
Genetic Deletion of Polo-Like Kinase 2 Induces a Pro-Fibrotic Pulmonary Phenotype

 ,
,
Abstract
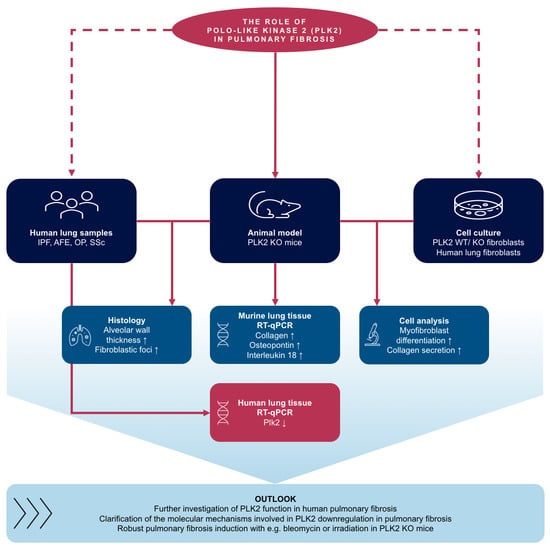
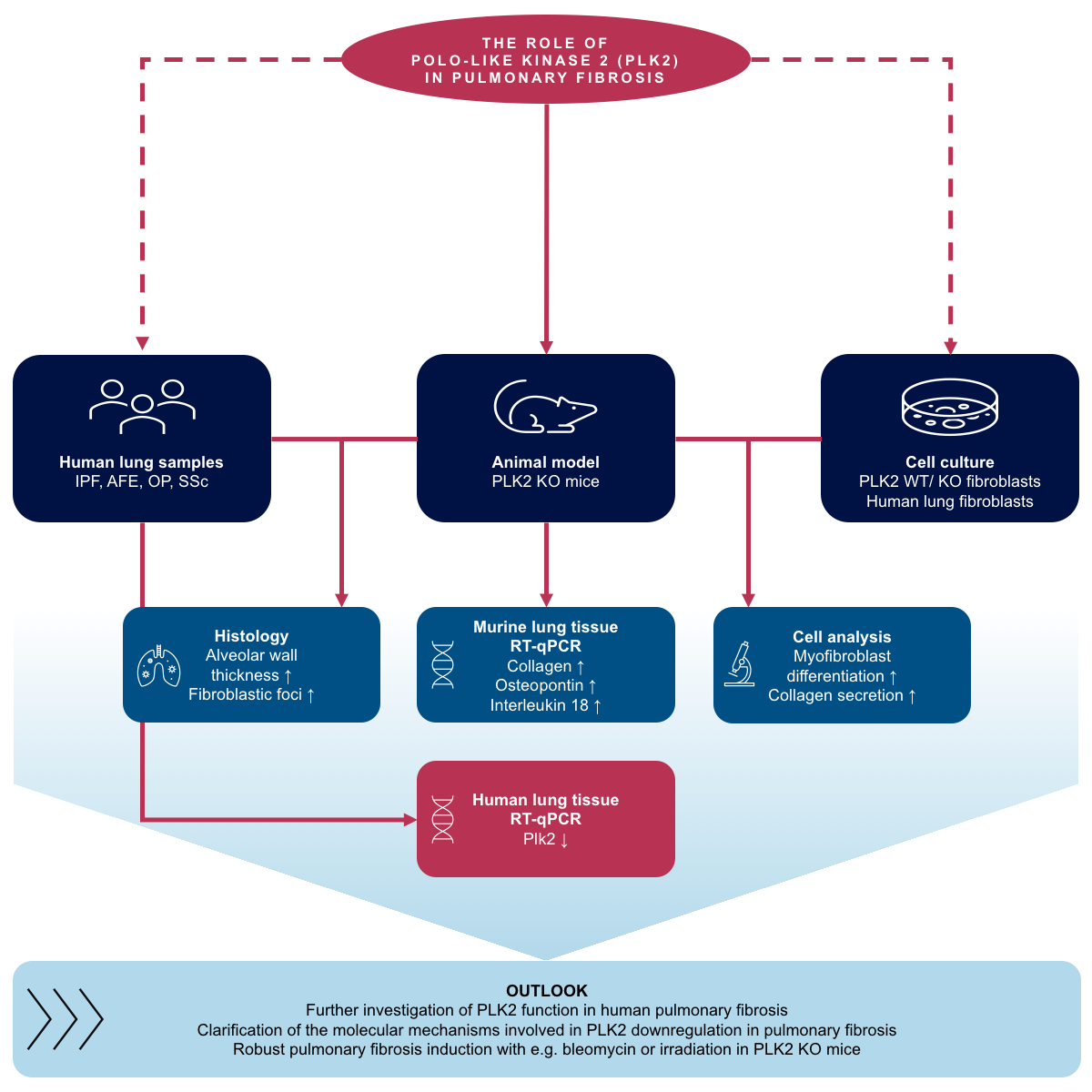
1. Introduction
2. Materials and Methods
2.1. PLK2 Mice
2.2. Human Sample Acquisition
2.3. SDS-PAGE, Western Blotting and Immunodetection
2.4. RNA Isolation, cDNA Synthesis and qPCR
2.5. Primary Fibroblast Isolation
2.6. Human Pulmonary MRC5 Fibroblasts
2.7. Cell Culture
2.8. Fibroblast Immunofluorescence Staining
2.9. Extracellular Collagen Deposition
2.10. Apoptosis Detection
2.11. Fibroblast Proliferation
2.12. Histology and Imaging
2.13. Units
2.14. Statistical Analysis
3. Results and Discussion
3.1. PLK2 Expression Is Reduced in Human Pulmonary Fibrosis
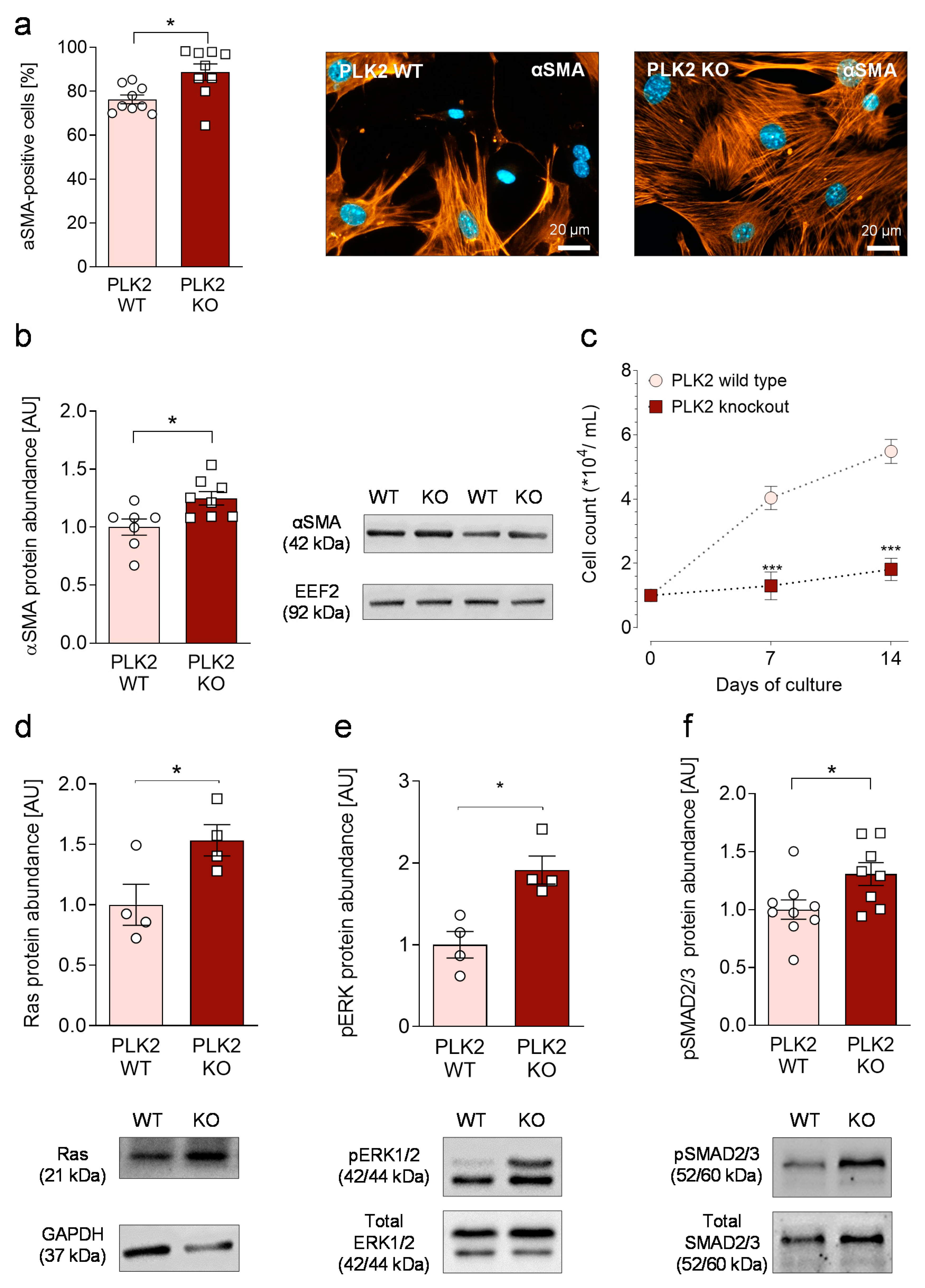
3.2. Primary PLK2 KO Fibroblasts Display a Myofibroblast Phenotype
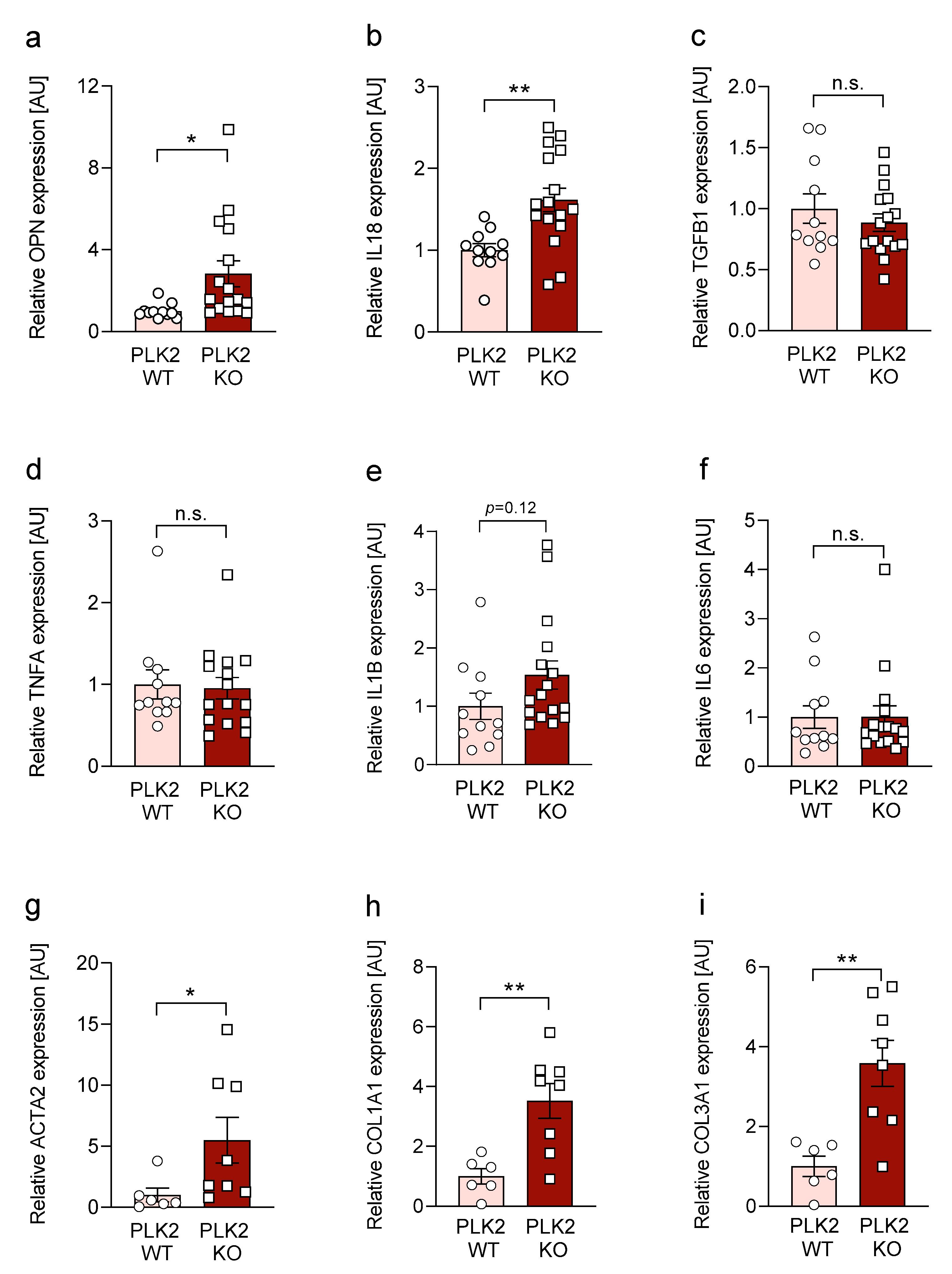
3.3. Expression of OPN and IL18 Is Elevated in PLK2 KO and Human IPF and AFE
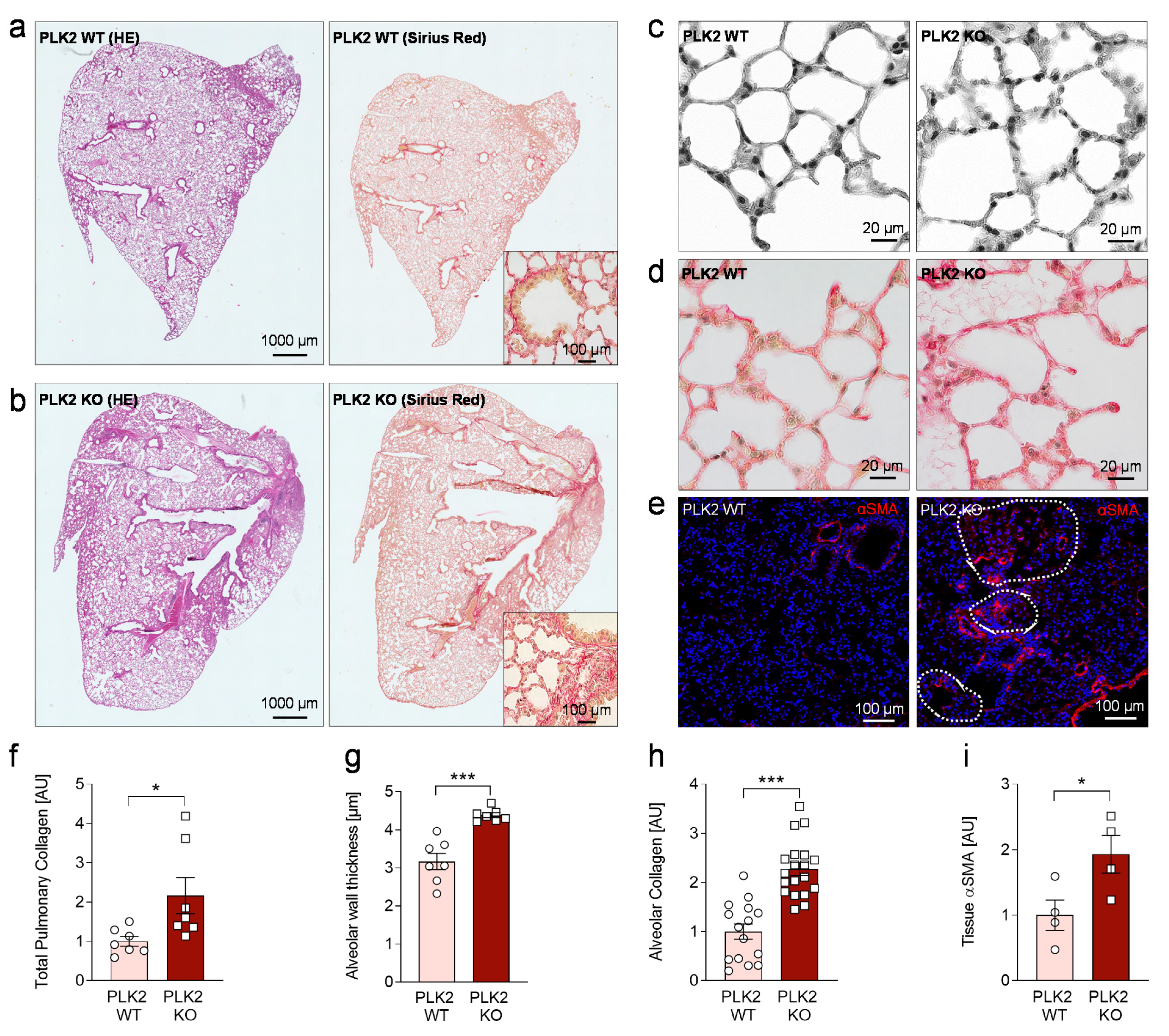
3.4. Histological Analysis of PLK2 KO Lungs
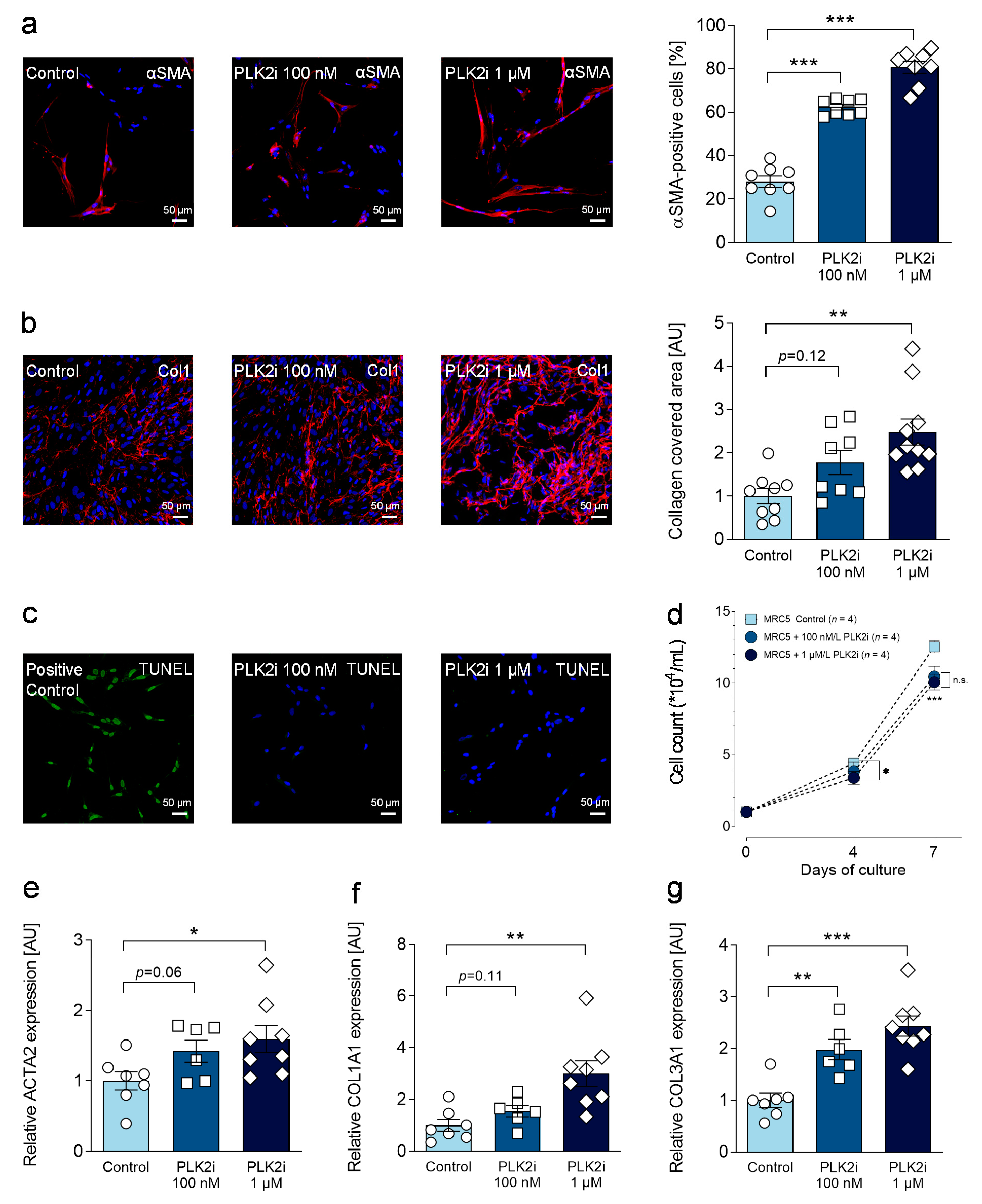
3.5. Pharmacological Inhibition of PLK2 Induces a Fibrotic Phenotype in Human Pulmonary Fibroblasts
3.6. Study Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wijsenbeek, M.; Cottin, V. Spectrum of Fibrotic Lung Diseases. N. Engl. J. Med. 2020, 383, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.B.; Hogaboam, C.M. Murine models of pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2008, 294, L152–L160. [Google Scholar] [CrossRef]
- Mathai, S.K.; Schwartz, D.A. Translational research in pulmonary fibrosis. Transl. Res. 2019, 209, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Distler, J.H.W.; Györfi, A.-H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef]
- Mosher, C.L.; Mentz, R.J. Cardiovascular implications of idiopathic pulmonary fibrosis: A way forward together? Am. Heart J. 2020, 226, 69–74. [Google Scholar] [CrossRef]
- Kizer, J.R.; Zisman, D.A.; Blumenthal, N.P.; Kotloff, R.M.; Kimmel, S.E.; Strieter, R.M.; Arcasoy, S.M.; Ferrari, V.A.; Hansen-Flaschen, J. Association Between Pulmonary Fibrosis and Coronary Artery Disease. Arch. Intern. Med. 2004, 164, 551–556. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kant, T.A.; Emig, R.; Rausch, J.S.E.; Newe, M.; Schubert, M.; Künzel, K.; Winter, L.; Klapproth, E.; Peyronnet, R.; et al. Repurposing mesalazine against cardiac fibrosis in vitro. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, M.; Lorenz, V.; Ivanek, R.; Della Verde, G.; Gaudiello, E.; Marsano, A.; Pfister, O.; Kuster, G.M. Polo-Like Kinase 2 is Dynamically Regulated to Coordinate Proliferation and Early Lineage Specification Downstream of Yes-Associated Protein 1 in Cardiac Progenitor Cells. J. Am. Heart Assoc. 2017, 6, e005920. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Charron, J.; Erikson, R.L. Role of Plk2 (Snk) in Mouse Development and Cell Proliferation. Mol. Cell. Biol. 2003, 23, 6936–6943. [Google Scholar] [CrossRef]
- Li, J.; Ma, W.; Wang, P.-Y.; Hurley, P.J.; Bunz, F.; Hwang, P.M. Polo-like kinase 2 activates an antioxidant pathway to promote the survival of cells with mitochondrial dysfunction. Free. Radic. Biol. Med. 2014, 73, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Wang, P.-Y.; Ma, W.; Sung, H.J.; Matoba, S.; Hwang, P.M. Polo-like kinases mediate cell survival in mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 14542–14546. [Google Scholar] [CrossRef] [PubMed]
- Kuenzel, S.; Klapproth, E.; Kuenzel, K.; Piorkowski, C.; Mayr, M.; Wagner, M.; Dobrev, D.; Rausch, J.; Ravens, U.; Weber, S.; et al. PLK2 is a novel regulator of osteopontin-driven fibrosis and diastolic dysfunction in permanent atrial fibrillation. Eur. Heart J. 2020, 41, ehaa946-3671. [Google Scholar] [CrossRef]
- Renzoni, E.A.; Abraham, D.J.; Howat, S.; Shi-Wen, X.; Sestini, P.; Bou-Gharios, G.; Wells, A.U.; Veeraraghavan, S.; Nicholson, A.G.; Denton, C.P.; et al. Gene expression profiling reveals novel TGFβ targets in adult lung fibroblasts. Respir. Res. 2004, 5, 24. [Google Scholar] [CrossRef]
- Mohamed, I.A.; Gadeau, A.-P.; Hasan, A.; Abdulrahman, N.; Mraiche, F. Osteopontin: A Promising Therapeutic Target in Cardiac Fibrosis. Cells 2019, 8, 1558. [Google Scholar] [CrossRef] [PubMed]
- Agnholt, J.; Kelsen, J.; Schack, L.; Hvas, C.L.; Dahlerup, J.F.; Sørensen, E.S. Osteopontin, a Protein with Cytokine-like Properties, is Associated with Inflammation in Crohn’s Disease. Scand. J. Immunol. 2007, 65, 453–460. [Google Scholar] [CrossRef]
- Kitasato, Y.; Hoshino, T.; Okamoto, M.; Kato, S.; Koda, Y.; Nagata, N.; Kinoshita, M.; Koga, H.; Yoon, D.-Y.; Asao, H.; et al. Enhanced Expression of Interleukin-18 and its Receptor in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2004, 31, 619–625. [Google Scholar] [CrossRef]
- Länger, F.; Stark, H.; Braubach, P.; Ackermann, M.; Hussein, K.; Teiken, K.; Maegel, L.; Kuehnel, M.; Jonigk, D. Schädigungsmuster interstitieller Lungenerkrankungen. Der. Pathol. 2018, 39, 262–271. [Google Scholar] [CrossRef]
- Ackermann, M.; Stark, H.; Neubert, L.; Schubert, S.; Borchert, P.; Linz, F.; Wagner, W.L.; Stiller, W.; Wielpütz, M.; Hoefer, A.; et al. Morphomolecular motifs of pulmonary neoangiogenesis in interstitial lung diseases. Eur. Respir. J. 2019, 55, 1900933. [Google Scholar] [CrossRef]
- El-Armouche, A.; Wittköpper, K.; Degenhardt, F.; Weinberger, F.; Didié, M.; Melnychenko, I.; Grimm, M.; Peeck, M.; Zimmermann, W.H.; Unsöld, B.; et al. Phosphatase inhibitor-1-deficient mice are protected from catecholamine-induced arrhythmias and myocardial hypertrophy. Cardiovasc. Res. 2008, 80, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Künzel, S.R.; Schaeffer, C.; Sekeres, K.; Mehnert, C.S.; Wall, S.M.S.; Newe, M.; Kämmerer, S.; El-Armouche, A. Ultrasonic-augmented Primary Adult Fibroblast Isolation. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Kamentsky, L.; Jones, T.R.; Fraser, A.; Bray, M.-A.; Logan, D.J.; Madden, K.L.; Ljosa, V.; Rueden, C.; Eliceiri, K.W.; Carpen-ter, A.E. Improved structure, function and compatibility for CellProfiler: Modular high-throughput image analysis software. Bioinformatics 2011, 27, 1179–1180. [Google Scholar] [CrossRef]
- Rangarajan, S.; Bone, N.B.; Zmijewska, A.A.; Jiang, S.; Park, D.W.; Bernard, K.; Locy, M.L.; Ravi, S.; Deshane, J.; Mannon, R.B.; et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat. Med. 2018, 24, 1121–1127. [Google Scholar] [CrossRef]
- Wernig, G.; Chen, S.-Y.; Cui, L.; Van Neste, C.; Tsai, J.M.; Kambham, N.; Vogel, H.; Natkunam, Y.; Gilliland, D.G.; Nolan, G.; et al. Unifying mechanism for different fibrotic diseases. Proc. Natl. Acad. Sci. USA 2017, 114, 4757–4762. [Google Scholar] [CrossRef] [PubMed]
- Baum, J.; Duffy, H.S. Fibroblasts and Myofibroblasts: What Are We Talking About? J. Cardiovasc. Pharmacol. 2011, 57, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Künzel, S.R.; Rausch, J.S.E.; Schäffer, C.; Hoffmann, M.; Künzel, K.; Klapproth, E.; Kant, T.; Herzog, N.; Küpper, J.; Lorenz, K.; et al. Modeling atrial fibrosis in vitro —Generation and characterization of a novel human atrial fibroblast cell line. FEBS Open Bio. 2020, 10, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-Smooth muscle actin expression upregulates Fi-broblast contractile activity. Mol. Biol. Cell 2001, 12, 2730–2741. [Google Scholar] [CrossRef]
- Pakshir, P.; Noskovicova, N.; Lodyga, M.; Son, D.O.; Schuster, R.; Goodwin, A.; Karvonen, H.; Hinz, B. The myofibroblast at a glance. J. Cell Sci. 2020, 133, jcs227900. [Google Scholar] [CrossRef]
- Hinz, B.; McCulloch, C.A.; Coelho, N.M. Mechanical regulation of myofibroblast phenoconversion and collagen contraction. Exp. Cell Res. 2019, 379, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Poulet, C.; Künzel, S.; Büttner, E.; Lindner, D.; Westermann, D.; Ravens, U. Altered physiological functions and ion currents in atrial fibroblasts from patients with chronic atrial fibrillation. Physiol. Rep. 2016, 4, e12681. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.-J.; et al. Fibroblast-specific TGF-β–Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Luo, S.; Hieu, T.B.; Ma, F.; Yu, Y.; Cao, Z.; Wang, M.; Wu, W.; Mao, Y.; Rose, P.; Law, B.Y.-K.; et al. ZYZ-168 alleviates cardiac fibrosis after myocardial infarction through inhibition of ERK1/2-dependent ROCK1 activation. Sci. Rep. 2017, 7, 43242. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.-Q.; Liu, T.; Cao, F.-J.; Xu, Y.-Q. Upregulated Ras/Raf/ERK1/2 signaling pathway: A new hope in the repair of spinal cord injury. Neural. Regen. Res. 2015, 10, 792–796. [Google Scholar] [CrossRef]
- Wang, A.-X.; Qi, X.-Y. Targeting RAS/RAF/MEK/ERK signaling in metastatic melanoma. IUBMB Life 2013, 65, 748–758. [Google Scholar] [CrossRef]
- Leivonen, S.-K.; Häkkinen, L.; Liu, D.; Kähäri, V.-M. Smad3 and Extracellular Signal-Regulated Kinase 1/2 Coordinately Mediate Transforming Growth Factor-β-Induced Expression of Connective Tissue Growth Factor in Human Fibroblasts. J. Investig. Dermatol. 2005, 124, 1162–1169. [Google Scholar] [CrossRef]
- Tee, J.K.; Peng, F.; Tan, Y.L.; Yu, B.; Ho, H.K. Magnesium Isoglycyrrhizinate Ameliorates Fibrosis and Disrupts TGF-β-Mediated SMAD Pathway in Activated Hepatic Stellate Cell Line LX2. Front. Pharmacol. 2018, 9, 1018. [Google Scholar] [CrossRef]
- Lee, K.J.; Hoe, H.-S.; Pak, D.T. Plk2 Raps up Ras to subdue synapses. Small GTPases 2011, 2, 162–166. [Google Scholar] [CrossRef]
- Lee, K.J.; Lee, Y.; Rozeboom, A.; Lee, J.-Y.; Udagawa, N.; Hoe, H.-S.; Pak, D.T. Requirement for Plk2 in Orchestrated Ras and Rap Signaling, Homeostatic Structural Plasticity, and Memory. Neuron 2011, 69, 957–973. [Google Scholar] [CrossRef]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Shochet, G.E.; Brook, E.; Israeli-Shani, L.; Edelstein, E.; Shitrit, D. Fibroblast paracrine TNF-α signaling elevates integrin A5 expression in idiopathic pulmonary fibrosis (IPF). Respir. Res. 2017, 18, 1–12. [Google Scholar] [CrossRef]
- Papiris, S.A.; Tomos, I.P.; Karakatsani, A.; Spathis, A.; Korbila, I.; Analitis, A.; Kolilekas, L.; Kagouridis, K.; Loukides, S.; Karakitsos, P.; et al. High levels of IL-6 and IL-8 characterize early-on idiopathic pulmonary fibrosis acute exacerbations. Cytokine 2018, 102, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Kolb, M.; Margetts, P.J.; Anthony, D.C.; Pitossi, F.; Gauldie, J. Transient expression of IL-1β induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Investig. 2001, 107, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Gibson, K.; Cisneros, J.; Richards, T.J.; Yang, Y.; Becerril, C.; Yousem, S.; Herrera, I.; Ruiz, V.; Selman, M.; et al. Up-Regulation and Profibrotic Role of Osteopontin in Human Idiopathic Pulmonary Fibrosis. PLoS Med. 2005, 2, e251. [Google Scholar] [CrossRef]
- Gui, X.; Qiu, X.; Xie, M.; Tian, Y.; Min, C.; Huang, M.; HongYan, W.; Chen, T.; Zhang, X.; Chen, J.; et al. Prognostic Value of Serum Osteopontin in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. BioMed Res. Int. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Jonigk, D.; Stark, H.; Braubach, P.; Neubert, L.; Shin, H.; Izykowski, N.; Welte, T.; Janciauskiene, S.; Warnecke, G.; Haverich, A.; et al. Morphological and molecular motifs of fibrosing pulmonary injury patterns. J. Pathol. Clin. Res. 2019, 5, 256–271. [Google Scholar] [CrossRef]
- Miller, E.R.; Putman, R.K.; Diaz, A.A.; Xu, H.; Estépar, R.S.J.; Araki, T.; Nishino, M.; De Frías, S.P.; Hida, T.; Ross, J.; et al. Increased Airway Wall Thickness in Interstitial Lung Abnormalities and Idiopathic Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2019, 16, 447–454. [Google Scholar] [CrossRef]
- Specks, U.; Nerlich, A.; Colby, T.V.; Wiest, I.; Timpl, R. Increased expression of type VI collagen in lung fibrosis. Am. J. Respir. Crit. Care Med. 1995, 151, 1956–1964. [Google Scholar] [CrossRef]
- Doyle, T.J.; Hunninghake, G.M.; Rosas, I.O. Subclinical Interstitial Lung Disease. Am. J. Respir. Crit. Care Med. 2012, 185, 1147–1153. [Google Scholar] [CrossRef]
- Moore, B.B.; Lawson, W.E.; Oury, T.D.; Sisson, T.H.; Raghavendran, K.; Hogaboam, C.M. Animal Models of Fibrotic Lung Disease. Am. J. Respir. Cell Mol. Biol. 2013, 49, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Lawson, W.E.; Cheng, D.-S.; Degryse, A.L.; Tanjore, H.; Polosukhin, V.V.; Xu, X.C.; Newcomb, D.C.; Jones, B.R.; Roldan, J.; Lane, K.B.; et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. USA 2011, 108, 10562–10567. [Google Scholar] [CrossRef]
- Burns, T.F.; Fei, P.; Scata, K.A.; Dicker, D.T.; El-Deiry, W.S. Silencing of the Novel p53 Target Gene Snk/Plk2 Leads to Mitotic Catastrophe in Paclitaxel (Taxol)-Exposed Cells. Mol. Cell. Biol. 2003, 23, 5556–5571. [Google Scholar] [CrossRef] [PubMed]
- Warnke, S.; Kemmler, S.; Hames, R.S.; Tsai, H.-L.; Hoffmann-Rohrer, U.; Fry, A.M.; Hoffmann, I. Polo-like Kinase-2 Is Required for Centriole Duplication in Mammalian Cells. Curr. Biol. 2004, 14, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Roach, K.M.; Sutcliffe, A.; Matthews, L.; Elliott, G.; Newby, C.; Amrani, Y.; Bradding, P. A model of human lung fibrogenesis for the assessment of anti-fibrotic strategies in idiopathic pulmonary fibrosis. Sci. Rep. 2018, 8, 342. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | IPF | AFE | OP | SSC | |
|---|---|---|---|---|---|
| Male | 3 | 3 | 3 | 3 | 3 |
| Female | 2 | 2 | 2 | 1 | 1 |
| Age [y] | 54.8 ± 7.8 | 53.4 ± 5.2 | 58.2 ± 1.9 | 58.25 ± 8.4 | 45.8 ± 1.9 |
| Primary Antibodies | ||||
|---|---|---|---|---|
| Protein | Dilution | Conjugate/Source | Product-Nr. | Usage |
| αSMA | 1:200 | Mouse | A5228 | ICC 1/ IHC 2 |
| SMAD2/3 | 1:1000 | Rabbit | #3102 | WB 3 |
| Phospho-SMAD2/3 | 1:1000 | Rabbit | #8828 | WB |
| ERK 1/2 | 1:1000 | Rabbit | #9102S | WB |
| Phospho-ERK 1/2 | 1:1000 | Rabbit | #9101S | WB |
| Ras | 1:1000 | Rabbit | #3339S | WB |
| Collagen 1 A1 | 1:100 | Goat | MBS316282 | ICC |
| Vimentin | 1:200 | Rabbit | ab137321 | ICC |
| Discoidin Domaine Receptor 2 (DDR2) | 1:200 | Mouse | ab63337 | ICC |
| Secondary Antibodies | ||||
| Goat-anti-rabbit | 1:10,000 | Peroxidase | 111-035-045 | WB |
| Alexa fluor 546 | 1:400 | Streptavidin | Z25004 | ICC |
| (Goat-anti-mouse) | ||||
| Alexa fluor 546 | 1:400 | Streptavidin | Z25304 | ICC |
| (Goat-anti-rabbit) | ||||
| Step | Action | Additional Information |
|---|---|---|
| 1 | Permeabilization (15 min, 0.1% Triton-X) | Room temperature |
| 2 | Washing (wash twice with PBS) | Room temperature |
| 3 | Blocking (1 h, 10% FCS) | Room temperature |
| 4 | Primary antibody (1 h in humidified chamber) | Room temperature or 4 °C overnight |
| 5 | Washing (wash twice with PBS) | Room temperature |
| 6 | Secondary antibody and DAPI (1 h in humidified chamber) | Room temperature |
| 7 | Washing (wash twice with PBS) | Room temperature |
| 8 | Mounting with 10 µL Fluoromount G | Room temperature |
| 9 | Storage until imaging | 4 °C |
| Control | IPF | AFE | OP | SSC | |
|---|---|---|---|---|---|
| n | 3 | 5 | 4 | 4 | 4 |
| Mean | 1.00 | 0.2 | 0.34 | 0.18 | 0.17 |
| SEM | 0.40 | 0.10 | 0.11 | 0.12 | 0.05 |
| p-Value (vs. Control) | n.a. | 0.020 | 0.038 | 0.023 | 0.021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kant, T.A.; Newe, M.; Winter, L.; Hoffmann, M.; Kämmerer, S.; Klapproth, E.; Künzel, K.; Kühnel, M.P.; Neubert, L.; El-Armouche, A.; et al. Genetic Deletion of Polo-Like Kinase 2 Induces a Pro-Fibrotic Pulmonary Phenotype. Cells 2021, 10, 617. https://doi.org/10.3390/cells10030617
Kant TA, Newe M, Winter L, Hoffmann M, Kämmerer S, Klapproth E, Künzel K, Kühnel MP, Neubert L, El-Armouche A, et al. Genetic Deletion of Polo-Like Kinase 2 Induces a Pro-Fibrotic Pulmonary Phenotype. Cells. 2021; 10(3):617. https://doi.org/10.3390/cells10030617
Chicago/Turabian StyleKant, Theresa A., Manja Newe, Luise Winter, Maximilian Hoffmann, Susanne Kämmerer, Erik Klapproth, Karolina Künzel, Mark P. Kühnel, Lavinia Neubert, Ali El-Armouche, and et al. 2021. "Genetic Deletion of Polo-Like Kinase 2 Induces a Pro-Fibrotic Pulmonary Phenotype" Cells 10, no. 3: 617. https://doi.org/10.3390/cells10030617
APA StyleKant, T. A., Newe, M., Winter, L., Hoffmann, M., Kämmerer, S., Klapproth, E., Künzel, K., Kühnel, M. P., Neubert, L., El-Armouche, A., & Künzel, S. R. (2021). Genetic Deletion of Polo-Like Kinase 2 Induces a Pro-Fibrotic Pulmonary Phenotype. Cells, 10(3), 617. https://doi.org/10.3390/cells10030617