
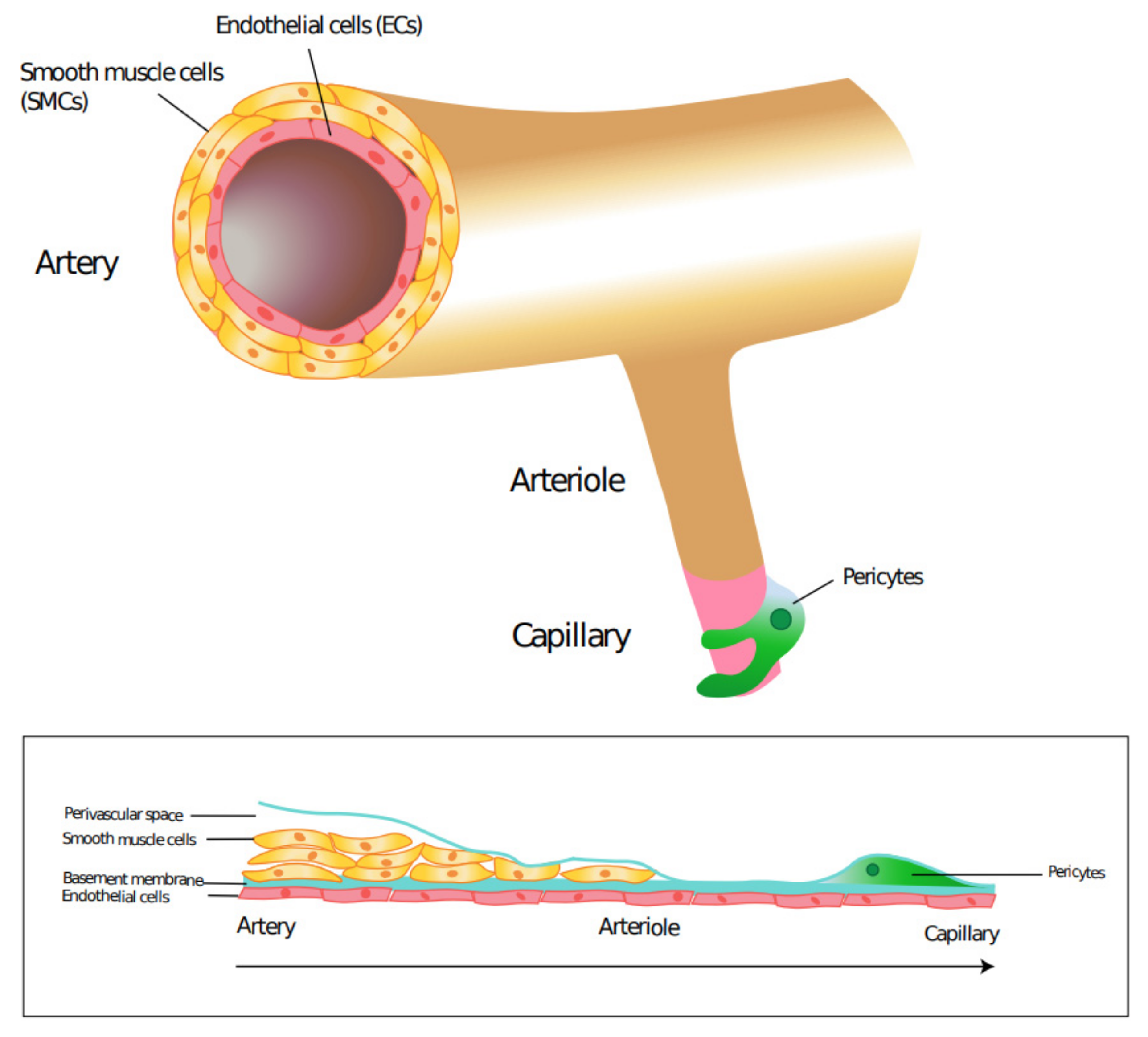
Mural Cells: Potential Therapeutic Targets to Bridge Cardiovascular Disease and Neurodegeneration

Abstract

1. Introduction
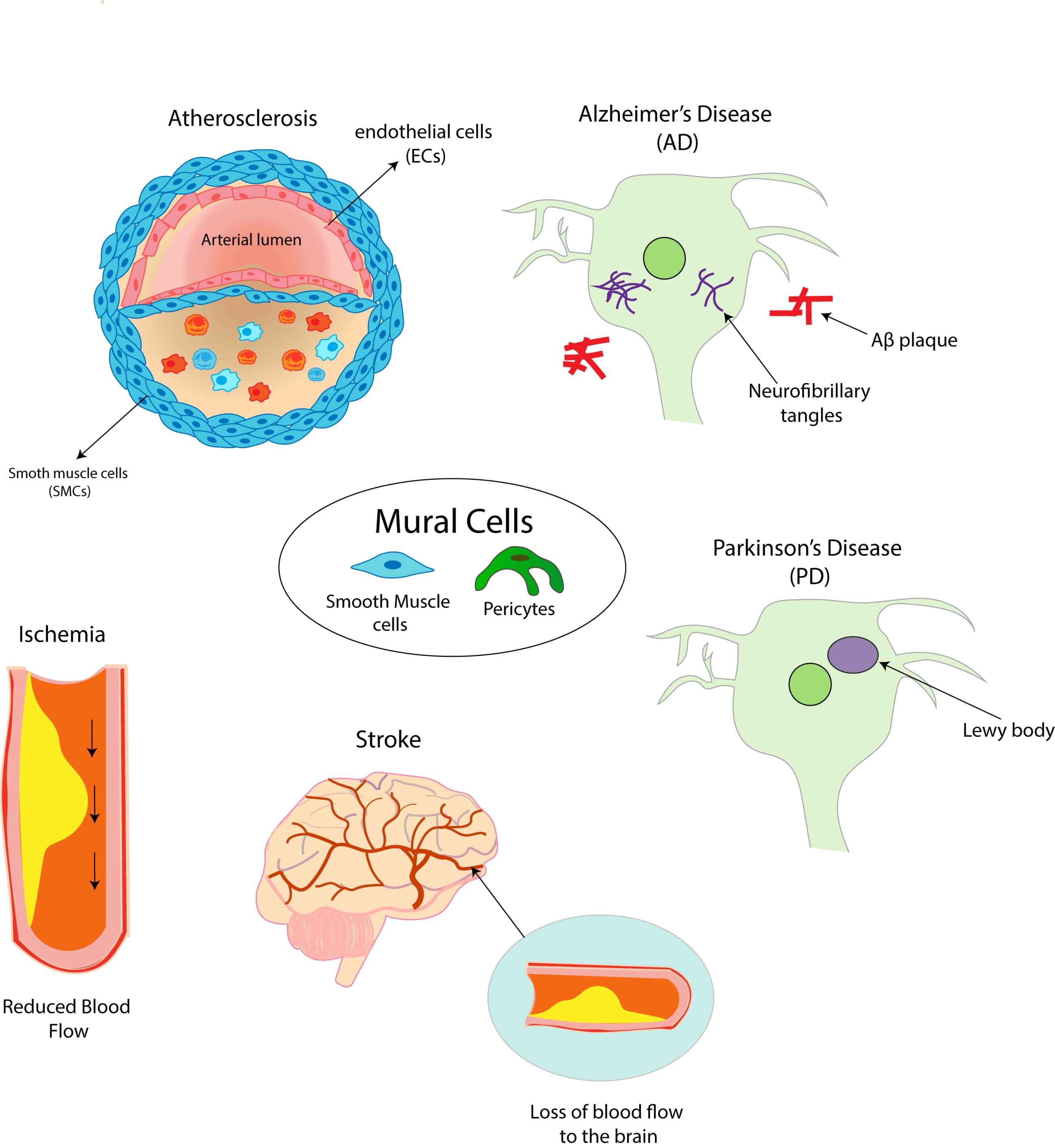
2. Mural Cells and Cardiovascular Disease
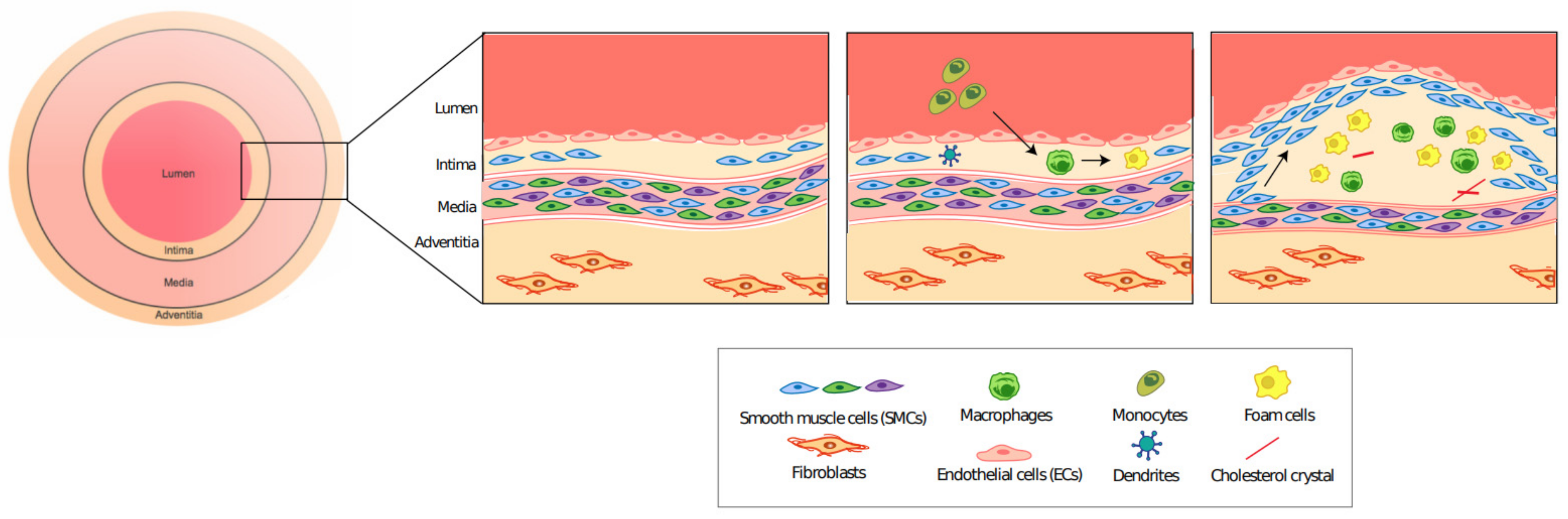
2.1. Atherosclerosis: A General Overview
2.2. VSMC Plasticity Plays an Important Role in the Progression of Atherosclerosis
2.3. PDGF Signaling in Atherosclerosis
2.4. Notch Signaling in Atherosclerosis
2.5. Role of Inflammation in Atherosclerosis
2.6. Ischemia
2.7. Stroke
3. Mural Cells and Neurological Disease
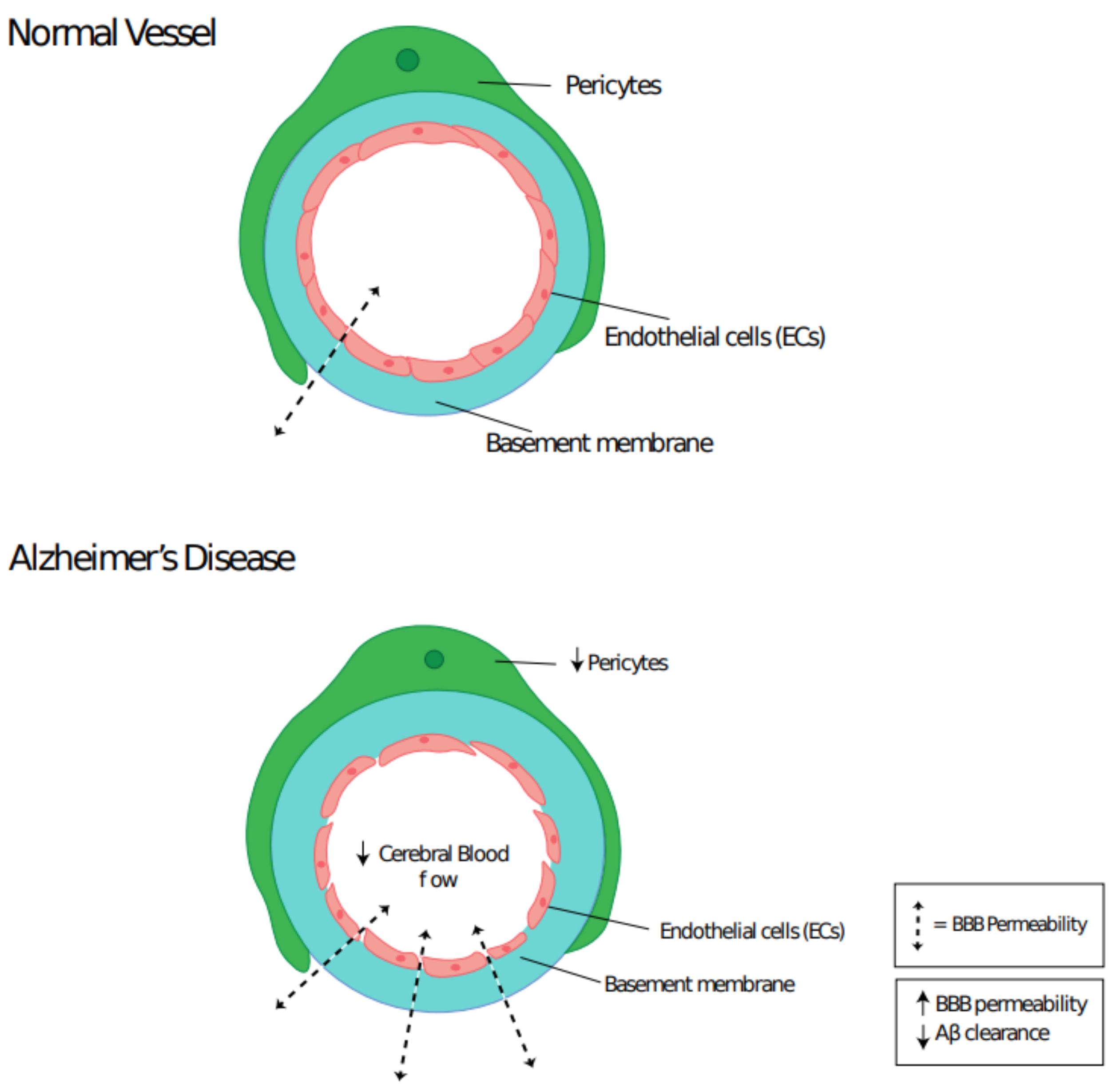
3.1. Pericyte Dysfunction and Neurodegenerative Disease
3.2. Role of Mural Cells in Alzheimer’s Disease
3.3. Role of Mural Cells in Parkinson’s Disease
3.4. Mural Cell Plasticity in Neurodegenerative Diseases
3.5. PDGF Signaling in Neurodegenerative Diseases
3.6. Notch Signaling in Neurodegenerative Diseases
3.7. Role of Neuroinflammation In Neurodegenerative Diseases
4. Conserved Therapeutic Targets: A Clinical Perspective
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Flamme, I.; Von Reutern, M.; Drexler, H.C.; Syed-Ali, S.; Risau, W. Overexpression of Vascular Endothelial Growth Factor in the Avian Embryo Induces Hypervascularization and Increased Vascular Permeability without Alterations of Embryonic Pattern Formation. Dev. Biol. 1995, 171, 399–414. [Google Scholar] [CrossRef]
- Flamme, I.; Risau, W. Induction of vasculogenesis and hematopoiesis in vitro. Development 1992, 116, 435–439. [Google Scholar] [PubMed]
- Mazurek, R.; Dave, J.M.; Chandran, R.R.; Misra, A.; Sheikh, A.Q.; Greif, D.M. Vascular Cells in Blood Vessel Wall Development and Disease. Adv. Pharmacol. 2017, 78, 323–350. [Google Scholar] [CrossRef]
- Delong, C.; Sharma, S. Physiology, Peripheral Vascular Resistance. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Zhuge, Y.; Zhang, J.; Qian, F.; Wen, Z.; Niu, C.; Xu, K.; Ji, H.; Rong, X.; Chu, M.; Jia, C. Role of smooth muscle cells in Cardiovascular Disease. Int. J. Biol. Sci. 2020, 16, 2741–2751. [Google Scholar] [CrossRef]
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Feng, Z.; Chandran, R.R.; Kabir, I.; Rotllan, N.; Aryal, B.; Sheikh, A.Q.; Ding, L.; Qin, L.; Fernandez-Hernando, C.; et al. Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nat. Commun. 2018, 9, 2073. [Google Scholar] [CrossRef]
- De La Torre, J. Impaired brain microcirculation may trigger Alzheimer’s disease. Neurosci. Biobehav. Rev. 1994, 18, 397–401. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Dehay, B. Alpha-synuclein spreading in Parkinson’s disease. Front. Neuroanat. 2014, 8. [Google Scholar] [CrossRef]
- Hofman, A.; Ott, A.; Breteler, M.M.; Bots, M.L.; Slooter, A.J.; van Harskamp, F.; van Duijn, C.N.; Van Broeckhoven, C.; Grobbee, D.E. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet 1997, 349, 151–154. [Google Scholar] [CrossRef]
- World Health Organization. “Cardiovascular Diseases (CVDs)”. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 24 December 2020).
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Basatemur, G.L.; Jørgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef] [PubMed]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of Plaque Formation and Rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Frostegård, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 1–117. [Google Scholar] [CrossRef]
- Rognoni, A.; Cavallino, C.; Veia, A.; Bacchini, S.; Rosso, R.; Facchini, M.; Secco, G.G.; Lupi, A.; Nardi, F.; Rametta, F.; et al. Pathophysiology of Atherosclerotic Plaque Development. Cardiovasc. Hematol. Agents Med. Chem. 2015, 13, 10–13. [Google Scholar] [CrossRef]
- Wingo, A.P.; Fan, W.; Duong, D.M.; Gerasimov, E.S.; Dammer, E.B.; White, B.; Thambisetty, M.; Troncoso, J.C.; Schneider, J.A.; Lah, J.J.; et al. Cerebral atherosclerosis contributes to Alzheimer’s dementia independently of its hallmark amyloid and tau pathologies. bioRxiv 2019, 793349. [Google Scholar] [CrossRef]
- Lusis, A.J. Genetics of atherosclerosis. Trends Genet. 2012, 28, 267–275. [Google Scholar] [CrossRef]
- Hussain, M.M.; Mahley, R.W.; Boyles, J.K.; Fainaru, M.; Brecht, W.J.; Lindquist, P.A. Chylomicron-chylomicron remnant clearance by liver and bone marrow in rabbits. Factors that modify tissue-specific uptake. J. Biol. Chem. 1989, 264, 9571–9582. [Google Scholar] [CrossRef]
- Tamminen, M.; Mottino, G.; Qiao, J.H.; Breslow, J.L.; Frank, J.S. Ultrastructure of Early Lipid Accumulation in ApoE-Deficient Mice. Arter. Thromb. Vasc. Biol. 1999, 19, 847–853. [Google Scholar] [CrossRef]
- Ishibashi, S.; Goldstein, J.L.; Brown, M.S.; Herz, J.; Burns, D.K. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J. Clin. Investig. 1994, 93, 1885–1893. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Bergheanu, S.C.; Bodde, M.C.; Jukema, J.W. Pathophysiology and treatment of atherosclerosis. Neth. Heart J. 2017, 25, 231–242. [Google Scholar] [CrossRef]
- Brophy, M.L.; Dong, Y.; Wu, H.; Rahman, H.N.A.; Song, K.; Chen, H. Eating the Dead to Keep Atherosclerosis at Bay. Front. Cardiovasc. Med. 2017, 4, 2. [Google Scholar] [CrossRef]
- Majesky, M.W. Developmental Basis of Vascular Smooth Muscle Diversity. Arter. Thromb. Vasc. Biol. 2007, 27, 1248–1258. [Google Scholar] [CrossRef]
- DeBakey, M.E.; Glaeser, D.H. Patterns of atherosclerosis: Effect of risk factors on recurrence and survival-analysis of 11,890 cases with more than 25-year follow-up. Am. J. Cardiol. 2000, 85, 1045–1053. [Google Scholar] [CrossRef]
- Dobnikar, L.; Taylor, A.L.; Chappell, J.; Oldach, P.; Harman, J.L.; Oerton, E.; Dzierzak, E.; Bennett, M.R.; Spivakov, M.; Jørgensen, H.F. Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Tang, J.; Wang, H.; Huang, X.; Li, F.; Zhu, H.; Li, Y.; He, L.; Zhang, H.; Pu, W.; Liu, K.; et al. Arterial Sca1+ Vascular Stem Cells Generate De Novo Smooth Muscle for Artery Repair and Regeneration. Cell Stem Cell 2020, 26, 81–96.e4. [Google Scholar] [CrossRef]
- Tang, Z.; Wang, A.; Yuan, F.; Yan, Z.; Liu, B.; Chu, J.S.; Helms, J.A.; Li, S. Differentiation of multipotent vascular stem cells contributes to vascular diseases. Nat. Commun. 2012, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Wang, D.; Xu, K.; Wang, J.; Zhang, Z.; Yang, L.; Yang, G.-Y.; Li, S. Contribution of Vascular Cells to Neointimal Formation. PLoS ONE 2017, 12, e0168914. [Google Scholar] [CrossRef]
- Kramann, R.; Goettsch, C.; Wongboonsin, J.; Iwata, H.; Schneider, R.K.; Kuppe, C.; Kaesler, N.; Chang-Panesso, M.; Machado, F.G.; Gratwohl, S.; et al. Adventitial MSC-like Cells Are Progenitors of Vascular Smooth Muscle Cells and Drive Vascular Calcification in Chronic Kidney Disease. Cell Stem Cell 2016, 19, 628–642. [Google Scholar] [CrossRef]
- Alencar, G.F.; Owsiany, K.M.; Haskins, R.; Baylis, R.A.; Finn, A.V.; McNamara, C.A.; Zunder, E.R.; Venkata, V.; Pasterkamp, G.; Björkegren, J.; et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 2020, 142, 2045–2059. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, A.S.; Vellarikkal, S.K.; Edelman, E.R.; Nguyen, L.; Subramanian, A.; Ellinor, P.T.; Regev, A.; Kathiresan, S.; Gupta, R.M. Single-Cell Analysis of the Normal Mouse Aorta Reveals Functionally Distinct Endothelial Cell Populations. Circulation 2019, 140, 147–163. [Google Scholar] [CrossRef]
- Newman, A.A.C.; Serbulea, V.; Baylis, R.A.; Shankman, L.S.; Bradley, X.; Alencar, G.F.; Owsiany, K.; Deaton, R.A.; Karnewar, S.; Shamsuzzaman, S.; et al. Multiple cell types contribute to the atherosclerotic lesion fibrous cap by PDGFRβ and bioenergetic mechanisms. Nat. Metab. 2021, 3, 166–181. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Fisher, E.A. Tipping the cap away from danger. Nat. Metab. 2021, 3, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Pahk, K.; Joung, C.; Jung, W.B.; Song, H.Y.; Park, J.Y.; Byun, J.W.; Lee, Y.-S.; Paeng, J.C.; Kim, C.; Kim, S.; et al. Visualization of Synthetic Vascular Smooth Muscle Cells in Atherosclerotic Carotid Rat Arteries by F-18 FDG PET. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl. Acad. Sci. USA 2003, 100, 13531–13536. [Google Scholar] [CrossRef]
- Speer, M.Y.; Yang, H.-Y.; Brabb, T.; Leaf, E.; Look, A.; Lin, W.-L.; Frutkin, A.; Dichek, D.; Giachelli, C.M. Smooth Muscle Cells Give Rise to Osteochondrogenic Precursors and Chondrocytes in Calcifying Arteries. Circ. Res. 2009, 104, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of Intimal Smooth Muscle Cells to Cholesterol Accumulation and Macrophage-Like Cells in Human Atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.-Y.; Haynes, P.A.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth Muscle Cell Phenotypic Transition Associated with Calcification. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef]
- Vengrenyuk, Y.; Nishi, H.; Long, X.; Ouimet, M.; Savji, N.; Martinez, F.O.; Cassella, C.P.; Moore, K.J.; Ramsey, S.A.; Miano, J.M.; et al. Cholesterol Loading Reprograms the MicroRNA-143/145–Myocardin Axis to Convert Aortic Smooth Muscle Cells to a Dysfunctional Macrophage-Like Phenotype. Arter. Thromb. Vasc. Biol. 2015, 35, 535–546. [Google Scholar] [CrossRef]
- Naik, V.; Leaf, E.M.; Hu, J.H.; Yang, H.-Y.; Nguyen, N.B.; Giachelli, C.M.; Speer, M.Y. Sources of cells that contribute to atherosclerotic intimal calcification: An in vivo genetic fate mapping study. Cardiovasc. Res. 2012, 94, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.C.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Cherepanova, O.A.; Gomez, D.; Shankman, L.S.; Swiatlowska, P.; Williams, J.; Sarmento, O.F.; Alencar, G.F.; Hess, D.L.; Bevard, M.H.; Greene, E.S.; et al. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat. Med. 2016, 22, 657–665. [Google Scholar] [CrossRef]
- Hannink, M.; Donoghue, D.J. Structure and function of platelet-derived growth factor (PDGF) and related proteins. Biochim. Biophys. Acta (BBA) Rev. Cancer 1989, 989, 1–10. [Google Scholar] [CrossRef]
- Ross, R.; Glomset, J.; Kariya, B.; Harker, L. A Platelet-Dependent Serum Factor That Stimulates the Proliferation of Arterial Smooth Muscle Cells In Vitro. Proc. Natl. Acad. Sci. USA 1974, 71, 1207–1210. [Google Scholar] [CrossRef]
- Sano, H.; Sudo, T.; Yokode, M.; Murayama, T.; Kataoka, H.; Takakura, N.; Nishikawa, S.; Nishikawa, S.-I.; Kita, T. Functional Blockade of Platelet-Derived Growth Factor Receptor-β but Not of Receptor-α Prevents Vascular Smooth Muscle Cell Accumulation in Fibrous Cap Lesions in Apolipoprotein E–Deficient Mice. Circulation 2001, 103, 2955–2960. [Google Scholar] [CrossRef]
- Wan, W.; Ding, Y.; Xie, Z.; Li, Q.; Yan, F.; Budbazar, E.; Pearce, W.J.; Hartman, R.; Obenaus, A.; Zhang, J.H.; et al. PDGFR-β modulates vascular smooth muscle cell phenotype via IRF-9/SIRT-1/NF-κB pathway in subarachnoid hemorrhage rats. Br. J. Pharmacol. 2018, 39, 1369–1380. [Google Scholar] [CrossRef]
- He, C.; Medley, S.C.; Hu, T.; Hinsdale, M.E.; Lupu, F.; Virmani, R.; Olson, L.E. PDGFRβ signalling regulates local inflammation and synergizes with hypercholesterolaemia to promote atherosclerosis. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef]
- D’Souza, B.; Meloty-Kapella, L.; Weinmaster, G. Canonical and Non-Canonical Notch Ligands. Protein Kinases Dev. Dis. 2010, 92, 73–129. [Google Scholar]
- Iso, T.; Hamamori, Y.; Kedes, L. Notch Signaling in Vascular Development. Arter. Thromb. Vasc. Biol. 2003, 23, 543–553. [Google Scholar] [CrossRef]
- Xue, Y.; Gao, X.; Lindsell, C.E.; Norton, C.R.; Chang, B.; Hicks, C.; Gendron-Maguire, M.; Rand, E.B.; Weinmaster, G.; Gridley, T. Embryonic Lethality and Vascular Defects in Mice Lacking the Notch Ligand Jagged1. Hum. Mol. Genet. 1999, 8, 723–730. [Google Scholar] [CrossRef]
- Krebs, L.T.; Xue, Y.; Norton, C.R.; Shutter, J.R.; Maguire, M.; Sundberg, J.P.; Gallahan, D.; Closson, V.; Kitajewski, J.; Callahan, R.; et al. Notch signaling is essential for vascular morphogenesis in mice. Genome Res. 2000, 14, 1343–1352. [Google Scholar]
- Lindner, V.; Booth, C.; Prudovsky, I.; Small, D.; Maciag, T.; Liaw, L. Members of the Jagged/Notch Gene Families Are Expressed in Injured Arteries and Regulate Cell Phenotype via Alterations in Cell Matrix and Cell-Cell Interaction. Am. J. Pathol. 2001, 159, 875–883. [Google Scholar] [CrossRef]
- Li, Y.; Takeshita, K.; Gridley, T.; Liao, J.K.; Liu, P.-Y.; Satoh, M.; Oyama, N.; Mukai, Y.; Chin, M.T.; Krebs, L.; et al. Smooth Muscle Notch1 Mediates Neointimal Formation After Vascular Injury. Circulation 2009, 119, 2686–2692. [Google Scholar] [CrossRef] [PubMed]
- Redmond, E.M.; Liu, W.; Hamm, K.; Hatch, E.; Cahill, P.A.; Morrow, D. Perivascular Delivery of Notch 1 siRNA Inhibits Injury-Induced Arterial Remodeling. PLoS ONE 2014, 9, e84122. [Google Scholar] [CrossRef]
- Quillard, T.; Devallière, J.; Coupel, S.; Charreau, B. Inflammation dysregulates Notch signaling in endothelial cells: Implication of Notch2 and Notch4 to endothelial dysfunction. Biochem. Pharmacol. 2010, 80, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Briot, A.; Civelek, M.; Seki, A.; Hoi, K.; Mack, J.J.; Lee, S.D.; Kim, J.; Hong, C.; Yu, J.; Fishbein, G.A.; et al. Endothelial NOTCH1 is suppressed by circulating lipids and antagonizes inflammation during atherosclerosis. J. Exp. Med. 2015, 212, 2147–2163. [Google Scholar] [CrossRef] [PubMed]
- Keuylian, Z.; De Baaij, J.H.F.; Glorian, M.; Rouxel, C.; Merlet, E.; Lipskaia, L.; Blaise, R.; Mateo, V.; Limon, I. The Notch Pathway Attenuates Interleukin 1β (IL1β)-mediated Induction of Adenylyl Cyclase 8 (AC8) Expression during Vascular Smooth Muscle Cell (VSMC) Trans-differentiation. J. Biol. Chem. 2012, 287, 24978–24989. [Google Scholar] [CrossRef] [PubMed]
- Ragot, H.; Monfort, A.; Chatziantoniou, C.; Samuel, J.-L.; Baudet, M.; Azibani, F.; Fazal, L.; Merval, R.; Polidano, E.; Cohen-Solal, A.; et al. Loss of Notch3 Signaling in Vascular Smooth Muscle Cells Promotes Severe Heart Failure Upon Hypertension. Hypertension 2016, 68, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.; Sweeney, C.; Birney, Y.A.; Cummins, P.M.; Walls, D.; Redmond, E.M.; Cahill, P.A. Cyclic Strain Inhibits Notch Receptor Signaling in Vascular Smooth Muscle Cells In Vitro. Circ. Res. 2005, 96, 567–575. [Google Scholar] [CrossRef]
- Davis-Knowlton, J.; Turner, J.E.; Turner, A.; Damian-Loring, S.; Hagler, N.; Henderson, T.; Emery, I.F.; Bond, K.; Duarte, C.W.; Vary, C.P.H.; et al. Characterization of smooth muscle cells from human atherosclerotic lesions and their responses to Notch signaling. Lab. Investig. 2019, 99, 290–304. [Google Scholar] [CrossRef]
- Fukuda, D.; Aikawa, E.; Aster, J.C.; Hotamisligil, G.S.; Yagita, H.; Aikawa, M.; Swirski, F.K.; Novobrantseva, T.I.; Kotelianski, V.; Gorgun, C.Z.; et al. Notch ligand Delta-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proc. Natl. Acad. Sci. USA 2012, 109, E1868–E1877. [Google Scholar] [CrossRef]
- Karasawa, T.; Takahashi, M. Role of NLRP3 Inflammasomes in Atherosclerosis. J. Atheroscler. Thromb. 2017, 24, 443–451. [Google Scholar] [CrossRef]
- Alexander, M.R.; Murgai, M.; Moehle, C.W.; Owens, G.K. Interleukin-1β modulates smooth muscle cell phenotype to a distinct inflammatory state relative to PDGF-DD via NF-κB-dependent mechanisms. Physiol. Genom. 2012, 44, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Vromman, A.; Ruvkun, V.; Shvartz, E.; Wojtkiewicz, G.; Masson, G.S.; Tesmenitsky, Y.; Folco, E.; Gram, H.; Nahrendorf, M.; Swirski, F.K.; et al. Stage-dependent differential effects of interleukin-1 isoforms on experimental atherosclerosis. Eur. Heart J. 2019, 40, 2482–2491. [Google Scholar] [CrossRef] [PubMed]
- Baylis, R.A.; Gomez, D.; Mallat, Z.; Pasterkamp, G.; Owens, G.K. The CANTOS Trial. Arter. Thromb. Vasc. Biol. 2017, 37, e174–e177. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.; Bernardo, A.S.; Trotter, M.W.B.; Pedersen, R.A.; Sinha, S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin–dependent disease susceptibility. Nat. Biotechnol. 2012, 30, 165–173. [Google Scholar] [CrossRef]
- Ornellas, F.M.; Ornellas, D.S.; Martini, S.V.; Castiglione, R.C.; Ventura, G.M.; Rocco, P.R.; Gutfilen, B.; De Souza, S.A.; Takiya, C.M.; Morales, M.M. Bone Marrow–Derived Mononuclear Cell Therapy Accelerates Renal Ischemia-Reperfusion Injury Recovery by Modulating Inflammatory, Antioxidant and Apoptotic Related Molecules. Cell. Physiol. Biochem. 2017, 41, 1736–1752. [Google Scholar] [CrossRef]
- Ye, G.; Fu, Q.; Jiang, L.; Li, Z. Vascular smooth muscle cells activate PI3K/Akt pathway to attenuate myocardial ischemia/reperfusion-induced apoptosis and autophagy by secreting bFGF. Biomed. Pharmacother. 2018, 107, 1779–1785. [Google Scholar] [CrossRef] [PubMed]
- Tadi, P.; Lui, F. Acute Stroke. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Khaku, A.S.; Tadi, P. Cerebrovascular Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Saver, J.L. Time Is Brain—Quantified. Stroke 2006, 37, 263–266. [Google Scholar] [CrossRef]
- Maddahi, A.; Edvinsson, L. Enhanced expressions of microvascular smooth muscle receptors after focal cerebral ischemia occur via the MAPK MEK/ERK pathway. BMC Neurosci. 2008, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Arimura, K.; Ago, T.; Kamouchi, M.; Nakamura, K.; Ishitsuka, K.; Kuroda, J.; Sugimori, H.; Ooboshi, H.; Sasaki, T.; Kitazono, T. PDGF Receptor β Signaling in Pericytes Following Ischemic Brain Injury. Curr. Neurovasc. Res. 2012, 9, 1–9. [Google Scholar] [CrossRef]
- Yemisci, M.; Gursoy-Ozdemir, Y.; Vural, A.; Can, A.; Topalkara, K.; Dalkara, T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 2009, 15, 1031–1037. [Google Scholar] [CrossRef]
- Cai, W.; Liu, H.; Zhao, J.; Chen, L.Y.; Chen, J.; Lu, Z.; Hu, X. Pericytes in Brain Injury and Repair After Ischemic Stroke. Transl. Stroke Res. 2017, 8, 107–121. [Google Scholar] [CrossRef]
- Duz, B.; Oztas, E.; Erginay, T.; Erdogan, E.; Gonul, E. The effect of moderate hypothermia in acute ischemic stroke on pericyte migration: An ultrastructural study. Cryobiology 2007, 55, 279–284. [Google Scholar] [CrossRef]
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef]
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Rink, C.; Khanna, S. Significance of Brain Tissue Oxygenation and the Arachidonic Acid Cascade in Stroke. Antioxid. Redox Signal. 2011, 14, 1889–1903. [Google Scholar] [CrossRef]
- Meschia, J.F.; Brott, T. Ischaemic stroke. Eur. J. Neurol. 2018, 25, 35–40. [Google Scholar] [CrossRef]
- Jersey, A.M.; Foster, D.M. Cerebral Aneurysm. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Wang, Y.; Meng, R.; Liu, G.; Cao, C.; Chen, F.; Jin, K.; Ji, X.; Cao, G. Intracranial atherosclerotic disease. Neurobiol. Dis. 2019, 124, 118–132. [Google Scholar] [CrossRef]
- Huntington disease: A single-gene degenerative disorder of the striatum. Dialog. Clin. Neurosci. 2016, 18, 91–98. [CrossRef]
- Lista, S.; O’Bryant, S.E.; Blennow, K.; Dubois, B.; Hugon, J.; Zetterberg, H.; Hampel, H. Biomarkers in Sporadic and Familial Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 47, 291–317. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.; Lim, K.-L. Genetic Insights into Sporadic Parkinson’s Disease Pathogenesis. Curr. Genomics 2014, 14, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Kumar, S.; Walter, J. Phosphorylation of amyloid beta (Aβ) peptides—A trigger for formation of toxic aggregates in Alzheimer’s disease. Aging 2011, 3, 803–812. [Google Scholar] [CrossRef]
- Nikfarjam, S.; Jouravleva, E.V.; Anisimov, M.A.; Woehl, T.J. Effects of Protein Unfolding on Aggregation and Gelation in Lysozyme Solutions. Biomolecules 2020, 10, 1262. [Google Scholar] [CrossRef]
- Wanga, J.; Dickson, D.W.; Trojanowski, J.Q.; Lee, V.M.-Y. The Levels of Soluble versus Insoluble Brain Aβ Distinguish Alzheimer’s Disease from Normal and Pathologic Aging. Exp. Neurol. 1999, 158, 328–337. [Google Scholar] [CrossRef]
- Araki, K.; Yagi, N.; Aoyama, K.; Choong, C.-J.; Hayakawa, H.; Fujimura, H.; Nagai, Y.; Goto, Y.; Mochizuki, H. Parkinson’s disease is a type of amyloidosis featuring accumulation of amyloid fibrils of α-synuclein. Proc. Natl. Acad. Sci. USA 2019, 116, 17963–17969. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Mishra, A.; Hall, C.N.; O’Farrell, F.M.; Dalkara, T. What is a pericyte? Br. J. Pharmacol. 2016, 36, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Sagare, A.P.; Zlokovic, B.V. The Pericyte: A Forgotten Cell Type with Important Implications for Alzheimer’s Disease? Brain Pathol. 2014, 24, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Mukouyama, Y.-S. Tissue Specific Origin, Development, and Pathological Perspectives of Pericytes. Front. Cardiovasc. Med. 2018, 5, 78. [Google Scholar] [CrossRef]
- Crouch, E.E.; Liu, C.; Silva-Vargas, V.; Doetsch, F. Regional and Stage-Specific Effects of Prospectively Purified Vascular Cells on the Adult V-SVZ Neural Stem Cell Lineage. J. Neurosci. 2015, 35, 4528–4539. [Google Scholar] [CrossRef]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef]
- Hill, R.A.; Tong, L.; Yuan, P.; Murikinati, S.; Gupta, S.; Grutzendler, J. Regional Blood Flow in the Normal and Ischemic Brain Is Controlled by Arteriolar Smooth Muscle Cell Contractility and Not by Capillary Pericytes. Neuron 2015, 87, 95–110. [Google Scholar] [CrossRef]
- Damisah, E.C.; Hill, R.A.; Tong, L.; Murray, K.N.; Grutzendler, J. A fluoro-Nissl dye identifies pericytes as distinct vascular mural cells during in vivo brain imaging. Nat. Neurosci. 2017, 20, 1023–1032. [Google Scholar] [CrossRef]
- Nehls, V.; Drenckhahn, D. Heterogeneity of microvascular pericytes for smooth muscle type alpha-actin. J. Cell Biol. 1991, 113, 147–154. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 2018, 21, 1318–1331. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef]
- Farrall, A.J.; Wardlaw, J.M. Blood–brain barrier: Ageing and microvascular disease–systematic review and meta-analysis. Neurobiol. Aging 2009, 30, 337–352. [Google Scholar] [CrossRef]
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of Amyloid-β Peptide Across the Blood-Brain Barrier: Implication for Therapies in Alzheimers Disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.D.; Zlokovic, B.V. Deficiency in Mural Vascular Cells Coincides with Blood-Brain Barrier Disruption in Alzheimer’s Disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Debbins, J.P.; Malek-Ahmadi, M.; Pipe, J.G.; Maze, S.; Belden, C.; Maarouf, C.L.; Hunter, J.M.; Kokjohn, T.A.; Walker, D.G.; et al. Cerebral blood flow in Alzheimer’s disease. Vasc. Health Risk Manag. 2012, 8, 599–611. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, J.; Mussivan, T. Can disturbed brain microcirculation cause Alzheimer’s disease? Neurol. Res. 1993, 15, 146–153. [Google Scholar] [CrossRef]
- Tublin, J.M.; Adelstein, J.M.; Del Monte, F.; Combs, C.K.; Wold, L.E. Getting to the Heart of Alzheimer Disease. Circ. Res. 2019, 124, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Review Article: Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef]
- Sims, R.; Hill, M.; Williams, J. The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 2020, 23, 311–322. [Google Scholar] [CrossRef]
- De Roeck, A.; Van Broeckhoven, C.; Sleegers, K. The role of ABCA7 in Alzheimer’s disease: Evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 2019, 138, 201–220. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. apoE isoform–specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Investig. 2008, 118, 4002–4013. [Google Scholar] [CrossRef] [PubMed]
- Sakae, N.; Liu, C.-C.; Shinohara, M.; Frisch-Daiello, J.; Ma, L.; Yamazaki, Y.; Tachibana, M.; Younkin, L.; Kurti, A.; Carrasquillo, M.M.; et al. ABCA7 Deficiency Accelerates Amyloid-β Generation and Alzheimer’s Neuronal Pathology. J. Neurosci. 2016, 36, 3848–3859. [Google Scholar] [CrossRef]
- Tcw, J.; Goate, A.M. Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2016, 7, a024539. [Google Scholar] [CrossRef]
- Guerreiro, R.J.; Lohmann, E.; Kinsella, E.; Brás, J.M.; Luu, N.; Gurunlian, N.; Dursun, B.; Bilgic, B.; Santana, I.; Hanagasi, H.; et al. Exome sequencing reveals an unexpected genetic cause of disease: NOTCH3 mutation in a Turkish family with Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1008.e17–1008.e23. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Mez, J.; Vardarajan, B.N.; Staley, L.; Chung, J.; Zhang, X.; Farrell, J.J.; Rynkiewicz, M.J.; Cannon-Albright, L.A.; Teerlink, C.C.; et al. Association of Rare Coding Mutations with Alzheimer Disease and Other Dementias Among Adults of European Ancestry. JAMA Netw. Open 2019, 2, e191350. [Google Scholar] [CrossRef]
- DeMaagd, G.; Philip, A. Parkinson’s Disease and Its Management: Part 1: Disease Entity, Risk Factors, Pathophysiology, Clinical Presentation, and Diagnosis. Pharm. Ther. 2015, 40, 504–532. [Google Scholar] [PubMed]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef]
- Austefjord, M.W.; Gerdes, H.-H.; Wang, X. Tunneling nanotubes. Commun. Integr. Biol. 2014, 7, e27934. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Zhang, J.; Tu, J.; Wang, X.-J.; Su, X.-D.; Wang, L.; Zhang, Y. Tunneling-nanotube direction determination in neurons and astrocytes. Cell Death Dis. 2012, 3, e438. [Google Scholar] [CrossRef]
- Costanzo, M.; Abounit, S.; Marzo, L.; Danckaert, A.; Chamoun, Z.; Roux, P.; Zurzolo, C. Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J. Cell Sci. 2013, 126, 3678–3685. [Google Scholar] [CrossRef] [PubMed]
- Dieriks, B.V.; Park, T.I.-H.; Fourie, C.; Faull, R.L.M.; Dragunow, M.; Curtis, M.A. α-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson’s disease patients. Sci. Rep. 2017, 7, srep42984. [Google Scholar] [CrossRef] [PubMed]
- Gousset, K.; Schiff, E.; Langevin, C.; Marijanovic, Z.; Caputo, A.; Browman, D.T.; Chenouard, N.; De Chaumont, F.; De Martino, A.; Enninga, J.; et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 2009, 11, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal Blood–Brain Barrier Permeability in Parkinson’s Disease. Br. J. Pharmacol. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Dohgu, S.; Takata, F.; Matsumoto, J.; Kimura, I.; Yamauchi, A.; Kataoka, Y. Monomeric α-synuclein induces blood-brain barrier dysfunction through activated brain pericytes releasing inflammatory mediators in vitro. Microvasc. Res. 2019, 124, 61–66. [Google Scholar] [CrossRef]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef]
- Siddiqui, I.J.; Pervaiz, N.; Abbasi, A.A. The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Sci. Rep. 2016, 6, 24475. [Google Scholar] [CrossRef]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef]
- Maekawa, T.; Sasaoka, T.; Azuma, S.; Ichikawa, T.; Melrose, H.L.; Farrer, M.J.; Obata, F. Leucine-rich repeat kinase 2 (LRRK2) regulates α-synuclein clearance in microglia. BMC Neurosci. 2016, 17, 1–12. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2010, 25, S32–S39. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, J.; Wang, J.; Yang, B.; He, Q.; Weng, Q. Role of DJ-1 in Immune and Inflammatory Diseases. Front. Immunol. 2020, 11, 994. [Google Scholar] [CrossRef]
- Park, J.-S.; Blair, N.F.; Sue, C.M. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Mov. Disord. 2015, 30, 770–779. [Google Scholar] [CrossRef]
- Orimo, S.; Uchihara, T.; Nakamura, A.; Mori, F.; Ikeuchi, T.; Onodera, O.; Nishizawa, M.; Ishikawa, A.; Kakita, A.; Wakabayashi, K.; et al. Cardiac sympathetic denervation in Parkinson’s disease linked to SNCA duplication. Acta Neuropathol. 2008, 116, 575–577. [Google Scholar] [CrossRef]
- Visanji, N.P.; Bhudhikanok, G.S.; Mestre, T.A.; Ghate, T.; Udupa, K.; Aldakheel, A.; Connolly, B.S.; Gasca-Salas, C.; Kern, D.S.; Jain, J.; et al. Heart rate variability in leucine-rich repeat kinase 2-associated Parkinson’s disease. Mov. Disord. 2017, 32, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, T.; Li, Z.; Liu, N.; Yan, Y.; Liu, B. Role of Mitophagy in Cardiovascular Disease. Aging Dis. 2020, 11, 419–437. [Google Scholar] [CrossRef]
- He, L.; Vanlandewijck, M.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. Single-cell RNA sequencing of mouse brain and lung vascular and vessel-associated cell types. Sci. Data 2018, 5, 180160. [Google Scholar] [CrossRef] [PubMed]
- Vanlandewijck, M.; He, L.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018, 554, 475–480. [Google Scholar] [CrossRef]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.-W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A Perivascular Origin for Mesenchymal Stem Cells in Multiple Human Organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef]
- Guimarães-Camboa, N.; Cattaneo, P.; Sun, Y.; Moore-Morris, T.; Gu, Y.; Dalton, N.D.; Rockenstein, E.; Masliah, E.; Peterson, K.L.; Stallcup, W.B.; et al. Pericytes of Multiple Organs Do Not Behave as Mesenchymal Stem Cells In Vivo. Cell Stem Cell 2017, 20, 345–359.e5. [Google Scholar] [CrossRef]
- Sakuma, R.; Kawahara, M.; Nakano-Doi, A.; Takahashi, A.; Tanaka, Y.; Narita, A.; Kuwahara-Otani, S.; Hayakawa, T.; Yagi, H.; Matsuyama, T.; et al. Brain pericytes serve as microglia-generating multipotent vascular stem cells following ischemic stroke. J. Neuroinflamm. 2016, 13, 1–13. [Google Scholar] [CrossRef]
- Murgai, M.; Ju, W.; Eason, M.; Kline, J.; Beury, D.W.; Kaczanowska, S.; Miettinen, M.M.; Kruhlak, M.; Lei, H.; Shern, J.F.; et al. KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat. Med. 2017, 23, 1176–1190. [Google Scholar] [CrossRef]
- Hess, D.L.; Kelly-Goss, M.R.; Cherepanova, O.A.; Nguyen, A.T.; Baylis, R.A.; Tkachenko, S.; Annex, B.H.; Peirce, S.M.; Owens, G.K. Perivascular cell-specific knockout of the stem cell pluripotency gene Oct4 inhibits angiogenesis. Nat. Commun. 2019, 10, 967. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Zou, X.; Jin, Y.; Gao, S.; Lv, J.; Li, B.; Cui, R. The Role of KLF4 in Alzheimer’s Disease. Front. Cell. Neurosci. 2018, 12, 325. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Periyasamy, P.; Thangaraj, A.; Chivero, E.T.; Buch, S. PDGF/PDGFR axis in the neural systems. Mol. Asp. Med. 2018, 62, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, P. Pericyte Loss and Microaneurysm Formation in PDGF-B-Deficient Mice. Science 1997, 277, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Tallquist, M.D.; French, W.J.; Soriano, P. Additive Effects of PDGF Receptor β Signaling Pathways in Vascular Smooth Muscle Cell Development. PLoS Biol. 2003, 1, e52. [Google Scholar] [CrossRef] [PubMed]
- Sagare, A.P.; Sweeney, M.D.; Makshanoff, J.; Zlokovic, B.V. Shedding of soluble platelet-derived growth factor receptor-β from human brain pericytes. Neurosci. Lett. 2015, 607, 97–101. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mattson, M.P.; Cheng, A. Notch: From neural development to neurological disorders. J. Neurochem. 2008, 107, 1471–1481. [Google Scholar] [CrossRef]
- Louvi, A.; Arboleda-Velasquez, J.F.; Artavanis-Tsakonas, S. CADASIL: A Critical Look at a Notch Disease. Dev. Neurosci. 2006, 28, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, I.; Bianchi, S.; De Stefano, N.; Dichgans, M.; Dotti, M.T.; Duering, M.; Jouvent, E.; Korczyn, A.D.; Lesnik-Oberstein, S.A.J.; Malandrini, A.; et al. Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) as a model of small vessel disease: Update on clinical, diagnostic, and management aspects. BMC Med. 2017, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Henshall, T.L.; Keller, A.; He, L.; Johansson, B.R.; Wallgard, E.; Raschperger, E.; Mäe, M.A.; Jin, S.; Betsholtz, C.; Lendahl, U. Notch3 Is Necessary for Blood Vessel Integrity in the Central Nervous System. Arter. Thromb. Vasc. Biol. 2015, 35, 409–420. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, L.; Moens, C.B.; Appel, B. Notch3 establishes brain vascular integrity by regulating pericyte number. Development 2013, 141, 307–317. [Google Scholar] [CrossRef]
- Nadeem, T.; Bogue, W.; Bigit, B.; Cuervo, H. Deficiency of Notch signaling in pericytes results in arteriovenous malformations. JCI Insight 2020, 5, e125940. [Google Scholar] [CrossRef]
- Jin, S.; Hansson, E.M.; Tikka, S.; Lanner, F.; Sahlgren, C.; Farnebo, F.; Baumann, M.; Kalimo, H.; Lendahl, U. Notch Signaling Regulates Platelet-Derived Growth Factor Receptor- Expression in Vascular Smooth Muscle Cells. Circ. Res. 2008, 102, 1483–1491. [Google Scholar] [CrossRef]
- Drachman, D.A.; Smith, T.W.; Alkamachi, B.; Kane, K. Microvascular changes in Down syndrome with Alzheimer’s-type pathology: Insights into a potential vascular mechanism for Down syndrome and Alzheimer’s disease. Alzheimer’s Dement. 2017, 13, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
- Placanica, L.; Zhu, L.; Li, Y.-M. Gender- and Age-Dependent γ-Secretase Activity in Mouse Brain and Its Implication in Sporadic Alzheimer Disease. PLoS ONE 2009, 4, e5088. [Google Scholar] [CrossRef] [PubMed]
- Caggiu, E.; Arru, G.; Hosseini, S.; Niegowska, M.; Sechi, G.; Zarbo, I.R.; Sechi, L.A. Inflammation, Infectious Triggers, and Parkinson’s Disease. Front. Neurol. 2019, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; BeMiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dementia Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Puxeddu, I.; Napoletano, S.; Scala, E.; Melillo, D.; Manocchio, S.; Angiolillo, A.; Migliorini, P.; Boraschi, D.; Vitale, E.; et al. Circulating levels of IL-1 family cytokines and receptors in Alzheimer’s disease: New markers of disease progression? J. Neuroinflamm. 2018, 15, 342. [Google Scholar] [CrossRef]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 1–14. [Google Scholar] [CrossRef]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; De Bernard, M. Triggering of Inflammasome by Aggregated α–Synuclein, an Inflammatory Response in Synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef]
- Quan, W.; Luo, Q.; Tang, Q.; Furihata, T.; Li, D.; Fassbender, K.; Liu, Y. NLRP3 Is Involved in the Maintenance of Cerebral Pericytes. Front. Cell. Neurosci. 2020, 14, 276. [Google Scholar] [CrossRef]
- Kin, K.; Yasuhara, T.; Kameda, M.; Date, I. Animal Models for Parkinson’s Disease Research: Trends in the 2000s. Int. J. Mol. Sci. 2019, 20, 5402. [Google Scholar] [CrossRef]
- Veseli, B.E.; Perrotta, P.; De Meyer, G.R.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Kowala, M.C.; Recce, R.; Beyer, S.; Gu, C.; Valentine, M. Characterization of atherosclerosis in LDL receptor knockout mice: Macrophage accumulation correlates with rapid and sustained expression of aortic MCP-1/JE. Atherosclerosis 2000, 149, 323–330. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Bolli, R.; Canty, J.M.; Du, X.-J.; Frangogiannis, N.G.; Frantz, S.; Gourdie, R.G.; Holmes, J.W.; Jones, S.P.; Kloner, R.A.; et al. Guidelines for experimental models of myocardial ischemia and infarction. Am. J. Physiol. Circ. Physiol. 2018, 314, H812–H838. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Fluri, F.; Schuhmann, M.K. Animal models of ischemic stroke and their application in clinical research. Drug Des. Dev. Ther. 2015, 9, 3445–3454. [Google Scholar] [CrossRef] [PubMed]
- Elder, G.A.; Sosa, M.A.G.; De Gasperi, R. Transgenic Mouse Models of Alzheimer’s Disease. Mt. Sinai J. Med. J. Transl. Personal. Med. 2010, 77, 69–81. [Google Scholar] [CrossRef]
- Schaeffer, E.L.; Figueiró, M.; Gattaz, W.F. Insights into Alzheimer disease pathogenesis from studies in transgenic animal models. Clinics 2011, 66, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, M.E.; Burgermeister, P.; Phinney, A.L.; Stalder, M.; Tolnay, M.; Wiederhold, K.-H.; Abramowski, D.; Sturchler-Pierrat, C.; Sommer, B.; Staufenbiel, M.; et al. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc. Natl. Acad. Sci. USA 1999, 96, 14088–14093. [Google Scholar] [CrossRef]
- Kumar, S.; Lemere, C.A.; Walter, J. Phosphorylated Aβ peptides in human Down syndrome brain and different Alzheimer’s-like mouse models. Acta Neuropathol. Commun. 2020, 8, 1–14. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.-Y. Neuronal α-Synucleinopathy with Severe Movement Disorder in Mice Expressing A53T Human α-Synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef]
- Plaas, M.; Karis, A.; Innos, J.; Rebane, E.; Baekelandt, V.; Vaarmann, A.; Luuk, H.; Vasar, E.; Koks, S. Alpha-synuclein A30P point-mutation generates age-dependent nigrostriatal deficiency in mice. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2008, 59, 205–216. [Google Scholar]
- Emmer, K.L.; Waxman, E.A.; Covy, J.P.; Giasson, B.I. E46K Human α-Synuclein Transgenic Mice Develop Lewy-like and Tau Pathology Associated with Age-dependent, Detrimental Motor Impairment*. J. Biol. Chem. 2011, 286, 35104–35118. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Kulshrestha, R.; Singh, N.; Jaggi, A.S. Expanding spectrum of anticancer drug, imatinib, in the disorders affecting brain and spinal cord. Pharmacol. Res. 2019, 143, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, T.; Kubota, N. Protective Role of Imatinib in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2004, 24, 801–803. [Google Scholar] [CrossRef] [PubMed]
- Estrada, L.D.; Chamorro, D.; Yañez, M.J.; Gonzalez, M.; Leal, N.; Von Bernhardi, R.; Dulcey, A.E.; Marugan, J.; Ferrer, M.; Soto, C.; et al. Reduction of Blood Amyloid-β Oligomers in Alzheimer’s Disease Transgenic Mice by c-Abl Kinase Inhibition. J. Alzheimer’s Dis. 2016, 54, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Gianni, D.; Zambrano, N.; Bimonte, M.; Minopoli, G.; Mercken, L.; Talamo, F.; Scaloni, A.; Russo, T. Platelet-derived Growth Factor Induces the β-γ-Secretase-mediated Cleavage of Alzheimer’s Amyloid Precursor Protein through a Src-Rac-dependent Pathway. J. Biol. Chem. 2003, 278, 9290–9297. [Google Scholar] [CrossRef]
- Olsson, B.; Legros, L.; Guilhot, F.; Strömberg, K.; Smith, J.; Livesey, F.J.; Wilson, D.H.; Zetterberg, H.; Blennow, K. Imatinib treatment and Aβ42 in humans. Alzheimer’s Dement. 2014, 10, S374–S380. [Google Scholar] [CrossRef]
- Lee, S.J.; Wang, J.Y. Exploiting the promiscuity of imatinib. J. Biol. 2009, 8, 30. [Google Scholar] [CrossRef]
- Rizzo, P.; Ferrari, R. The Notch pathway: A new therapeutic target in atherosclerosis? Eur. Heart J. Suppl. 2015, 17, A74–A76. [Google Scholar] [CrossRef][Green Version]
- Fava, C.; Montagnana, M. Atherosclerosis Is an Inflammatory Disease which Lacks a Common Anti-inflammatory Therapy: How Human Genetics Can Help to This Issue. A Narrative Review. Front. Pharmacol. 2018, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Jaturapatporn, D.; Isaac, M.G.E.K.N.; Mccleery, J.; Tabet, N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst. Rev. 2012, CD006378. [Google Scholar] [CrossRef]
- Jacobsson, L.T.H.; Turesson, C.; Gülfe, A.; Kapetanovic, M.C.; Petersson, I.F.; Saxne, T.; Geborek, P. Treatment with tumor necrosis factor blockers is associated with a lower incidence of first cardiovascular events in patients with rheumatoid arthritis. J. Rheumatol. 2005, 32, 1213–1218. [Google Scholar] [PubMed]
- Shadfar, S.; Hwang, C.J.; Lim, M.-S.; Choi, D.-Y.; Hong, J.T. Involvement of inflammation in Alzheimer’s disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch. Pharmacal. Res. 2015, 38, 2106–2119. [Google Scholar] [CrossRef]
- Velican, C.; Velican, D. Intimal thickening in developing coronary arteries and its relevance to atherosclerotic involvement. Atherosclerosis 1976, 23, 345–355. [Google Scholar] [CrossRef]
- Gomez, D.; Baylis, R.A.; Durgin, B.G.; Newman, A.A.C.; Alencar, G.F.; Mahan, S.; Hilaire, C.S.; Müller, W.; Waisman, A.; Francis, S.E.; et al. Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat. Med. 2018, 24, 1418–1429. [Google Scholar] [CrossRef]
- Jacobsen, K.; Lund, M.B.; Shim, J.; Gunnersen, S.; Füchtbauer, E.-M.; Kjolby, M.; Carramolino, L.; Bentzon, J.F. Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Model | Phenotype | Reference |
|---|---|---|---|
| Atherosclerosis | Apoe-/- | High cholesterol levels | [169] |
| Increased sensitivity to fat and cholesterol-based dietsExtensive atherosclerosis by 3 months | |||
| Ldlr-/- | Plaque development only in high fat diets | [169,170] | |
| Better mimics human pathogenesis (lipoprotein profile) | |||
| Atherosclerosis induction by 6 months | |||
| Apoe-/- + Ldlr-/- | More severe atherosclerosis than Apoe-/- and Ldlr-/- individual knockouts | [169] | |
| Lipoprotein profile similar to Apoe-/- | |||
| Ischemia | Suture occlusion of artery | Depends on which affected tissue is being modelled | [171,172] |
| Stroke | Suture to occlude the middle cerebral artery | Infarction (size dependent on occlusion time, suture size, suture material, etc.) | [172] |
| Striatum blood flow normalizes after 2 h | |||
| Cortical blood flow remains low | |||
| Endothelin-1 injection directly to middle cerebral artery | Infarction (size dependent on dose) | [172] | |
| Reduced cerebral blood flow–reperfusion takes hours | |||
| Microsphere insertion into the middle cerebral artery | Infarction (size dependent on microsphere size, material) | [172] | |
| Alzheimer’s | App-Indiana mutation with PDGF promoter | 18× APP RNA | [173,174] |
| 10× APP protein | |||
| Amyloid deposition | |||
| apparent at 9 months | |||
| Cerebral amyloid angiopathy | |||
| App-Swedish mutation with hamster prion protein promoter | 5× APP protein | [9,173,175] | |
| Amyloid deposition apparent at 11–13 months | |||
| Cerebral amyloid angiopathy | |||
| App-Swedish + Indiana mutation with hamster prion protein promoter | Amyloid deposition apparent at 3 months | [173,176] | |
| Cerebral amyloid angiopathy | |||
| Parkinson’s | Snca-a53t mutation with mouse prion protein promoter | Initial motor control degradation and α-Syn inclusions at 8 months | [177] |
| Snca-a30p mutation with hamster prion protein promoter | Initial motor control degradation at 13 months | [178] | |
| Snca-e46k mutation with mouse prion protein promoter | Initial motor degradation at 16 months | [179] | |
| Slower disease progression than other mutations |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, A.; Peiris, N.J.; Dhaliwal, H.; Hakim, M.; Li, W.; Ganesh, S.; Ramaswamy, Y.; Patel, S.; Misra, A. Mural Cells: Potential Therapeutic Targets to Bridge Cardiovascular Disease and Neurodegeneration. Cells 2021, 10, 593. https://doi.org/10.3390/cells10030593
Lin A, Peiris NJ, Dhaliwal H, Hakim M, Li W, Ganesh S, Ramaswamy Y, Patel S, Misra A. Mural Cells: Potential Therapeutic Targets to Bridge Cardiovascular Disease and Neurodegeneration. Cells. 2021; 10(3):593. https://doi.org/10.3390/cells10030593
Chicago/Turabian StyleLin, Alexander, Niridu Jude Peiris, Harkirat Dhaliwal, Maria Hakim, Weizhen Li, Subramaniam Ganesh, Yogambha Ramaswamy, Sanjay Patel, and Ashish Misra. 2021. "Mural Cells: Potential Therapeutic Targets to Bridge Cardiovascular Disease and Neurodegeneration" Cells 10, no. 3: 593. https://doi.org/10.3390/cells10030593
APA StyleLin, A., Peiris, N. J., Dhaliwal, H., Hakim, M., Li, W., Ganesh, S., Ramaswamy, Y., Patel, S., & Misra, A. (2021). Mural Cells: Potential Therapeutic Targets to Bridge Cardiovascular Disease and Neurodegeneration. Cells, 10(3), 593. https://doi.org/10.3390/cells10030593