goebel, H.H. The neuronal ceroid-lipofuscinoses: A historical introduction. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1795–1800. [Google Scholar] [CrossRef]
- Kousi, M.; Lehesjoki, A.-E.; Mole, S.E. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum. Mutat. 2011, 33, 42–63. [Google Scholar] [CrossRef] [PubMed]
- Kohlschütter, A.; Schulz, A.; Bartsch, U.; Storch, S. Current and Emerging Treatment Strategies for Neuronal Ceroid Lipofuscinoses. CNS Drugs 2019, 33, 315–325. [Google Scholar] [CrossRef]
- Di Fruscio, G.; Schulz, A.; De Cegli, R.; Savarese, M.; Mutarelli, M.; Parenti, G.; Banfi, S.; Braulke, T.; Nigro, V.; Ballabio, A. Lysoplex: An efficient toolkit to detect DNA sequence variations in the autophagy-lysosomal pathway. Autophagy 2015, 11, 928–938. [Google Scholar] [CrossRef]
- Boustany, R.-M.N. Lysosomal storage diseases—The horizon expands. Nat. Rev. Neurol. 2013, 9, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Onyenwoke, R.U.; Brenman, J.E. Lysosomal Storage Diseases-Regulating Neurodegeneration. J. Exp. Neurosci. 2015, 9, JEN-S25475. [Google Scholar] [CrossRef] [PubMed]
- Getty, A.L.; Rothberg, P.G.; Pearce, D.A. Diagnosis of neuronal ceroid lipofuscinosis: Mutation detection strategies. Expert Opin. Med. Diagn. 2007, 1, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.W.; Goebel, H.H.; Simonati, A. Human pathology in NCL. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1807–1826. [Google Scholar] [CrossRef] [PubMed]
- Varvagiannis, K.; Hanquinet, S.; Billieux, M.H.; De Luca, R.; Rimensberger, P.; Lidgren, M.; Guipponi, M.; Makrythanasis, P.; Blouin, J.L.; Antonarakis, S.E.; et al. Congenital Neuronal Ceroid Lipofuscinosis with a Novel CTSD Gene Mutation: A Rare Cause of Neonatal-Onset Neurodegenerative Disorder. Neuropediatrics 2017, 49, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Benes, P.; Vetvicka, V.; Fusek, M. Cathepsin D—Many functions of one aspartic protease. Crit. Rev. Oncol. 2008, 68, 12–28. [Google Scholar] [CrossRef]
- Pereira, H.; Oliveira, C.; Castro, L.; Preto, A.; Chaves, S.R.; Côrte-Real, M. Yeast as a tool to explore cathepsin D function. Microb. Cell 2015, 2, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Siintola, E.; Partanen, S.; Strömme, P.; Haapanen, A.; Haltia, M.; Maehlen, J.; Lehesjoki, A.-E.; Tyynelä, J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain 2006, 129, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Fritchie, K.; Siintola, E.; Armao, D.; Lehesjoki, A.-E.; Marino, T.; Powell, C.; Tennison, M.; Booker, J.M.; Koch, S.; Partanen, S.; et al. Novel mutation and the first prenatal screening of cathepsin D deficiency (CLN10). Acta Neuropathol. 2008, 117, 201–208. [Google Scholar] [CrossRef]
- Meyer, S.; Yilmaz, U.; Kim, Y.-J.; Steinfeld, R.; Meyberg-Solomayer, G.; Oehl-Jaschkowitz, B.; Tzschach, A.; Gortner, L.; Igel, J.; Schofer, O. Congenital CLN disease in two siblings. Wien. Med. Wochenschr. 2015, 165, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Steinfeld, R.; Reinhardt, K.; Schreiber, K.; Hillebrand, M.; Kraetzner, R.; Brück, W.; Saftig, P.; Gärtner, J. Cathepsin D Deficiency Is Associated with a Human Neurodegenerative Disorder. Am. J. Hum. Genet. 2006, 78, 988–998. [Google Scholar] [CrossRef]
- Hersheson, J.; Burke, D.; Clayton, R.; Anderson, G.; Jacques, T.S.; Mills, P.; Wood, N.W.; Gissen, P.; Clayton, P.; Fearnley, J.; et al. Cathepsin D deficiency causes juvenile-onset ataxia and distinctive muscle pathology. Neurology 2014, 83, 1873–1875. [Google Scholar] [CrossRef]
- Doccini, S.; Sartori, S.; Maeser, S.; Pezzini, F.; Rossato, S.; Moro, F.; Toldo, I.; Przybylski, M.; Santorelli, F.M.; Simonati, A. Early infantile neuronal ceroid lipofuscinosis (CLN10 disease) associated with a novel mutation in CTSD. J. Neurol. 2016, 263, 1029–1032. [Google Scholar] [CrossRef]
- Bond, M.; Holthaus, S.-M.K.; Tammen, I.; Tear, G.; Russell, C. Use of model organisms for the study of neuronal ceroid lipofuscinosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1842–1865. [Google Scholar] [CrossRef] [PubMed]
- Faller, K.M.; Gutierrez-Quintana, R.; Mohammed, A.; Rahim, A.A.; Tuxworth, R.I.; Wager, K.; Bond, M. The neuronal ceroid lipofuscinoses: Opportunities from model systems. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Hetman, M.; Schmahl, W.; Weber, K.; Heine, L.; Mossmann, H.; Köster, A.; Hess, B.; Evers, M.; Von Figura, K. Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO J. 1995, 14, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Tyynelä, J.; Sohar, I.; Sleat, D.E.; Gin, R.M.; Nelly, R.J.D.; Baumann, M.; Haltia, M.; Lobel, P. Congenital ovine neuronal ceroid lipofuscinosis—Acathepsin D deficiency with increased levels of the inactive enzyme. Eur. J. Paediatr. Neurol. 2001, 5, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Rakoczy, P.E.; Zhang, D.; Robertson, T.; Barnett, N.L.; Papadimitriou, J.; Constable, I.J.; Lai, C.-M. Progressive Age-Related Changes Similar to Age-Related Macular Degeneration in a Transgenic Mouse Model. Am. J. Pathol. 2002, 161, 1515–1524. [Google Scholar] [CrossRef]
- Awano, T.; Katz, M.L.; O’Brien, D.P.; Taylor, J.F.; Evans, J.; Khan, S.; Sohar, I.; Lobel, P.; Johnson, G.S. A mutation in the cathepsin D gene (CTSD) in American Bulldogs with neuronal ceroid lipofuscinosis. Mol. Genet. Metab. 2006, 87, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Nakanishi, H.; Säftig, P.; Ezaki, J.; Isahara, K.; Ohsawa, Y.; Schulz-Schaeffer, W.; Watanabe, T.; Waguri, S.; Kametaka, S.; et al. Cathepsin D Deficiency Induces Lysosomal Storage with Ceroid Lipofuscin in Mouse CNS Neurons. J. Neurosci. 2000, 20, 6898–6906. [Google Scholar] [CrossRef] [PubMed]
- Götzl, J.K.; Mori, K.; Damme, M.; Fellerer, K.; Tahirovic, S.; Kleinberger, G.; Janssens, J.; Van Der Zee, J.; Lang, C.M.; Kremmer, E.; et al. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 2014, 127, 845–860. [Google Scholar] [CrossRef]
- Haapanen, A.; Ramadan, U.A.; Autti, T.; Joensuu, R.; Tyynelä, J. In vivo MRI reveals the dynamics of pathological changes in the brains of cathepsin D-deficient mice and correlates changes in manganese-enhanced MRI with microglial activation. Magn. Reson. Imaging 2007, 25, 1024–1031. [Google Scholar] [CrossRef]
- Partanen, S.; Haapanen, A.; Kielar, C.; Pontikis, C.; Alexander, N.; Inkinen, T.; Saftig, P.; Gillingwater, T.H.; Cooper, J.D.; Tyynelä, J. Synaptic Changes in the Thalamocortical System of Cathepsin D-Deficient Mice. J. Neuropathol. Exp. Neurol. 2008, 67, 16–29. [Google Scholar] [CrossRef]
- Nakanishi, H.; Zhang, J.; Koike, M.; Nishioku, T.; Okamoto, Y.; Kominami, E.; Von Figura, K.; Peters, C.; Yamamoto, K.; Saftig, P.; et al. Involvement of Nitric Oxide Released from Microglia-Macrophages in Pathological Changes of Cathepsin D-Deficient Mice. J. Neurosci. 2001, 21, 7526–7533. [Google Scholar] [CrossRef]
- Radke, J.; Stenzel, W.; Goebel, H.H. Human NCL Neuropathology. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2262–2266. [Google Scholar] [CrossRef] [PubMed]
- Thottath, J.; Vellarikkal, S.K.; Jayarajan, R.; Verma, A.; Manamel, M.; Singh, A.; Rajendran, V.R.; Sivasubbu, S.; Scaria, V. A novel cathepsin D mutation in 2 siblings with late infantile neuronal ceroid lipofuscinosis. Neurol. Genet. 2019, 5, e302. [Google Scholar] [CrossRef]
- Koike, M.; Shibata, M.; Ohsawa, Y.; Nakanishi, H.; Koga, T.; Kametaka, S.; Waguri, S.; Momoi, T.; Kominami, E.; Peters, C.; et al. Involvement of two different cell death pathways in retinal atrophy of cathepsin D-deficient mice. Mol. Cell. Neurosci. 2003, 22, 146–161. [Google Scholar] [CrossRef]
- Flachsbarth, K.; Kruszewski, K.; Jung, G.; Jankowiak, W.; Riecken, K.; Wagenfeld, L.; Richard, G.; Fehse, B.; Bartsch, U. Neural Stem Cell–Based Intraocular Administration of Ciliary Neurotrophic Factor Attenuates the Loss of Axotomized Ganglion Cells in Adult Mice. Investig. Opthalmol. Vis. Sci. 2014, 55, 7029–7039. [Google Scholar] [CrossRef]
- Dulz, S.; Bassal, M.; Flachsbarth, K.; Riecken, K.; Fehse, B.; Schlichting, S.; Bartsch, S.; Bartsch, U. Intravitreal Co-Administration of GDNF and CNTF Confers Synergistic and Long-Lasting Protection against Injury-Induced Cell Death of Retinal Ganglion Cells in Mice. Cells 2020, 9, 2082. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Henseler, M.; Klein, C.; Suzuki, K.; Harzer, K.; Sandhoff, K. Sphingolipid Activator Protein D (sap-D) Stimulates the Lysosomal Degradation of Ceramide in Vivo. Biochem. Biophys. Res. Commun. 1994, 200, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Jeon, C.-J.; Strettoi, E.; Masland, R.H. The Major Cell Populations of the Mouse Retina. J. Neurosci. 1998, 18, 8936–8946. [Google Scholar] [CrossRef]
- Palmer, D.N.; Barry, L.A.; Tyynelä, J.; Cooper, J.D. NCL disease mechanisms. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1882–1893. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.E.; Ekielian, T. Neuroinflammatory paradigms in lysosomal storage diseases. Front. Neurosci. 2015, 9, 417. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, R.; Zhang, J.; Koshiishi, I.; Suniarti, D.F.S.; Wu, Z.; Peters, C.; Schwake, M.; Uchiyama, Y.; Kira, J.-I.; Saftig, P.; et al. Involvement of lysosomal storage-induced p38 MAP kinase activation in the overproduction of nitric oxide by microglia in cathepsin D-deficient mice. Mol. Cell. Neurosci. 2007, 35, 573–584. [Google Scholar] [CrossRef]
- Groh, J.; Kühl, T.G.; Ip, C.W.; Nelvagal, H.R.; Sri, S.; Duckett, S.; Mirza, M.; Langmann, T.; Cooper, J.D.; Martini, R. Immune cells perturb axons and impair neuronal survival in a mouse model of infantile neuronal ceroid lipofuscinosis. Brain 2013, 136, 1083–1101. [Google Scholar] [CrossRef][Green Version]
- Groh, J.; Ribechini, E.; Stadler, D.; Schilling, T.; Lutz, M.B.; Martini, R. Sialoadhesin promotes neuroinflammation-related disease progression in two mouse models of CLN disease. Glia 2016, 64, 792–809. [Google Scholar] [CrossRef] [PubMed]
- Groh, J.; Berve, K.; Martini, R. Fingolimod and Teriflunomide Attenuate Neurodegeneration in Mouse Models of Neuronal Ceroid Lipofuscinosis. Mol. Ther. 2017, 25, 1889–1899. [Google Scholar] [CrossRef]
- Mirza, M.; Volz, C.; Karlstetter, M.; Langiu, M.; Somogyi, A.; Ruonala, M.O.; Tamm, E.R.; Jägle, H.; Langmann, T. Progressive Retinal Degeneration and Glial Activation in the CLN6nclf Mouse Model of Neuronal Ceroid Lipofuscinosis: A Beneficial Effect of DHA and Curcumin Supplementation. PLoS ONE 2013, 8, e75963. [Google Scholar] [CrossRef]
- Dannhausen, K.; Möhle, C.; Langmann, T. Immunomodulation with minocycline rescues retinal degeneration in juvenile neuronal ceroid lipofuscinosis mice highly susceptible to light damage. Dis. Model. Mech. 2018, 11, dmm033597. [Google Scholar] [CrossRef]
- Tyynelä, J.; Suopanki, J.; Baumann, M.; Haltia, M. Sphingolipid Activator Proteins (SAPs) in Neuronal Ceroid Lipofuscinoses (NCL). Neuropediatrics 1997, 28, 49–52. [Google Scholar] [CrossRef]
- Oswald, M.J.; Palmer, D.N.; Kay, G.W.; Shemilt, S.; Rezaie, P.; Cooper, J.D.; Shemilt, S.J.A. Glial activation spreads from specific cerebral foci and precedes neurodegeneration in presymptomatic ovine neuronal ceroid lipofuscinosis (CLN6). Neurobiol. Dis. 2005, 20, 49–63. [Google Scholar] [CrossRef]
- Kay, G.W.; Jay, N.P.; Palmer, D.N. The specific loss of GnRH-positive neurons from the hypothalamus of sheep with CLN6 neuronal ceroid lipofuscinosis occurs without glial activation and has only minor effects on reproduction. Neurobiol. Dis. 2011, 41, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Shevtsova, Z.; Garrido, M.; Weishaupt, J.; Saftig, P.; Bähr, M.; Lühder, F.; Kügler, S. CNS-Expressed Cathepsin D Prevents Lymphopenia in a Murine Model of Congenital Neuronal Ceroid Lipofuscinosis. Am. J. Pathol. 2010, 177, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.A.; Di Spiezio, A.; Thießen, N.; Schmidt, L.; Grötzinger, J.; Lüllmann-Rauch, R.; Damme, M.; Storck, S.E.; Pietrzik, C.U.; Fogh, J.; et al. Enzyme replacement therapy with recombinant pro-CTSD (cathepsin D) corrects defective proteolysis and autophagy in neuronal ceroid lipofuscinosis. Autophagy 2020, 16, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, U.; Galliciotti, G.; Jofre, G.F.; Jankowiak, W.; Hagel, C.; Braulke, T. Apoptotic Photoreceptor Loss and Altered Expression of Lysosomal Proteins in thenclfMouse Model of Neuronal Ceroid Lipofuscinosis. Investig. Opthalmol. Vis. Sci. 2013, 54, 6952–6959. [Google Scholar] [CrossRef]
- Jankowiak, W.; Brandenstein, L.; Dulz, S.; Hagel, C.; Storch, S.; Bartsch, U. Retinal Degeneration in Mice Deficient in the Lysosomal Membrane Protein CLN7. Investig. Opthalmol. Vis. Sci. 2016, 57, 4989. [Google Scholar] [CrossRef] [PubMed]
- Atiskova, Y.; Bartsch, S.; Danyukova, T.; Becker, E.; Hagel, C.; Storch, S.; Bartsch, U. Mice deficient in the lysosomal enzyme palmitoyl-protein thioesterase 1 (PPT1) display a complex retinal phenotype. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Settembre, C.; Ballabio, A. Lysosomal Adaptation: How the Lysosome Responds to External Cues. Cold Spring Harb. Perspect. Biol. 2014, 6, a016907. [Google Scholar] [CrossRef]
- Ballabio, A. The awesome lysosome. EMBO Mol. Med. 2016, 8, 73–76. [Google Scholar] [CrossRef]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017, 61, 733–749. [Google Scholar] [CrossRef]
- Marques, A.R.A.; Saftig, P. Lysosomal storage disorders—Challenges, concepts and avenues for therapy: Beyond rare diseases. J. Cell Sci. 2019, 132, jcs221739. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Medina, D.L.; Ballabio, A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol. Med. 2021, 13, e12836. [Google Scholar] [CrossRef] [PubMed]
- Koike, M.; Shibata, M.; Waguri, S.; Yoshimura, K.; Tanida, I.; Kominami, E.; Gotow, T.; Peters, C.; von Figura, K.; Mizushima, N.; et al. Participation of Autophagy in Storage of Lysosomes in Neurons from Mouse Models of Neuronal Ceroid-Lipofuscinoses (Batten Disease). Am. J. Pathol. 2005, 167, 1713–1728. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Thelen, M.; Daμμe, M.; Schweizer, M.; Hagel, C.; Wong, A.M.; Cooper, J.D.; Braulke, T.; Galliciotti, G. Disruption of the Autophagy-Lysosome Pathway Is Involved in Neuropathology of the nclf Mouse Model of Neuronal Ceroid Lipofuscinosis. PLoS ONE 2012, 7, e35493. [Google Scholar] [CrossRef]
- Carter-Dawson, L.D.; Lavail, M.M. Rods and cones in the mouse retina. II. Autoradiographic analysis of cell generation using tritiated thymidine. J. Comp. Neurol. 1979, 188, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Young, R.W. Cell differentiation in the retina of the mouse. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 1985, 212, 199–205. [Google Scholar] [CrossRef]
- Schmitz, F.; Tabares, L.; Khimich, D.; Strenzke, N.; De La Villa-Polo, P.; Castellano-Muñoz, M.; Bulankina, A.; Moser, T.; Fernández-Chacón, R.; Südhof, T.C. CSP -deficiency causes massive and rapid photoreceptor degeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 2926–2931. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, H.; Keksa-Goldsteine, V.; Ragauskas, S.; Kohlmann, P.; Singh, Y.; Savchencko, E.; Puranen, J.; Malm, T.; Kalesnykas, G.; Koistinaho, J.; et al. Retinal Degeneration In A Mouse Model Of CLN5 Disease Is Associated With Compromised Autophagy. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Bronson, R.T.; Donahue, L.R.; Johnson, K.R.; Tanner, A.; Lane, P.W.; Faust, J.R. Neuronal ceroid lipofuscinosis (nclf), a new disorder of the mouse linked to chromosome 9. Am. J. Med. Genet. 1998, 77, 289–297. [Google Scholar] [CrossRef]
- Bronson, R.T.; Lake, B.D.; Cook, S.; Taylor, S.; Davisson, M.T. Motor neuron degeneration of mice is a model of neuronal ceroid lipofuscinosis (Batten’s disease). Ann. Neurol. 1993, 33, 381–385. [Google Scholar] [CrossRef]
- Messer, A.; Plummer, J.; Wong, V.; Lavail, M. Retinal Degeneration in Motor Neuron Degeneration (mnd) Mutant Mice. Exp. Eye Res. 1993, 57, 637–641. [Google Scholar] [CrossRef]
- Guarneri, R.; Russo, D.; Cascio, C.; D’Agostino, S.; Galizzi, G.; Bigini, P.; Mennini, T.; Guarneri, P. Retinal oxidation, apoptosis and age- and sex-differences in the mnd mutant mouse, a model of neuronal ceroid lipofuscinosis. Brain Res. 2004, 1014, 209–220. [Google Scholar] [CrossRef]
- Wang, F.; Wang, H.; Tuan, H.-F.; Nguyen, D.H.; Sun, V.; Keser, V.; Bowne, S.J.; Sullivan, L.S.; Luo, H.; Zhao, L.; et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: Identification of a novel genotype-phenotype correlation and clinical refinements. Qual. Life Res. 2014, 133, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Roosing, S.; Born, L.I.V.D.; Sangermano, R.; Banfi, S.; Koenekoop, R.K.; Zonneveld-Vrieling, M.N.; Klaver, C.C.; Van Lith-Verhoeven, J.J.; Cremers, F.P.; Hollander, A.I.D.; et al. Mutations in MFSD8, Encoding a Lysosomal Membrane Protein, Are Associated with Nonsyndromic Autosomal Recessive Macular Dystrophy. Ophthalmology 2015, 122, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.N.; El-Asrag, M.E.; Ku, C.A.; Holder, G.E.; McKibbin, M.; Arno, G.; Poulter, J.A.; Carss, K.; Bommireddy, T.; Bagheri, S.; et al. Specific Alleles of CLN7/MFSD8, a Protein That Localizes to Photoreceptor Synaptic Terminals, Cause a Spectrum of Nonsyndromic Retinal Dystrophy. Investig. Opthalmol. Vis. Sci. 2017, 58, 2906–2914. [Google Scholar] [CrossRef]
- Ku, C.A.; Hull, S.; Arno, G.; Vincent, A.; Carss, K.; Kayton, R.; Weeks, D.; Anderson, G.W.; Geraets, R.; Parker, C.; et al. Detailed Clinical Phenotype and Molecular Genetic Findings in CLN3-Associated Isolated Retinal Degeneration. JAMA Ophthalmol. 2017, 135, 749–760. [Google Scholar] [CrossRef]
- Birtel, J.; Gliem, M.; Mangold, E.; Müller, P.L.; Holz, F.G.; Neuhaus, C.; Lenzner, S.; Zahnleiter, D.; Betz, C.; Eisenberger, T.; et al. Next-generation sequencing identifies unexpected genotype-phenotype correlations in patients with retinitis pigmentosa. PLoS ONE 2018, 13, e0207958. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.K.; Zhang, X.; Eintracht, J.; Zhang, D.; Arunachalam, S.; Thompson, J.A.; Chelva, E.; Mallon, D.; Chen, S.-C.; McLaren, T.; et al. Clinical and molecular characterization of non-syndromic retinal dystrophy due to c.175G>A mutation in ceroid lipofuscinosis neuronal 3 (CLN3). Doc. Ophthalmol. 2018, 138, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, V.M.; Nassisi, M.; Hernandez, C.S.; Méjécase, C.; El Shamieh, S.; Condroyer, C.; Antonio, A.; Meunier, I.; Andrieu, C.; Defoort-Dhellemmes, S.; et al. Retinal Phenotype of Patients With Isolated Retinal Degeneration Due to CLN3 Pathogenic Variants in a French Retinitis Pigmentosa Cohort. JAMA Ophthalmol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Strauss, O. The Retinal Pigment Epithelium. In Webvision: The Organization of the Retina and Visual System; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 1995. [Google Scholar]
- Zhong, Y.; Mohan, K.; Liu, J.; Al-Attar, A.; Lin, P.; Flight, R.M.; Sun, Q.; Warmoes, M.O.; Deshpande, R.R.; Liu, H.; et al. Loss of CLN3, the gene mutated in juvenile neuronal ceroid lipofuscinosis, leads to metabolic impairment and autophagy induction in retinal pigment epithelium. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165883. [Google Scholar] [CrossRef]
- Katz, M.L.; Johnson, G.S.; Tullis, G.E.; Lei, B. Phenotypic characterization of a mouse model of juvenile neuronal ceroid lipofuscinosis. Neurobiol. Dis. 2008, 29, 242–253. [Google Scholar] [CrossRef]
- Goebel, H.H.; Dopfmer, I. An ultrastructural study on retinal neural and pigment epithelial cells in ovine neuronal ceroid-lipofuscinosis. Ophthalmic Paediatr. Genet. 1990, 11, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Wavre-Shapton, S.T.; Calvi, A.A.; Turmaine, M.; Seabra, M.C.; Cutler, D.F.; Futter, C.E.; Mitchison, H.M. Photoreceptor phagosome processing defects and disturbed autophagy in retinal pigment epithelium of Cln3Δex1-6 mice modelling juvenile neuronal ceroid lipofuscinosis (Batten disease). Hum. Mol. Genet. 2015, 24, 7060–7074. [Google Scholar] [CrossRef]
- Von Eisenhart-Rothe, P.; Grubman, A.; Greferath, U.; Fothergill, L.J.; Jobling, A.I.; Phipps, J.A.; White, A.R.; Fletcher, E.L.; Vessey, K.A. Failure of Autophagy–Lysosomal Pathways in Rod Photoreceptors Causes the Early Retinal Degeneration Phenotype Observed inCln6nclfMice. Investig. Opthalmol. Vis. Sci. 2018, 59, 5082–5097. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.; Tullis, G.E.; Kirk, M.D.; Zhang, K.; Katz, M.L. Ocular phenotype in a mouse gene knockout model for infantile neuronal ceroid lipofuscinosis. J. Neurosci. Res. 2006, 84, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.L.; Coates, J.R.; Cooper, J.J.; O’Brien, D.P.; Jeong, M.; Narfström, K. Retinal Pathology in a Canine Model of Late Infantile Neuronal Ceroid Lipofuscinosis. Investig. Opthalmol.Vis. Sci. 2008, 49, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Groh, J.; Stadler, D.; Buttmann, M.; Martini, R. Non-invasive assessment of retinal alterations in mouse models of infantile and juvenile neuronal ceroid lipofuscinosis by spectral domain optical coherence tomography. Acta Neuropathol. Commun. 2014, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Volz, C.; Mirza, M.; Langmann, T.; Jägle, H. Retinal Function in Aging Homozygous Cln3 Δex7/8 Knock-In Mice. In Retinal Degenerative Diseases; Advances in Experimental Medicine and Biology Series; Springer: Berlin/Heidelberg, Germany, 2014; Volume 801, pp. 495–501. [Google Scholar] [CrossRef]
- Holthaus, S.-M.K.; Ribeiro, J.; Abelleira-Hervas, L.; Pearson, R.A.; Duran, Y.; Georgiadis, A.; Sampson, R.D.; Rizzi, M.; Hoke, J.; Maswood, R.; et al. Prevention of Photoreceptor Cell Loss in a Cln6 Mouse Model of Batten Disease Requires CLN6 Gene Transfer to Bipolar Cells. Mol. Ther. 2018, 26, 1343–1353. [Google Scholar] [CrossRef]
- Seigel, G.M.; Wagner, J.; Wronska, A.; Campbell, L.; Ju, W.; Zhong, N. Progression of early postnatal retinal pathology in a mouse model of neuronal ceroid lipofuscinosis. Eye 2004, 19, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Hafler, B.P.; Klein, Z.A.; Zhou, Z.J.; Strittmatter, S.M. Progressive retinal degeneration and accumulation of autofluorescent lipopigments in Progranulin deficient mice. Brain Res. 2014, 1588, 168–174. [Google Scholar] [CrossRef]
- Sappington, R.M.; Pearce, D.A.; Calkins, D.J. Optic Nerve Degeneration in a Murine Model of Juvenile Ceroid Lipofuscinosis. Investig. Opthalmol.Vis. Sci. 2003, 44, 3725–3731. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weimer, J.M.; Custer, A.W.; Benedict, J.W.; Alexander, N.A.; Kingsley, E.; Federoff, H.J.; Cooper, J.D.; Pearce, D.A. Visual deficits in a mouse model of Batten disease are the result of optic nerve degeneration and loss of dorsal lateral geniculate thalamic neurons. Neurobiol. Dis. 2006, 22, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Mutka, A.-L.; Haapanen, A.; Käkelä, R.; Lindfors, M.; Wright, A.K.; Inkinen, T.; Hermansson, M.; Rokka, A.; Corthals, G.; Jauhiainen, M.; et al. Murine cathepsin D deficiency is associated with dysmyelination/myelin disruption and accumulation of cholesteryl esters in the brain. J. Neurochem. 2009, 112, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.-Z.; Xiao, L.; Liu, Y.-J.; Shen, C.; Lou, H.-F.; Lv, Y.; Pan, S.-Y. Cathepsin D deficiency delays central nervous system myelination by inhibiting proteolipid protein trafficking from late endosome/lysosome to plasma membrane. Exp. Mol. Med. 2018, 50, e457. [Google Scholar] [CrossRef] [PubMed]
- Neverman, N.J.; Best, H.L.; Hofmann, S.L.; Hughes, S.M. Experimental therapies in the neuronal ceroid lipofuscinoses. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 2292–2300. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Geraets, R.D.; Koh, S.Y.; Hastings, M.L.; Kielian, T.; Pearce, D.A.; Weimer, J.M. Moving towards effective therapeutic strategies for Neuronal Ceroid Lipofuscinosis. Orphanet J. Rare Dis. 2016, 11, 1–13. [Google Scholar] [CrossRef]
- Johnson, T.B.; Cain, J.T.; White, K.A.; Ramirez-Montealegre, D.; Pearce, D.A.; Weimer, J.M. Therapeutic landscape for Batten disease: Current treatments and future prospects. Nat. Rev. Neurol. 2019, 15, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Mole, S.E.; Anderson, G.; Band, H.A.; Berkovic, S.F.; Cooper, J.D.; Holthaus, S.-M.K.; McKay, T.R.; Medina, D.L.; Rahim, A.A.; Schulz, A.; et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol. 2019, 18, 107–116. [Google Scholar] [CrossRef]
- Schulz, A.; Ajayi, T.; Specchio, N.; Reyes, E.D.L.; Gissen, P.; Ballon, D.; Dyke, J.P.; Cahan, H.; Slasor, P.; Jacoby, D.; et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N. Engl. J. Med. 2018, 378, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.S.; Tannous, B.A.; Deliolanis, N.C.; Hsich, G.; Morse, D.; Tung, C.-H.; Sena-Esteves, M.; Breakefield, X.O. Imaging gene delivery in a mouse model of congenital neuronal ceroid lipofuscinosis. Gene Ther. 2011, 18, 1173–1178. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Dilution | Company/Reference | Catalog Number | RRID |
|---|---|---|---|---|
| brain-specific homeobox/POU domain protein 3A (BRN-3A) | 1:200 | Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA | Sc-31984 | AB_2167511 |
| calbindin | 1:2000 | Sigma-Aldrich, St. Louis, MO, USA | C 9848 | AB_476894 |
| cathepsin D (CTSD) | 1:2000 | Santa Cruz Biotechnology, Inc. | Sc-6486 | AB_637896 |
| cathepsin X/Z/P (CTSZ) | 1:100 | R&D Systems GmbH | AF1033 | AB_2088116 |
| cluster of differentiation 68 (CD68) | 1:1000 | Bio Rad Laboratories, Kidlington, UK | MCA1957 | AB_322219 |
| arrestin | 1:5000 | Millipore, Temecula, CA, USA | AB15282 | AB_1163387 |
| glial fibrillary acidic protein (GFAP) | 1:500 | Dako Cytomation GmbH, Hamburg, Germany | Z0334 | AB_10013382 |
| ionized calcium-binding adapter molecule 1 (IBA1) | 1:500 | Wako Chemicals GmbH, Neuss, Germany | 019-19741 | AB_839504 |
| lysosomal-associated membrane protein 1 (LAMP1) | 1:2000 | Santa Cruz Biotechnology, Inc. | Sc-19992 | AB_2134495 |
| lysosomal-associated membrane protein 2 (LAMP2) | 1:200 | Developmental Studies Hybridoma Bank, lowa City, IA, USA | ABL93 | AB_2134767 |
| protein kinase C alpha (PKCα) | 1:500 | Santa Cruz Biotechnology, Inc. | Sc-208 | AB_2168668 |
| recoverin | 1:3000 | Milllipore, Temecula, CA, USA | AB5585 | AB_2253622 |
| saposin D | 1:4000 | Konrad Sandhoff, Bonn, Germany [39] | N/A | N/A |
| secretagogin (SCGN) | 1:2000 | BioVendor Research and Diagnostic Products | RD184120100 | AB_2034062 |
| sequestosome 1/p62 (SQSTM1/p62) | 1:1000 | Enzo Life Sciences GmbH, Lörrach, Germany | BML-PW9860 | AB_2196009 |
| subunit c of mitochondrial ATP synthase (SCMAS) | 1:1000 | Abcam, Cambridge, UK | Ab181243 | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bassal, M.; Liu, J.; Jankowiak, W.; Saftig, P.; Bartsch, U. Rapid and Progressive Loss of Multiple Retinal Cell Types in Cathepsin D-Deficient Mice—An Animal Model of CLN10 Disease . Cells 2021, 10, 696. https://doi.org/10.3390/cells10030696
Bassal M, Liu J, Jankowiak W, Saftig P, Bartsch U. Rapid and Progressive Loss of Multiple Retinal Cell Types in Cathepsin D-Deficient Mice—An Animal Model of CLN10 Disease . Cells. 2021; 10(3):696. https://doi.org/10.3390/cells10030696
Chicago/Turabian StyleBassal, Mahmoud, Junling Liu, Wanda Jankowiak, Paul Saftig, and Udo Bartsch. 2021. "Rapid and Progressive Loss of Multiple Retinal Cell Types in Cathepsin D-Deficient Mice—An Animal Model of CLN10 Disease " Cells 10, no. 3: 696. https://doi.org/10.3390/cells10030696
APA StyleBassal, M., Liu, J., Jankowiak, W., Saftig, P., & Bartsch, U. (2021). Rapid and Progressive Loss of Multiple Retinal Cell Types in Cathepsin D-Deficient Mice—An Animal Model of CLN10 Disease . Cells, 10(3), 696. https://doi.org/10.3390/cells10030696