
Targeting the Activin Receptor Signaling to Counteract the Multi-Systemic Complications of Cancer and Its Treatments



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
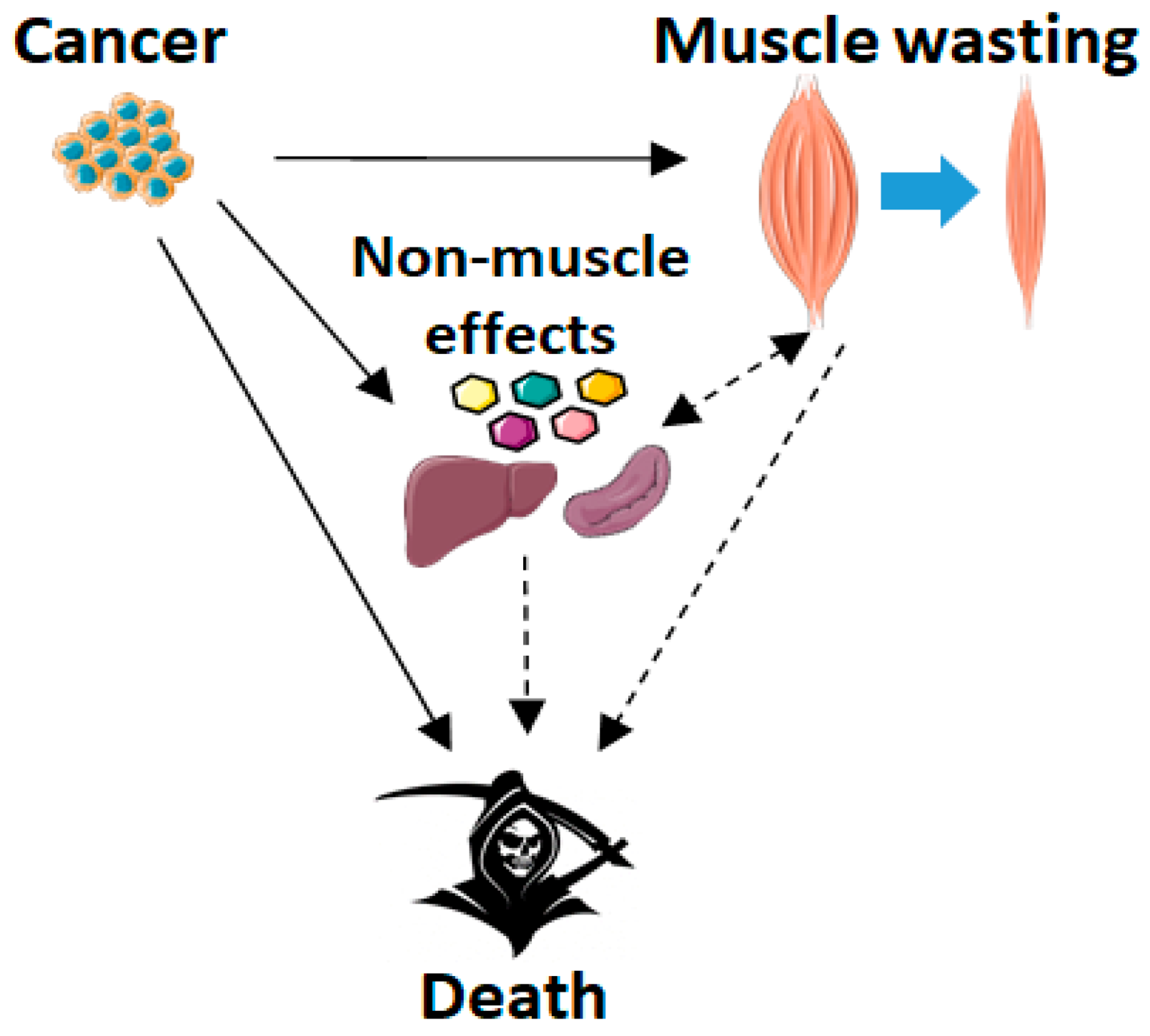
2. Cachexia Induced by Cancer or Chemotherapy
2.1. Cancer Cachexia and Skeletal Muscle Wasting
2.2. Chemotherapy and Skeletal Muscle Wasting
2.3. Cachexia and Survival: The Role of Skeletal Muscle Wasting
3. Activin Receptor Ligands in Cancer- and Chemotherapy-Induced Cachexia
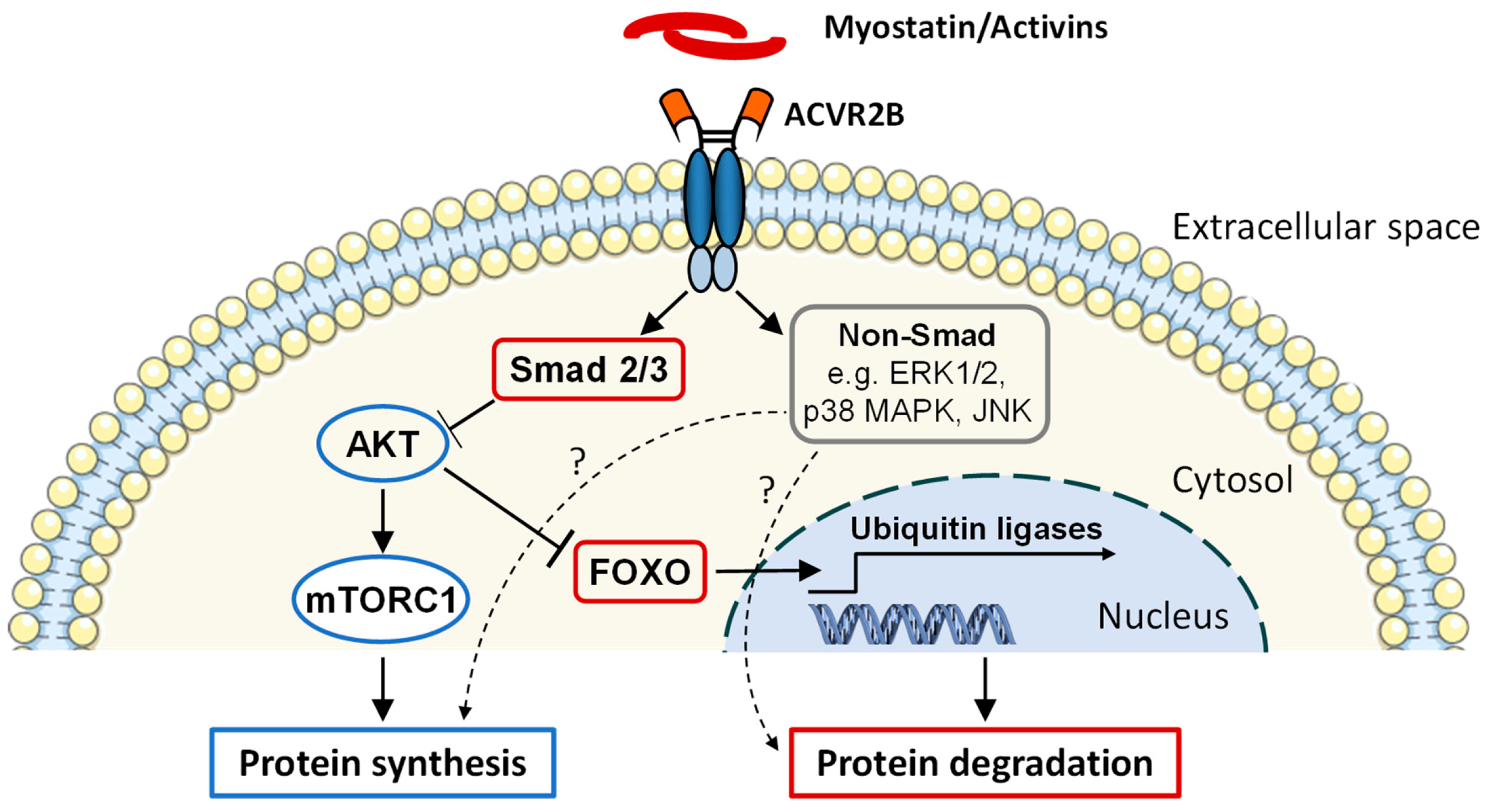
3.1. Discovery and Function
3.2. Levels of ACVR2 Ligands in Cancer Cachexia and in Cancer Treatment
3.3. Strategies to Block ACVR2 Signaling
3.4. Effects of Blocking ACVR2 Signaling on Skeletal Muscle
3.5. Blocking ACVR2 Ligands Improves Survival in Pre-Clinical Cancer Cachexia: Are the Effects Mediated by Skeletal Muscle?
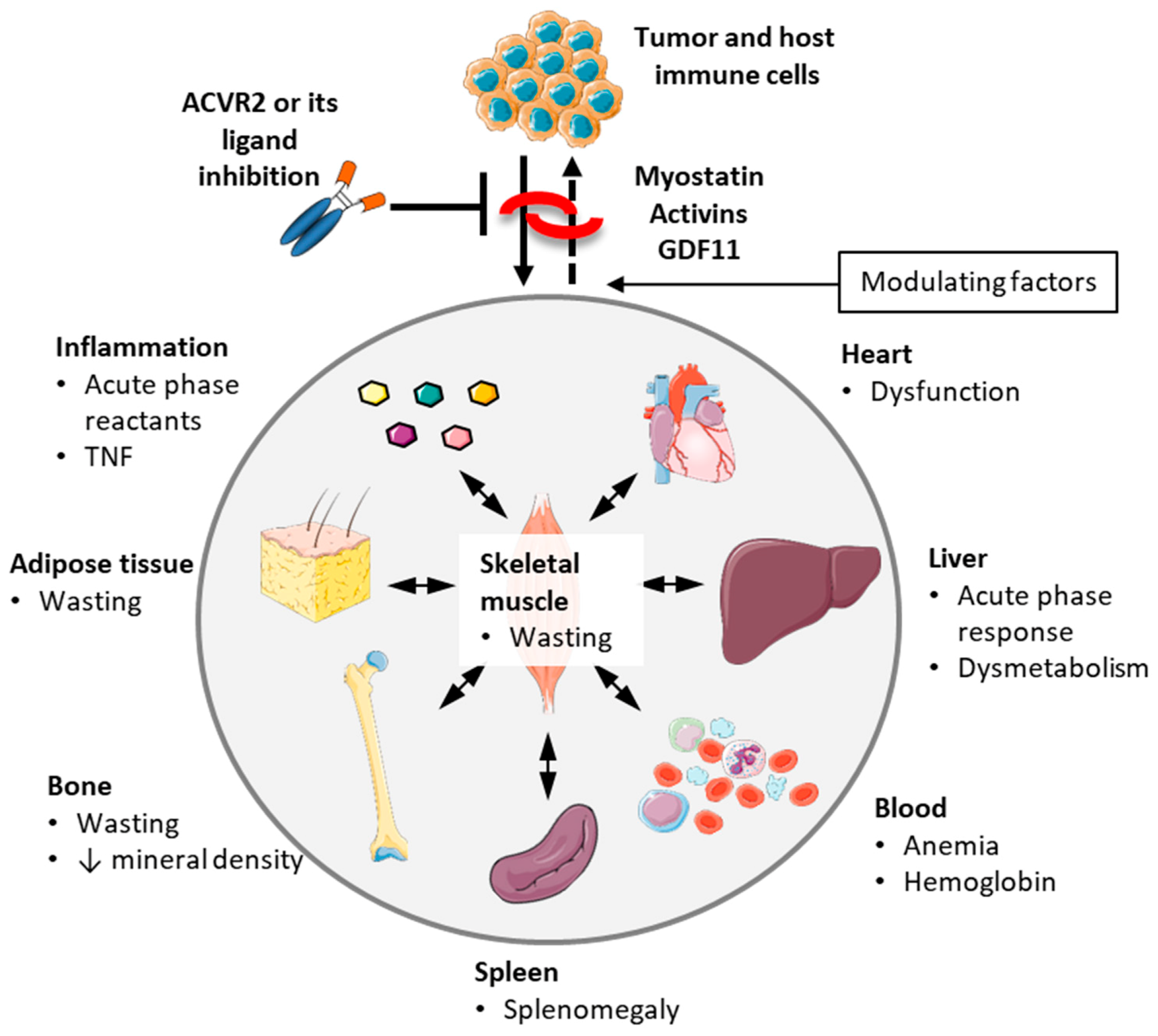
3.6. Non-Muscle Effects of Blocking ACVR2 Ligands
3.6.1. Heart
3.6.2. Adipose Tissue
3.6.3. Blood Cells and Anemia
3.6.4. Inflammation, Splenomegaly, Acute Phase Response and Tumor/Metastasis
3.6.5. Bone
3.6.6. Negative Side Effects of Blocking Myostatin, Activins and GDF11 and Other ACVR2 Ligands
3.6.7. Effects of Blocking ACVR2 Ligands in Cancer: Omics Approach
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fanzani, A.; Conraads, V.M.; Penna, F.; Martinet, W. Molecular and cellular mechanisms of skeletal muscle atrophy: An update. J. Cachexia Sarcopenia Muscle 2012, 3, 163–179. [Google Scholar] [CrossRef]
- Wolfe, R.R. The underappreciated role of muscle in health and disease. Am. J. Clin. Nutr. 2006, 84, 475–482. [Google Scholar] [CrossRef]
- Cooper, R.; Kuh, D.; Hardy, R.; Mortality Review Group; FALCon and HALCyon Study Teams. Objectively measured physical capability levels and mortality: Systematic review and meta-analysis. BMJ 2010, 341, c4467. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Arends, J.; Baracos, V. Understanding the mechanisms and treatment options in cancer cachexia. Nat. Rev. Clin. Oncol. 2013, 10, 90–99. [Google Scholar] [CrossRef]
- Martin, L.; Birdsell, L.; Macdonald, N.; Reiman, T.; Clandinin, M.T.; McCargar, L.J.; Murphy, R.; Ghosh, S.; Sawyer, M.B.; Baracos, V.E. Cancer cachexia in the age of obesity: Skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. J. Clin. Oncol. 2013, 31, 1539–1547. [Google Scholar] [CrossRef]
- Argiles, J.M.; Busquets, S.; Stemmler, B.; Lopez-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Warren, S. The immediate causes of death in cancer. Am. J. Med Sci. 1932, 184, 610–615. [Google Scholar] [CrossRef]
- Benny Klimek, M.E.; Aydogdu, T.; Link, M.J.; Pons, M.; Koniaris, L.G.; Zimmers, T.A. Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem. Biophys. Res. Commun. 2010, 391, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S.; Summermatter, S.; Jourdain, M.; Melly, S.; Minetti, G.C.; Lach-Trifilieff, E. ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti-cancer treatments. Skelet. Muscle 2016, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Toledo, M.; Busquets, S.; Penna, F.; Zhou, X.; Marmonti, E.; Betancourt, A.; Massa, D.; Lopez-Soriano, F.J.; Han, H.Q.; Argiles, J.M. Complete reversal of muscle wasting in experimental cancer cachexia: Additive effects of activin type II receptor inhibition and beta-2 agonist. Int. J. Cancer 2016, 138, 2021–2029. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, T.A.; Hentila, J.; Penna, F.; Lampinen, A.; Lautaoja, J.H.; Fachada, V.; Holopainen, T.; Ritvos, O.; Kivela, R.; Hulmi, J.J. Treating cachexia using soluble ACVR2B improves survival, alters mTOR localization, and attenuates liver and spleen responses. J. Cachexia Sarcopenia Muscle 2018, 9, 514–529. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, T.A.; Degerman, J.; Rasanen, M.; Poikonen, A.R.; Koskinen, S.; Mervaala, E.; Pasternack, A.; Ritvos, O.; Kivela, R.; Hulmi, J.J. Systemic blockade of ACVR2B ligands prevents chemotherapy-induced muscle wasting by restoring muscle protein synthesis without affecting oxidative capacity or atrogenes. Sci. Rep. 2016, 6, 32695. [Google Scholar] [CrossRef]
- Hulmi, J.J.; Nissinen, T.A.; Rasanen, M.; Degerman, J.; Lautaoja, J.H.; Hemanthakumar, K.A.; Backman, J.T.; Ritvos, O.; Silvennoinen, M.; Kivela, R. Prevention of chemotherapy-induced cachexia by ACVR2B ligand blocking has different effects on heart and skeletal muscle. J. Cachexia Sarcopenia Muscle 2018, 9, 417–432. [Google Scholar] [CrossRef]
- O’Connell, T.M.; Pin, F.; Couch, M.E.; Bonetto, A. Treatment with Soluble Activin Receptor Type IIB Alters Metabolic Response in Chemotherapy-Induced Cachexia. Cancers 2019, 11, 1222. [Google Scholar] [CrossRef]
- Barreto, R.; Waning, D.L.; Gao, H.; Liu, Y.; Zimmers, T.A.; Bonetto, A. Chemotherapy-related cachexia is associated with mitochondrial depletion and the activation of ERK1/2 and p38 MAPKs. Oncotarget 2016, 7, 43442–43460. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Lee, Y.S. Myostatin Inhibitors: Panacea or Predicament for Musculoskeletal Disorders? J. Bone Metab. 2020, 27, 151–165. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Debruin, D.A.; Bagaric, R.M.; Campelj, D.G.; Hayes, A. The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle. Cells 2020, 9, 2657. [Google Scholar] [CrossRef]
- Loberg, R.D.; Bradley, D.A.; Tomlins, S.A.; Chinnaiyan, A.M.; Pienta, K.J. The lethal phenotype of cancer: The molecular basis of death due to malignancy. CA Cancer J. Clin. 2007, 57, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef]
- Aulino, P.; Berardi, E.; Cardillo, V.M.; Rizzuto, E.; Perniconi, B.; Ramina, C.; Padula, F.; Spugnini, E.P.; Baldi, A.; Faiola, F.; et al. Molecular, cellular and physiological characterization of the cancer cachexia-inducing C26 colon carcinoma in mouse. BMC Cancer 2010, 10, 363. [Google Scholar] [CrossRef]
- Bonetto, A.; Rupert, J.E.; Barreto, R.; Zimmers, T.A. The Colon-26 Carcinoma Tumor-bearing Mouse as a Model for the Study of Cancer Cachexia. J. Vis. Exp. 2016, 117, e54893. [Google Scholar] [CrossRef]
- Murphy, K.T.; Chee, A.; Trieu, J.; Naim, T.; Lynch, G.S. Importance of functional and metabolic impairments in the characterization of the C-26 murine model of cancer cachexia. Dis. Models Mech. 2012, 5, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Huot, J.R.; Novinger, L.J.; Pin, F.; Bonetto, A. HCT116 colorectal liver metastases exacerbate muscle wasting in a mouse model for the study of colorectal cancer cachexia. Dis. Models Mech. 2020, 13, dmm043166. [Google Scholar] [CrossRef]
- Pin, F.; Barreto, R.; Kitase, Y.; Mitra, S.; Erne, C.E.; Novinger, L.J.; Zimmers, T.A.; Couch, M.E.; Bonewald, L.F.; Bonetto, A. Growth of ovarian cancer xenografts causes loss of muscle and bone mass: A new model for the study of cancer cachexia. J. Cachexia Sarcopenia Muscle 2018, 9, 685–700. [Google Scholar] [CrossRef]
- Kazemi-Bajestani, S.M.; Mazurak, V.C.; Baracos, V. Computed tomography-defined muscle and fat wasting are associated with cancer clinical outcomes. Semin. Cell Dev. Biol. 2016, 54, 2–10. [Google Scholar] [CrossRef]
- Prado, C.M.; Baracos, V.E.; McCargar, L.J.; Reiman, T.; Mourtzakis, M.; Tonkin, K.; Mackey, J.R.; Koski, S.; Pituskin, E.; Sawyer, M.B. Sarcopenia as a determinant of chemotherapy toxicity and time to tumor progression in metastatic breast cancer patients receiving capecitabine treatment. Clin. Cancer Res. 2009, 15, 2920–2926. [Google Scholar] [CrossRef]
- Prado, C.M.; Baracos, V.E.; McCargar, L.J.; Mourtzakis, M.; Mulder, K.E.; Reiman, T.; Butts, C.A.; Scarfe, A.G.; Sawyer, M.B. Body composition as an independent determinant of 5-fluorouracil-based chemotherapy toxicity. Clin. Cancer Res. 2007, 13, 3264–3268. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Ballaro, R.; Beltra, M.; De Lucia, S.; Garcia Castillo, L.; Costelli, P. The Skeletal Muscle as an Active Player against Cancer Cachexia. Front. Physiol. 2019, 10, 41. [Google Scholar] [CrossRef]
- Smith, K.L.; Tisdale, M.J. Increased protein degradation and decreased protein synthesis in skeletal muscle during cancer cachexia. Br. J. Cancer 1993, 67, 680–685. [Google Scholar] [CrossRef]
- Samuels, S.E.; Knowles, A.L.; Tilignac, T.; Debiton, E.; Madelmont, J.C.; Attaix, D. Higher skeletal muscle protein synthesis and lower breakdown after chemotherapy in cachectic mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, 133. [Google Scholar] [CrossRef]
- White, J.P.; Baynes, J.W.; Welle, S.L.; Kostek, M.C.; Matesic, L.E.; Sato, S.; Carson, J.A. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS ONE 2011, 6, e24650. [Google Scholar] [CrossRef] [PubMed]
- Horstman, A.M.; Olde Damink, S.W.; Schols, A.M.; van Loon, L.J. Is Cancer Cachexia Attributed to Impairments in Basal or Postprandial Muscle Protein Metabolism? Nutrients 2016, 8, 499. [Google Scholar] [CrossRef] [PubMed]
- Hulmi, J.J.; Penna, F.; Pollanen, N.; Nissinen, T.A.; Hentila, J.; Euro, L.; Lautaoja, J.H.; Ballaro, R.; Soliymani, R.; Baumann, M.; et al. Muscle NAD(+) depletion and Serpina3n as molecular determinants of murine cancer cachexia-the effects of blocking myostatin and activins. Mol. Metab. 2020, 41, 101046. [Google Scholar] [CrossRef] [PubMed]
- Talbert, E.E.; Guttridge, D.C. Impaired regeneration: A role for the muscle microenvironment in cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Tilignac, T.; Temparis, S.; Combaret, L.; Taillandier, D.; Pouch, M.N.; Cervek, M.; Cardenas, D.M.; Le Bricon, T.; Debiton, E.; Samuels, S.E.; et al. Chemotherapy inhibits skeletal muscle ubiquitin-proteasome-dependent proteolysis. Cancer Res. 2002, 62, 2771–2777. [Google Scholar]
- Le Bricon, T.; Gugins, S.; Cynober, L.; Baracos, V.E. Negative impact of cancer chemotherapy on protein metabolism in healthy and tumor-bearing rats. Metabolism 1995, 44, 1340–1348. [Google Scholar] [CrossRef]
- Argiles, J.M.; Lopez-Soriano, F.J.; Busquets, S. Mediators of cachexia in cancer patients. Nutrition 2019, 66, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Vejpongsa, P.; Yeh, E.T. Prevention of anthracycline-induced cardiotoxicity: Challenges and opportunities. J. Am. Coll. Cardiol. 2014, 64, 938–945. [Google Scholar] [CrossRef]
- Gilliam, L.A.; St Clair, D.K. Chemotherapy-induced weakness and fatigue in skeletal muscle: The role of oxidative stress. Antioxid. Redox Signal. 2011, 15, 2543–2563. [Google Scholar] [CrossRef]
- Rasanen, M.; Degerman, J.; Nissinen, T.A.; Miinalainen, I.; Kerkela, R.; Siltanen, A.; Backman, J.T.; Mervaala, E.; Hulmi, J.J.; Kivela, R.; et al. VEGF-B gene therapy inhibits doxorubicin-induced cardiotoxicity by endothelial protection. Proc. Natl. Acad. Sci. USA 2016, 113, 13144–13149. [Google Scholar] [CrossRef]
- Hayward, R.; Hydock, D.; Gibson, N.; Greufe, S.; Bredahl, E.; Parry, T. Tissue retention of doxorubicin and its effects on cardiac, smooth, and skeletal muscle function. J. Physiol. Biochem. 2013, 69, 177–187. [Google Scholar] [CrossRef]
- Braun, T.P.; Szumowski, M.; Levasseur, P.R.; Grossberg, A.J.; Zhu, X.; Agarwal, A.; Marks, D.L. Muscle atrophy in response to cytotoxic chemotherapy is dependent on intact glucocorticoid signaling in skeletal muscle. PLoS ONE 2014, 9, e106489. [Google Scholar] [CrossRef]
- Ertunc, M.; Sara, Y.; Korkusuz, P.; Onur, R. Differential contractile impairment of fast- and slow-twitch skeletal muscles in a rat model of doxorubicin-induced congestive heart failure. Pharmacology 2009, 84, 240–248. [Google Scholar] [CrossRef]
- Gilliam, L.A.; Ferreira, L.F.; Bruton, J.D.; Moylan, J.S.; Westerblad, H.; St Clair, D.K.; Reid, M.B. Doxorubicin acts through tumor necrosis factor receptor subtype 1 to cause dysfunction of murine skeletal muscle. J. Appl. Physiol. 2009, 107, 1935–1942. [Google Scholar] [CrossRef]
- Gilliam, L.A.; Moylan, J.S.; Callahan, L.A.; Sumandea, M.P.; Reid, M.B. Doxorubicin causes diaphragm weakness in murine models of cancer chemotherapy. Muscle Nerve 2011, 43, 94–102. [Google Scholar] [CrossRef]
- Gilliam, L.A.; Moylan, J.S.; Ferreira, L.F.; Reid, M.B. TNF/TNFR1 signaling mediates doxorubicin-induced diaphragm weakness. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L225–L231. [Google Scholar] [CrossRef]
- Gouspillou, G.; Scheede-Bergdahl, C.; Spendiff, S.; Vuda, M.; Meehan, B.; Mlynarski, H.; Archer-Lahlou, E.; Sgarioto, N.; Purves-Smith, F.M.; Konokhova, Y.; et al. Anthracycline-containing chemotherapy causes long-term impairment of mitochondrial respiration and increased reactive oxygen species release in skeletal muscle. Sci. Rep. 2015, 5, 8717. [Google Scholar] [CrossRef]
- Hydock, D.S.; Lien, C.Y.; Jensen, B.T.; Schneider, C.M.; Hayward, R. Characterization of the effect of in vivo doxorubicin treatment on skeletal muscle function in the rat. Anticancer Res. 2011, 31, 2023–2028. [Google Scholar]
- Min, K.; Kwon, O.S.; Smuder, A.J.; Wiggs, M.P.; Sollanek, K.J.; Christou, D.D.; Yoo, J.K.; Hwang, M.H.; Szeto, H.H.; Kavazis, A.N.; et al. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J. Physiol. 2015, 593, 2017–2036. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, J.H.; Tallent, C.; Schechter, J.E. Ultrastructural features of Adriamycin-induced skeletal and cardiac muscle toxicity. Am. J. Pathol. 1985, 118, 288–297. [Google Scholar]
- Hiensch, A.E.; Bolam, K.A.; Mijwel, S.; Jeneson, J.A.L.; Huitema, A.D.R.; Kranenburg, O.; van der Wall, E.; Rundqvist, H.; Wengstrom, Y.; May, A.M. Doxorubicin-induced skeletal muscle atrophy: Elucidating the underlying molecular pathways. Acta Physiol. 2019, 229, e13400. [Google Scholar] [CrossRef]
- Smuder, A.J.; Kavazis, A.N.; Min, K.; Powers, S.K. Exercise protects against doxorubicin-induced markers of autophagy signaling in skeletal muscle. J. Appl. Physiol. 2011, 111, 1190–1198. [Google Scholar] [CrossRef]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Sacheck, J.M.; Hyatt, J.P.; Raffaello, A.; Jagoe, R.T.; Roy, R.R.; Edgerton, V.R.; Lecker, S.H.; Goldberg, A.L. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 2007, 21, 140–155. [Google Scholar] [CrossRef]
- Barreto, R.; Kitase, Y.; Matsumoto, T.; Pin, F.; Colston, K.C.; Couch, K.E.; O’Connell, T.M.; Couch, M.E.; Bonewald, L.F.; Bonetto, A. ACVR2B/Fc counteracts chemotherapy-induced loss of muscle and bone mass. Sci. Rep. 2017, 7, 14470-1. [Google Scholar] [CrossRef]
- Barreto, R.; Mandili, G.; Witzmann, F.A.; Novelli, F.; Zimmers, T.A.; Bonetto, A. Cancer and Chemotherapy Contribute to Muscle Loss by Activating Common Signaling Pathways. Front. Physiol. 2016, 7, 472. [Google Scholar] [CrossRef]
- Pin, F.; Barreto, R.; Couch, M.E.; Bonetto, A.; O’Connell, T.M. Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism. J. Cachexia Sarcopenia Muscle 2019, 10, 140–154. [Google Scholar] [CrossRef]
- Huot, J.R.; Essex, A.L.; Gutierrez, M.; Barreto, R.; Wang, M.; Waning, D.L.; Plotkin, L.I.; Bonetto, A. Chronic Treatment with Multi-Kinase Inhibitors Causes Differential Toxicities on Skeletal and Cardiac Muscles. Cancers 2019, 11, 571. [Google Scholar] [CrossRef]
- Garcia, J.M.; Cata, J.P.; Dougherty, P.M.; Smith, R.G. Ghrelin prevents cisplatin-induced mechanical hyperalgesia and cachexia. Endocrinology 2008, 149, 455–460. [Google Scholar] [CrossRef]
- Hain, B.A.; Xu, H.; Wilcox, J.R.; Mutua, D.; Waning, D.L. Chemotherapy-induced loss of bone and muscle mass in a mouse model of breast cancer bone metastases and cachexia. JCSM Rapid Commun. 2019, 2, 1–12. [Google Scholar] [CrossRef]
- Essex, A.L.; Pin, F.; Huot, J.R.; Bonewald, L.F.; Plotkin, L.I.; Bonetto, A. Bisphosphonate Treatment Ameliorates Chemotherapy-Induced Bone and Muscle Abnormalities in Young Mice. Front. Endocrinol. 2019, 10, 809. [Google Scholar] [CrossRef]
- Barret, M.; Antoun, S.; Dalban, C.; Malka, D.; Mansourbakht, T.; Zaanan, A.; Latko, E.; Taieb, J. Sarcopenia is linked to treatment toxicity in patients with metastatic colorectal cancer. Nutr. Cancer 2014, 66, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.H.; Brammer, K.; Randhawa, N.; Welch, N.T.; Parsons, S.L.; James, E.J.; Catton, J.A. Sarcopenia is associated with toxicity in patients undergoing neo-adjuvant chemotherapy for oesophago-gastric cancer. Eur. J. Surg. Oncol. 2015, 41, 333–338. [Google Scholar] [CrossRef]
- Cooper, A.B.; Slack, R.; Fogelman, D.; Holmes, H.M.; Petzel, M.; Parker, N.; Balachandran, A.; Garg, N.; Ngo-Huang, A.; Varadhachary, G.; et al. Characterization of Anthropometric Changes that Occur During Neoadjuvant Therapy for Potentially Resectable Pancreatic Cancer. Ann. Surg. Oncol. 2015, 22, 2416–2423. [Google Scholar] [CrossRef] [PubMed]
- Dewys, W.D.; Begg, C.; Lavin, P.T.; Band, P.R.; Bennett, J.M.; Bertino, J.R.; Cohen, M.H.; Douglass, H.O.; Engstrom, P.F.; Ezdinli, E.Z.; et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am. J. Med. 1980, 69, 491–497. [Google Scholar] [CrossRef]
- Prado, C.M.; Baracos, V.E.; Xiao, J.; Birdsell, L.; Stuyckens, K.; Park, Y.C.; Parekh, T.; Sawyer, M.B. The association between body composition and toxicities from the combination of Doxil and trabectedin in patients with advanced relapsed ovarian cancer. Appl. Physiol. Nutr. Metab. 2014, 39, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Prado, C.M.; Lieffers, J.R.; McCargar, L.J.; Reiman, T.; Sawyer, M.B.; Martin, L.; Baracos, V.E. Prevalence and clinical implications of sarcopenic obesity in patients with solid tumours of the respiratory and gastrointestinal tracts: A population-based study. Lancet Oncol. 2008, 9, 629–635. [Google Scholar] [CrossRef]
- Stobaus, N.; Kupferling, S.; Lorenz, M.L.; Norman, K. Discrepancy between body surface area and body composition in cancer. Nutr. Cancer 2013, 65, 1151–1156. [Google Scholar] [CrossRef]
- Pin, F.; Couch, M.E.; Bonetto, A. Preservation of muscle mass as a strategy to reduce the toxic effects of cancer chemotherapy on body composition. Curr. Opin. Support. Palliat. Care 2018, 12, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Talbert, E.E.; Yang, J.; Mace, T.A.; Farren, M.R.; Farris, A.B.; Young, G.S.; Elnaggar, O.; Che, Z.; Timmers, C.D.; Rajasekera, P.; et al. Dual Inhibition of MEK and PI3K/Akt Rescues Cancer Cachexia through both Tumor-Extrinsic and -Intrinsic Activities. Mol. Cancer Ther. 2017, 16, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Arthur, S.T.; Noone, J.M.; Van Doren, B.A.; Roy, D.; Blanchette, C.M. One-year prevalence, comorbidities and cost of cachexia-related inpatient admissions in the USA. Drugs Context 2014, 3, 212265. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Senesse, P.; Gioulbasanis, I.; Antoun, S.; Bozzetti, F.; Deans, C.; Strasser, F.; Thoresen, L.; Jagoe, R.T.; Chasen, M.; et al. Diagnostic criteria for the classification of cancer-associated weight loss. J. Clin. Oncol. 2015, 33, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Fouladiun, M.; Korner, U.; Gunnebo, L.; Sixt-Ammilon, P.; Bosaeus, I.; Lundholm, K. Daily physical-rest activities in relation to nutritional state, metabolism, and quality of life in cancer patients with progressive cachexia. Clin. Cancer Res. 2007, 13, 6379–6385. [Google Scholar] [CrossRef]
- Bachmann, J.; Heiligensetzer, M.; Krakowski-Roosen, H.; Buchler, M.W.; Friess, H.; Martignoni, M.E. Cachexia worsens prognosis in patients with resectable pancreatic cancer. J. Gastrointest. Surg. 2008, 12, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, T.; Ilonen, I.; Kauppi, J.; Salo, J.; Rasanen, J. Loss of skeletal muscle mass during neoadjuvant treatments correlates with worse prognosis in esophageal cancer: A retrospective cohort study. World J. Surg. Oncol. 2018, 16, 27-4. [Google Scholar] [CrossRef]
- Jarvinen, T.; Ilonen, I.; Kauppi, J.; Volmonen, K.; Salo, J.; Rasanen, J. Low skeletal muscle mass in stented esophageal cancer predicts poor survival: A retrospective observational study. Thorac. Cancer 2018, 9, 1429–1436. [Google Scholar] [CrossRef]
- Dalal, S.; Hui, D.; Bidaut, L.; Lem, K.; Del Fabbro, E.; Crane, C.; Reyes-Gibby, C.C.; Bedi, D.; Bruera, E. Relationships among body mass index, longitudinal body composition alterations, and survival in patients with locally advanced pancreatic cancer receiving chemoradiation: A pilot study. J. Pain Symptom Manag. 2012, 44, 181–191. [Google Scholar] [CrossRef]
- Stene, G.B.; Helbostad, J.L.; Amundsen, T.; Sorhaug, S.; Hjelde, H.; Kaasa, S.; Gronberg, B.H. Changes in skeletal muscle mass during palliative chemotherapy in patients with advanced lung cancer. Acta Oncol. 2015, 54, 340–348. [Google Scholar] [CrossRef]
- Fogelman, D.R.; Holmes, H.; Mohammed, K.; Katz, M.H.; Prado, C.M.; Lieffers, J.; Garg, N.; Varadhachary, G.R.; Shroff, R.; Overman, M.J.; et al. Does IGFR1 inhibition result in increased muscle mass loss in patients undergoing treatment for pancreatic cancer? J. Cachexia Sarcopenia Muscle 2014, 5, 307–313. [Google Scholar] [CrossRef]
- Brown, J.C.; Caan, B.J.; Meyerhardt, J.A.; Weltzien, E.; Xiao, J.; Cespedes Feliciano, E.M.; Kroenke, C.H.; Castillo, A.; Kwan, M.L.; Prado, C.M. The deterioration of muscle mass and radiodensity is prognostic of poor survival in stage I-III colorectal cancer: A population-based cohort study (C-SCANS). J. Cachexia Sarcopenia Muscle 2018, 9, 664–672. [Google Scholar] [CrossRef]
- Camus, V.; Lanic, H.; Kraut, J.; Modzelewski, R.; Clatot, F.; Picquenot, J.M.; Contentin, N.; Lenain, P.; Groza, L.; Lemasle, E.; et al. Prognostic impact of fat tissue loss and cachexia assessed by computed tomography scan in elderly patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Eur. J. Haematol. 2014, 93, 9–18. [Google Scholar] [CrossRef]
- Harimoto, N.; Shirabe, K.; Yamashita, Y.I.; Ikegami, T.; Yoshizumi, T.; Soejima, Y.; Ikeda, T.; Maehara, Y.; Nishie, A.; Yamanaka, T. Sarcopenia as a predictor of prognosis in patients following hepatectomy for hepatocellular carcinoma. Br. J. Surg. 2013, 100, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Iritani, S.; Imai, K.; Takai, K.; Hanai, T.; Ideta, T.; Miyazaki, T.; Suetsugu, A.; Shiraki, M.; Shimizu, M.; Moriwaki, H. Skeletal muscle depletion is an independent prognostic factor for hepatocellular carcinoma. J. Gastroenterol. 2015, 50, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Meza-Junco, J.; Montano-Loza, A.J.; Baracos, V.E.; Prado, C.M.; Bain, V.G.; Beaumont, C.; Esfandiari, N.; Lieffers, J.R.; Sawyer, M.B. Sarcopenia as a prognostic index of nutritional status in concurrent cirrhosis and hepatocellular carcinoma. J. Clin. Gastroenterol. 2013, 47, 861–870. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Baba, Y.; Sakamoto, Y.; Ohuchi, M.; Tokunaga, R.; Kurashige, J.; Hiyoshi, Y.; Iwagami, S.; Yoshida, N.; Yoshida, M.; et al. Sarcopenia is a Negative Prognostic Factor After Curative Resection of Colorectal Cancer. Ann. Surg. Oncol. 2015, 22, 2663–2668. [Google Scholar] [CrossRef]
- Peng, P.; Hyder, O.; Firoozmand, A.; Kneuertz, P.; Schulick, R.D.; Huang, D.; Makary, M.; Hirose, K.; Edil, B.; Choti, M.A.; et al. Impact of sarcopenia on outcomes following resection of pancreatic adenocarcinoma. J. Gastrointest. Surg. 2012, 16, 1478–1486. [Google Scholar] [CrossRef]
- Psutka, S.P.; Carrasco, A.; Schmit, G.D.; Moynagh, M.R.; Boorjian, S.A.; Frank, I.; Stewart, S.B.; Thapa, P.; Tarrell, R.F.; Cheville, J.C.; et al. Sarcopenia in patients with bladder cancer undergoing radical cystectomy: Impact on cancer-specific and all-cause mortality. Cancer 2014, 120, 2910–2918. [Google Scholar] [CrossRef]
- Van Vledder, M.G.; Levolger, S.; Ayez, N.; Verhoef, C.; Tran, T.C.; Ijzermans, J.N. Body composition and outcome in patients undergoing resection of colorectal liver metastases. Br. J. Surg. 2012, 99, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Veasey Rodrigues, H.; Baracos, V.E.; Wheler, J.J.; Parsons, H.A.; Hong, D.S.; Naing, A.; Fu, S.; Falchoock, G.; Tsimberidou, A.M.; Piha-Paul, S.; et al. Body composition and survival in the early clinical trials setting. Eur. J. Cancer 2013, 49, 3068–3075. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Tselikas, L.; Pietrasz, D.; Pigneur, F.; Laurent, A.; Compagnon, P.; Salloum, C.; Luciani, A.; Azoulay, D. Sarcopenia Impacts on Short- and Long-term Results of Hepatectomy for Hepatocellular Carcinoma. Ann. Surg. 2015, 261, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, S.; Asghar, A.; Mott, S.L.; Johnson, B.E.; Button, A.M.; Clark, E.; Mezhir, J.J. Sarcopenia is an independent predictor of complications following pancreatectomy for adenocarcinoma. J. Surg. Oncol. 2015, 111, 771–775. [Google Scholar] [CrossRef]
- Lodewick, T.M.; van Nijnatten, T.J.; van Dam, R.M.; van Mierlo, K.; Dello, S.A.; Neumann, U.P.; Olde Damink, S.W.; Dejong, C.H. Are sarcopenia, obesity and sarcopenic obesity predictive of outcome in patients with colorectal liver metastases? HPB 2015, 17, 438–446. [Google Scholar] [CrossRef]
- Peng, P.D.; van Vledder, M.G.; Tsai, S.; de Jong, M.C.; Makary, M.; Ng, J.; Edil, B.H.; Wolfgang, C.L.; Schulick, R.D.; Choti, M.A.; et al. Sarcopenia negatively impacts short-term outcomes in patients undergoing hepatic resection for colorectal liver metastasis. HPB 2011, 13, 439–446. [Google Scholar] [CrossRef]
- Rollins, K.E.; Tewari, N.; Ackner, A.; Awwad, A.; Madhusudan, S.; Macdonald, I.A.; Fearon, K.C.; Lobo, D.N. The impact of sarcopenia and myosteatosis on outcomes of unresectable pancreatic cancer or distal cholangiocarcinoma. Clin. Nutr. 2016, 35, 1103–1109. [Google Scholar] [CrossRef]
- Tan, B.H.; Birdsell, L.A.; Martin, L.; Baracos, V.E.; Fearon, K.C. Sarcopenia in an overweight or obese patient is an adverse prognostic factor in pancreatic cancer. Clin. Cancer Res. 2009, 15, 6973–6979. [Google Scholar] [CrossRef] [PubMed]
- Huot, J.R.; Pin, F.; Narasimhan, A.; Novinger, L.J.; Keith, A.S.; Zimmers, T.A.; Willis, M.S.; Bonetto, A. ACVR2B antagonism as a countermeasure to multi-organ perturbations in metastatic colorectal cancer cachexia. J. Cachexia Sarcopenia Muscle 2020, 11, 1779–1798. [Google Scholar] [CrossRef]
- Roberts, B.M.; Ahn, B.; Smuder, A.J.; Al-Rajhi, M.; Gill, L.C.; Beharry, A.W.; Powers, S.K.; Fuller, D.D.; Ferreira, L.F.; Judge, A.R. Diaphragm and ventilatory dysfunction during cancer cachexia. FASEB J. 2013, 27, 2600–2610. [Google Scholar] [CrossRef]
- Schapira, D.V.; Studnicki, J.; Bradham, D.D.; Wolff, P.; Jarrett, A. Intensive care, survival, and expense of treating critically ill cancer patients. JAMA 1993, 269, 783–786. [Google Scholar] [CrossRef]
- Azoulay, E.; Thiery, G.; Chevret, S.; Moreau, D.; Darmon, M.; Bergeron, A.; Yang, K.; Meignin, V.; Ciroldi, M.; Le Gall, J.R.; et al. The prognosis of acute respiratory failure in critically ill cancer patients. Medicine 2004, 83, 360–370. [Google Scholar] [CrossRef]
- Anker, S.D.; Coats, A.J. Cardiac cachexia: A syndrome with impaired survival and immune and neuroendocrine activation. Chest 1999, 115, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Potsch, M.S.; Ishida, J.; Palus, S.; Tschirner, A.; von Haehling, S.; Doehner, W.; Anker, S.D.; Springer, J. MT-102 prevents tissue wasting and improves survival in a rat model of severe cancer cachexia. J. Cachexia Sarcopenia Muscle 2020, 11, 594–605. [Google Scholar] [CrossRef]
- Cai, D.; Frantz, J.D.; Tawa, N.E.; Melendez, P.A.; Oh, B.C.; Lidov, H.G.; Hasselgren, P.O.; Frontera, W.R.; Lee, J.; Glass, D.J.; et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 2004, 119, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Gallot, Y.S.; Durieux, A.C.; Castells, J.; Desgeorges, M.M.; Vernus, B.; Plantureux, L.; Remond, D.; Jahnke, V.E.; Lefai, E.; Dardevet, D.; et al. Myostatin gene inactivation prevents skeletal muscle wasting in cancer. Cancer Res. 2014, 74, 7344–7356. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.J.; Murphy, K.T.; Jenkinson, L.; Laine, D.; Emmrich, K.; Faou, P.; Weston, R.; Jayatilleke, K.M.; Schloegel, J.; Talbo, G.; et al. Targeting of Fn14 Prevents Cancer-Induced Cachexia and Prolongs Survival. Cell 2015, 162, 1365–1378. [Google Scholar] [CrossRef]
- Lerner, L.; Tao, J.; Liu, Q.; Nicoletti, R.; Feng, B.; Krieger, B.; Mazsa, E.; Siddiquee, Z.; Wang, R.; Huang, L.; et al. MAP3K11/GDF15 axis is a critical driver of cancer cachexia. J. Cachexia Sarcopenia Muscle 2016, 7, 467–482. [Google Scholar] [CrossRef]
- Pretto, F.; Ghilardi, C.; Moschetta, M.; Bassi, A.; Rovida, A.; Scarlato, V.; Talamini, L.; Fiordaliso, F.; Bisighini, C.; Damia, G.; et al. Sunitinib prevents cachexia and prolongs survival of mice bearing renal cancer by restraining STAT3 and MuRF-1 activation in muscle. Oncotarget 2015, 6, 3043–3054. [Google Scholar] [CrossRef]
- Tseng, Y.C.; Kulp, S.K.; Lai, I.L.; Hsu, E.C.; He, W.A.; Frankhouser, D.E.; Yan, P.S.; Mo, X.; Bloomston, M.; Lesinski, G.B.; et al. Preclinical Investigation of the Novel Histone Deacetylase Inhibitor AR-42 in the Treatment of Cancer-Induced Cachexia. J. Natl. Cancer Inst. 2015, 107, djv274. [Google Scholar] [CrossRef]
- Li, Q.; Kumar, R.; Underwood, K.; O’Connor, A.E.; Loveland, K.L.; Seehra, J.S.; Matzuk, M.M. Prevention of cachexia-like syndrome development and reduction of tumor progression in inhibin-deficient mice following administration of a chimeric activin receptor type II-murine Fc protein. Mol. Hum. Reprod. 2007, 13, 675–683. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Rhee, C.; Sim, J.J.; Stenvinkel, P.; Anker, S.D.; Kovesdy, C.P. Why cachexia kills: Examining the causality of poor outcomes in wasting conditions. J. Cachexia Sarcopenia Muscle 2013, 4, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Hill, C.S. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Grobet, L.; Martin, L.J.; Poncelet, D.; Pirottin, D.; Brouwers, B.; Riquet, J.; Schoeberlein, A.; Dunner, S.; Menissier, F.; Massabanda, J.; et al. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat. Genet. 1997, 17, 71–74. [Google Scholar] [CrossRef]
- Kambadur, R.; Sharma, M.; Smith, T.P.; Bass, J.J. Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res. 1997, 7, 910–916. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lee, S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. [Google Scholar] [CrossRef]
- Lee, S.J.; McPherron, A.C. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef]
- Whittemore, L.A.; Song, K.; Li, X.; Aghajanian, J.; Davies, M.; Girgenrath, S.; Hill, J.J.; Jalenak, M.; Kelley, P.; Knight, A.; et al. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem. Biophys. Res. Commun. 2003, 300, 965–971. [Google Scholar] [CrossRef]
- Yang, J.; Ratovitski, T.; Brady, J.P.; Solomon, M.B.; Wells, K.D.; Wall, R.J. Expression of myostatin pro domain results in muscular transgenic mice. Mol. Reprod. Dev. 2001, 60, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Durieux, A.C.; Amirouche, A.; Banzet, S.; Koulmann, N.; Bonnefoy, R.; Pasdeloup, M.; Mouret, C.; Bigard, X.; Peinnequin, A.; Freyssenet, D. Ectopic expression of myostatin induces atrophy of adult skeletal muscle by decreasing muscle gene expression. Endocrinology 2007, 148, 3140–3147. [Google Scholar] [CrossRef]
- Zimmers, T.A.; Davies, M.V.; Koniaris, L.G.; Haynes, P.; Esquela, A.F.; Tomkinson, K.N.; McPherron, A.C.; Wolfman, N.M.; Lee, S.J. Induction of cachexia in mice by systemically administered myostatin. Science 2002, 296, 1486–1488. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor. Nat. Genet. 1999, 22, 260–264. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, Y.; Liu, D.; Liu, F.; Li, X.; Pan, L.; Pang, Y.; Chen, D. Role of growth differentiation factor 11 in development, physiology and disease. Oncotarget 2017, 8, 81604–81616. [Google Scholar] [CrossRef] [PubMed]
- Hammers, D.W.; Merscham-Banda, M.; Hsiao, J.Y.; Engst, S.; Hartman, J.J.; Sweeney, H.L. Supraphysiological levels of GDF11 induce striated muscle atrophy. EMBO Mol. Med. 2017, 9, 531–544. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Huynh, T.V.; Lee, S.J. Redundancy of myostatin and growth/differentiation factor 11 function. BMC Dev. Biol. 2009, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.G.; Poggioli, T.; Katsimpardi, L.; Buchanan, S.M.; Oh, J.; Wattrus, S.; Heidecker, B.; Fong, Y.W.; Rubin, L.L.; Ganz, P.; et al. Biochemistry and Biology of GDF11 and Myostatin: Similarities, Differences, and Questions for Future Investigation. Circ. Res. 2016, 118, 1125–1141. [Google Scholar] [CrossRef] [PubMed]
- Bloise, E.; Ciarmela, P.; Dela Cruz, C.; Luisi, S.; Petraglia, F.; Reis, F.M. Activin A in Mammalian Physiology. Physiol. Rev. 2019, 99, 739–780. [Google Scholar] [CrossRef]
- Vale, W.; Rivier, J.; Vaughan, J.; McClintock, R.; Corrigan, A.; Woo, W.; Karr, D.; Spiess, J. Purification and characterization of an FSH releasing protein from porcine ovarian follicular fluid. Nature 1986, 321, 776–779. [Google Scholar] [CrossRef]
- Namwanje, M.; Brown, C.W. Activins and Inhibins: Roles in Development, Physiology, and Disease. Cold Spring Harb. Perspect. Biol. 2016, 8, a021881. [Google Scholar] [CrossRef]
- Matzuk, M.M.; Finegold, M.J.; Mather, J.P.; Krummen, L.; Lu, H.; Bradley, A. Development of cancer cachexia-like syndrome and adrenal tumors in inhibin-deficient mice. Proc. Natl. Acad. Sci. USA 1994, 91, 8817–8821. [Google Scholar] [CrossRef]
- Chen, J.L.; Walton, K.L.; Winbanks, C.E.; Murphy, K.T.; Thomson, R.E.; Makanji, Y.; Qian, H.; Lynch, G.S.; Harrison, C.A.; Gregorevic, P. Elevated expression of activins promotes muscle wasting and cachexia. FASEB J. 2014, 28, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Gilson, H.; Schakman, O.; Kalista, S.; Lause, P.; Tsuchida, K.; Thissen, J.P. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am. J. Physiol. Endocrinol. Metab. 2009, 297, 157. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, Y.S.; Zimmers, T.A.; Soleimani, A.; Matzuk, M.M.; Tsuchida, K.; Cohn, R.D.; Barton, E.R. Regulation of muscle mass by follistatin and activins. Mol. Endocrinol. 2010, 24, 1998–2008. [Google Scholar] [CrossRef]
- Chen, J.L.; Walton, K.L.; Hagg, A.; Colgan, T.D.; Johnson, K.; Qian, H.; Gregorevic, P.; Harrison, C.A. Specific targeting of TGF-beta family ligands demonstrates distinct roles in the regulation of muscle mass in health and disease. Proc. Natl. Acad. Sci. USA 2017, 114, E5266–E5275. [Google Scholar] [CrossRef]
- Chen, J.L.; Colgan, T.D.; Walton, K.L.; Gregorevic, P.; Harrison, C.A. The TGF-beta Signalling Network in Muscle Development, Adaptation and Disease. Adv. Exp. Med. Biol. 2016, 900, 97–131. [Google Scholar] [CrossRef] [PubMed]
- Hilden, K.; Tuuri, T.; Eramaa, M.; Ritvos, O. Expression of type II activin receptor genes during differentiation of human K562 cells and cDNA cloning of the human type IIB activin receptor. Blood 1994, 83, 2163–2170. [Google Scholar] [CrossRef]
- Mathews, L.S.; Vale, W.W. Expression cloning of an activin receptor, a predicted transmembrane serine kinase. Cell 1991, 65, 973–982. [Google Scholar] [CrossRef]
- Trendelenburg, A.U.; Meyer, A.; Rohner, D.; Boyle, J.; Hatakeyama, S.; Glass, D.J. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am. J. Physiol. Cell Physiol. 2009, 296, C1258–C1270. [Google Scholar] [CrossRef]
- Lautaoja, J.H.; Pekkala, S.; Pasternack, A.; Laitinen, M.; Ritvos, O.; Hulmi, J.J. Differentiation of Murine C2C12 Myoblasts Strongly Reduces the Effects of Myostatin on Intracellular Signaling. Biomolecules 2020, 10, 695. [Google Scholar] [CrossRef] [PubMed]
- Philip, B.; Lu, Z.; Gao, Y. Regulation of GDF-8 signaling by the p38 MAPK. Cell. Signal. 2005, 17, 365–375. [Google Scholar] [CrossRef]
- Yang, W.; Chen, Y.; Zhang, Y.; Wang, X.; Yang, N.; Zhu, D. Extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase pathway is involved in myostatin-regulated differentiation repression. Cancer Res. 2006, 66, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Sartori, R.; Sandri, M. Bone and morphogenetic protein signalling and muscle mass. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 215–220. [Google Scholar] [CrossRef]
- Hoda, M.A.; Rozsas, A.; Lang, E.; Klikovits, T.; Lohinai, Z.; Torok, S.; Berta, J.; Bendek, M.; Berger, W.; Hegedus, B.; et al. High circulating activin A level is associated with tumor progression and predicts poor prognosis in lung adenocarcinoma. Oncotarget 2016, 7, 13388–13399. [Google Scholar] [CrossRef]
- Loumaye, A.; de Barsy, M.; Nachit, M.; Lause, P.; van Maanen, A.; Trefois, P.; Gruson, D.; Thissen, J.P. Circulating Activin A predicts survival in cancer patients. J. Cachexia Sarcopenia Muscle 2017, 8, 768–777. [Google Scholar] [CrossRef]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Pons, M.; Poirier, C.; Jiang, Y.; Liu, J.; Sandusky, G.E.; Shahda, S.; Nakeeb, A.; Schmidt, C.M.; House, M.G.; et al. The systemic activin response to pancreatic cancer: Implications for effective cancer cachexia therapy. J. Cachexia Sarcopenia Muscle 2019, 10, 1083–1101. [Google Scholar] [CrossRef] [PubMed]
- Loumaye, A.; de Barsy, M.; Nachit, M.; Lause, P.; Frateur, L.; van Maanen, A.; Trefois, P.; Gruson, D.; Thissen, J.P. Role of Activin A and myostatin in human cancer cachexia. J. Clin. Endocrinol. Metab. 2015, 100, 2030–2038. [Google Scholar] [CrossRef]
- Paajanen, J.; Ilonen, I.; Lauri, H.; Jarvinen, T.; Sutinen, E.; Ollila, H.; Rouvinen, E.; Lemstrom, K.; Rasanen, J.; Ritvos, O.; et al. Elevated Circulating Activin A Levels in Patients With Malignant Pleural Mesothelioma Are Related to Cancer Cachexia and Reduced Response to Platinum-based Chemotherapy. Clin. Lung Cancer 2020, 21, e142–e150. [Google Scholar] [CrossRef]
- Talar-Wojnarowska, R.; Wozniak, M.; Borkowska, A.; Olakowski, M.; Malecka-Panas, E. Clinical significance of activin A and myostatin in patients with pancreatic adenocarcinoma and progressive weight loss. J. Physiol. Pharmacol. 2020, 71. [Google Scholar] [CrossRef]
- Ries, A.; Schelch, K.; Falch, D.; Pany, L.; Hoda, M.A.; Grusch, M. Activin A: An emerging target for improving cancer treatment? Expert Opin. Ther. Targets 2020, 24, 985–996. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Schirripa, M.; Suenaga, M.; Cao, S.; Zhang, W.; Okazaki, S.; Berger, M.D.; Matsusaka, S.; Yang, D.; Ning, Y.; et al. A polymorphism in the cachexia-associated gene INHBA predicts efficacy of regorafenib in patients with refractory metastatic colorectal cancer. PLoS ONE 2020, 15, e0239439. [Google Scholar] [CrossRef] [PubMed]
- Aversa, Z.; Bonetto, A.; Penna, F.; Costelli, P.; Di Rienzo, G.; Lacitignola, A.; Baccino, F.M.; Ziparo, V.; Mercantini, P.; Rossi Fanelli, F.; et al. Changes in myostatin signaling in non-weight-losing cancer patients. Ann. Surg. Oncol. 2012, 19, 1350–1356. [Google Scholar] [CrossRef]
- Bonetto, A.; Penna, F.; Aversa, Z.; Mercantini, P.; Baccino, F.M.; Costelli, P.; Ziparo, V.; Lucia, S.; Rossi Fanelli, F.; Muscaritoli, M. Early changes of muscle insulin-like growth factor-1 and myostatin gene expression in gastric cancer patients. Muscle Nerve 2013, 48, 387–392. [Google Scholar] [CrossRef]
- Burch, P.M.; Pogoryelova, O.; Palandra, J.; Goldstein, R.; Bennett, D.; Fitz, L.; Guglieri, M.; Bettolo, C.M.; Straub, V.; Evangelista, T.; et al. Reduced serum myostatin concentrations associated with genetic muscle disease progression. J. Neurol. 2017, 264, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Amthor, H.; Hoogaars, W.M. Interference with myostatin/ActRIIB signaling as a therapeutic strategy for Duchenne muscular dystrophy. Curr. Gene Ther. 2012, 12, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Walton, K.L.; Al-Musawi, S.L.; Kelly, E.K.; Qian, H.; La, M.; Lu, L.; Lovrecz, G.; Ziemann, M.; Lazarus, R.; et al. Development of novel activin-targeted therapeutics. Mol. Ther. 2015, 23, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, H.; Kuramochi, T.; Katada, H.; Ueyama, A.; Ruike, Y.; Ohmine, K.; Shida-Kawazoe, M.; Miyano-Nishizawa, R.; Shimizu, Y.; Okuda, M.; et al. Novel myostatin-specific antibody enhances muscle strength in muscle disease models. Sci. Rep. 2021, 11, 2160. [Google Scholar] [CrossRef]
- Murphy, K.T.; Chee, A.; Gleeson, B.G.; Naim, T.; Swiderski, K.; Koopman, R.; Lynch, G.S. Antibody-directed myostatin inhibition enhances muscle mass and function in tumor-bearing mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, 716. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, T.A.; Hentila, J.; Fachada, V.; Lautaoja, J.H.; Pasternack, A.; Ritvos, O.; Kivela, R.; Hulmi, J.J. Muscle follistatin gene delivery increases muscle protein synthesis independent of periodical physical inactivity and fasting. FASEB J. 2021, 35, e21387. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Reed, L.A.; Davies, M.V.; Girgenrath, S.; Goad, M.E.; Tomkinson, K.N.; Wright, J.F.; Barker, C.; Ehrmantraut, G.; Holmstrom, J.; et al. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 18117–18122. [Google Scholar] [CrossRef] [PubMed]
- Hulmi, J.J.; Oliveira, B.M.; Silvennoinen, M.; Hoogaars, W.M.; Ma, H.; Pierre, P.; Pasternack, A.; Kainulainen, H.; Ritvos, O. Muscle protein synthesis, mTORC1/MAPK/Hippo signaling, and capillary density are altered by blocking of myostatin and activins. Am. J. Physiol. Endocrinol. Metab. 2013, 304, 41. [Google Scholar] [CrossRef]
- Attie, K.M.; Borgstein, N.G.; Yang, Y.; Condon, C.H.; Wilson, D.M.; Pearsall, A.E.; Kumar, R.; Willins, D.A.; Seehra, J.S.; Sherman, M.L. A single ascending-dose study of muscle regulator ACE-031 in healthy volunteers. Muscle Nerve 2013, 47, 416–423. [Google Scholar] [CrossRef]
- Rooks, D.; Praestgaard, J.; Hariry, S.; Laurent, D.; Petricoul, O.; Perry, R.G.; Lach-Trifilieff, E.; Roubenoff, R. Treatment of Sarcopenia with Bimagrumab: Results from a Phase II, Randomized, Controlled, Proof-of-Concept Study. J. Am. Geriatr. Soc. 2017, 65, 1988–1995. [Google Scholar] [CrossRef]
- Han, H.Q.; Zhou, X.; Mitch, W.E.; Goldberg, A.L. Myostatin/activin pathway antagonism: Molecular basis and therapeutic potential. Int. J. Biochem. Cell Biol. 2013, 45, 2333–2347. [Google Scholar] [CrossRef] [PubMed]
- Relizani, K.; Mouisel, E.; Giannesini, B.; Hourde, C.; Patel, K.; Morales Gonzalez, S.; Julich, K.; Vignaud, A.; Pietri-Rouxel, F.; Fortin, D.; et al. Blockade of ActRIIB signaling triggers muscle fatigability and metabolic myopathy. Mol. Ther. 2014, 22, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, A.; Penna, F.; Minero, V.G.; Reffo, P.; Bonelli, G.; Baccino, F.M.; Costelli, P. Deacetylase inhibitors modulate the myostatin/follistatin axis without improving cachexia in tumor-bearing mice. Curr. Cancer Drug Targets 2009, 9, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Walton, K.L.; Chen, J.L.; Arnold, Q.; Kelly, E.; La, M.; Lu, L.; Lovrecz, G.; Hagg, A.; Colgan, T.D.; Qian, H.; et al. Activin A-Induced Cachectic Wasting Is Attenuated by Systemic Delivery of Its Cognate Propeptide in Male Mice. Endocrinology 2019, 160, 2417–2426. [Google Scholar] [CrossRef]
- Busquets, S.; Toledo, M.; Orpi, M.; Massa, D.; Porta, M.; Capdevila, E.; Padilla, N.; Frailis, V.; Lopez-Soriano, F.J.; Han, H.Q.; et al. Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J. Cachexia Sarcopenia Muscle 2012, 3, 37–43. [Google Scholar] [CrossRef]
- Toledo, M.; Penna, F.; Oliva, F.; Luque, M.; Betancourt, A.; Marmonti, E.; Lopez-Soriano, F.J.; Argiles, J.M.; Busquets, S. A multifactorial anti-cachectic approach for cancer cachexia in a rat model undergoing chemotherapy. J. Cachexia Sarcopenia Muscle 2016, 7, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Heymsfield, S.B.; Coleman, L.A.; Miller, R.; Rooks, D.S.; Laurent, D.; Petricoul, O.; Praestgaard, J.; Swan, T.; Wade, T.; Perry, R.G.; et al. Effect of Bimagrumab vs. Placebo on Body Fat Mass Among Adults With Type 2 Diabetes and Obesity: A Phase 2 Random-ized Clinical Trial. JAMA Netw. Open 2021, 4, e2033457. [Google Scholar] [CrossRef]
- Kays, J.K.; Shahda, S.; Stanley, M.; Bell, T.M.; O’Neill, B.H.; Kohli, M.D.; Couch, M.E.; Koniaris, L.G.; Zimmers, T.A. Three cachexia phenotypes and the impact of fat-only loss on survival in FOLFIRINOX therapy for pancreatic cancer. J. Cachexia Sarcopenia Muscle 2018, 9, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Taber, C.B.; Vigotsky, A.; Nuckols, G.; Haun, C.T. Exercise-Induced Myofibrillar Hypertrophy is a Contributory Cause of Gains in Muscle Strength. Sports Med. 2019, 49, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Anker, M.S.; von Haehling, S.; Springer, J. Blocking myostatin: Muscle mass equals muscle strength? J. Cachexia Sarcopenia Muscle 2020, 11, 1396–1398. [Google Scholar] [CrossRef]
- Rooks, D.; Swan, T.; Goswami, B.; Filosa, L.A.; Bunte, O.; Panchaud, N.; Coleman, L.A.; Miller, R.R.; Garcia Garayoa, E.; Praestgaard, J.; et al. Bimagrumab vs. Optimized Standard of Care for Treatment of Sarcopenia in Community-Dwelling Older Adults: A Randomized Clinical Trial. JAMA Netw. Open 2020, 3, e2020836. [Google Scholar] [CrossRef]
- Levolger, S.; Wiemer, E.A.C.; van Vugt, J.L.A.; Huisman, S.A.; van Vledder, M.G.; van Damme-van Engel, S.; Ambagtsheer, G.; IJzermans, J.N.M.; de Bruin, R.W.F. Inhibition of activin-like kinase 4/5 attenuates cancer cachexia associated muscle wasting. Sci. Rep. 2019, 9, 9826-9. [Google Scholar] [CrossRef]
- Kainulainen, H.; Papaioannou, K.G.; Silvennoinen, M.; Autio, R.; Saarela, J.; Oliveira, B.M.; Nyqvist, M.; Pasternack, A.; ‘t Hoen, P.A.; Kujala, U.M.; et al. Myostatin/activin blocking combined with exercise reconditions skeletal muscle expression profile of mdx mice. Mol. Cell. Endocrinol. 2015, 399, 131–142. [Google Scholar] [CrossRef]
- Baati, N.; Feillet-Coudray, C.; Fouret, G.; Vernus, B.; Goustard, B.; Jollet, M.; Bertrand-Gaday, C.; Coudray, C.; Lecomte, J.; Bonnieu, A.; et al. New evidence of exercise training benefits in myostatin-deficient mice: Effect on lipidomic abnormalities. Biochem. Biophys. Res. Commun. 2019, 516, 89–95. [Google Scholar] [CrossRef]
- Chiappalupi, S.; Sorci, G.; Vukasinovic, A.; Salvadori, L.; Sagheddu, R.; Coletti, D.; Renga, G.; Romani, L.; Donato, R.; Riuzzi, F. Targeting RAGE prevents muscle wasting and prolongs survival in cancer cachexia. J. Cachexia Sarcopenia Muscle 2020. [Google Scholar] [CrossRef]
- Parajuli, P.; Kumar, S.; Loumaye, A.; Singh, P.; Eragamreddy, S.; Nguyen, T.L.; Ozkan, S.; Razzaque, M.S.; Prunier, C.; Thissen, J.P.; et al. Twist1 Activation in Muscle Progenitor Cells Causes Muscle Loss Akin to Cancer Cachexia. Dev. Cell 2018, 45, 712–725.e6. [Google Scholar] [CrossRef]
- Argiles, J.M.; Stemmler, B.; Lopez-Soriano, F.J.; Busquets, S. Inter-tissue communication in cancer cachexia. Nat. Rev. Endocrinol. 2018, 15, 9–20. [Google Scholar] [CrossRef]
- Stephens, N.A.; Skipworth, R.J.; Fearon, K.C. Cachexia, survival and the acute phase response. Curr. Opin. Support. Palliat. Care 2008, 2, 267–274. [Google Scholar] [CrossRef]
- Bonetto, A.; Aydogdu, T.; Kunzevitzky, N.; Guttridge, D.C.; Khuri, S.; Koniaris, L.G.; Zimmers, T.A. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS ONE 2011, 6, e22538. [Google Scholar] [CrossRef] [PubMed]
- Lieffers, J.R.; Mourtzakis, M.; Hall, K.D.; McCargar, L.J.; Prado, C.M.; Baracos, V.E. A viscerally driven cachexia syndrome in patients with advanced colorectal cancer: Contributions of organ and tumor mass to whole-body energy demands. Am. J. Clin. Nutr. 2009, 89, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.T. The pathogenesis and treatment of cardiac atrophy in cancer cachexia. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, 466. [Google Scholar] [CrossRef]
- Genton, L.; Mareschal, J.; Charretier, Y.; Lazarevic, V.; Bindels, L.B.; Schrenzel, J. Targeting the Gut Microbiota to Treat Cachexia. Front. Cell. Infect. Microbiol. 2019, 9, 305. [Google Scholar] [CrossRef]
- Bindels, L.B.; Neyrinck, A.M.; Claus, S.P.; Le Roy, C.I.; Grangette, C.; Pot, B.; Martinez, I.; Walter, J.; Cani, P.D.; Delzenne, N.M. Synbiotic approach restores intestinal homeostasis and prolongs survival in leukaemic mice with cachexia. ISME J. 2016, 10, 1456–1470. [Google Scholar] [CrossRef] [PubMed]
- Herremans, K.M.; Riner, A.N.; Cameron, M.E.; Trevino, J.G. The Microbiota and Cancer Cachexia. Int. J. Mol. Sci. 2019, 20, 6267. [Google Scholar] [CrossRef]
- Bonetto, A.; Kays, J.K.; Parker, V.A.; Matthews, R.R.; Barreto, R.; Puppa, M.J.; Kang, K.S.; Carson, J.A.; Guise, T.A.; Mohammad, K.S.; et al. Differential Bone Loss in Mouse Models of Colon Cancer Cachexia. Front. Physiol. 2017, 7, 679. [Google Scholar] [CrossRef]
- Magga, J.; Vainio, L.; Kilpio, T.; Hulmi, J.J.; Taponen, S.; Lin, R.; Rasanen, M.; Szabo, Z.; Gao, E.; Rahtu-Korpela, L.; et al. Systemic Blockade of ACVR2B Ligands Protects Myocardium from Acute Ischemia-Reperfusion Injury. Mol. Ther. 2019, 27, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lehar, A.; Liu, Y.; Ly, C.H.; Pham, Q.M.; Michaud, M.; Rydzik, R.; Youngstrom, D.W.; Shen, M.M.; Kaartinen, V.; et al. Functional redundancy of type I and type II receptors in the regulation of skeletal muscle growth by myostatin and activin A. Proc. Natl. Acad. Sci. USA 2020, 117, 30907–30917. [Google Scholar] [CrossRef]
- Danai, L.V.; Babic, A.; Rosenthal, M.H.; Dennstedt, E.A.; Muir, A.; Lien, E.C.; Mayers, J.R.; Tai, K.; Lau, A.N.; Jones-Sali, P.; et al. Altered exocrine function can drive adipose wasting in early pancreatic cancer. Nature 2018, 558, 600–604. [Google Scholar] [CrossRef]
- Baracos, V.E.; Arribas, L. Sarcopenic obesity: Hidden muscle wasting and its impact for survival and complications of cancer therapy. Ann. Oncol. 2018, 29, ii1–ii9. [Google Scholar] [CrossRef]
- Braga, M.; Reddy, S.T.; Vergnes, L.; Pervin, S.; Grijalva, V.; Stout, D.; David, J.; Li, X.; Tomasian, V.; Reid, C.B.; et al. Follistatin promotes adipocyte differentiation, browning, and energy metabolism. J. Lipid Res. 2014, 55, 375–384. [Google Scholar] [CrossRef]
- Lautaoja, J.H.; Lalowski, M.; Nissinen, T.A.; Hentila, J.; Shi, Y.; Ritvos, O.; Cheng, S.; Hulmi, J.J. Muscle and serum metabolomes are dysregulated in colon-26 tumor-bearing mice despite amelioration of cachexia with activin receptor type 2B ligand blockade. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E852–E865. [Google Scholar] [CrossRef]
- Toledo, M.; Penna, F.; Busquets, S.; Lopez-Soriano, F.J.; Argiles, J.M. Distinct behaviour of sorafenib in experimental cachexia-inducing tumours: The role of STAT. PLoS ONE 2014, 9, e113931. [Google Scholar] [CrossRef]
- Pin, F.; Busquets, S.; Toledo, M.; Camperi, A.; Lopez-Soriano, F.J.; Costelli, P.; Argiles, J.M.; Penna, F. Combination of exercise training and erythropoietin prevents cancer-induced muscle alterations. Oncotarget 2015, 6, 43202–43215. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Busquets, S.; Toledo, M.; Pin, F.; Massa, D.; Lopez-Soriano, F.J.; Costelli, P.; Argiles, J.M. Erythropoietin administration partially prevents adipose tissue loss in experimental cancer cachexia models. J. Lipid Res. 2013, 54, 3045–3051. [Google Scholar] [CrossRef]
- Vayrynen, J.P.; Tuomisto, A.; Vayrynen, S.A.; Klintrup, K.; Karhu, T.; Makela, J.; Herzig, K.H.; Karttunen, T.J.; Makinen, M.J. Preoperative anemia in colorectal cancer: Relationships with tumor characteristics, systemic inflammation, and survival. Sci. Rep. 2018, 8, 1126. [Google Scholar] [CrossRef] [PubMed]
- Caro, J.J.; Salas, M.; Ward, A.; Goss, G. Anemia as an independent prognostic factor for survival in patients with cancer: A systemic, quantitative review. Cancer 2001, 91, 2214–2221. [Google Scholar] [CrossRef]
- Lewis, H.L.; Chakedis, J.M.; Talbert, E.; Haverick, E.; Rajasekera, P.; Hart, P.; Bloomston, M.; Dillhoff, M.; Pawlik, T.M.; Guttridge, D.; et al. Perioperative cytokine levels portend early death after pancreatectomy for ductal adenocarcinoma. J. Surg. Oncol. 2017, 117, 1260–1266. [Google Scholar] [CrossRef]
- Martin, F.; Santolaria, F.; Batista, N.; Milena, A.; Gonzalez-Reimers, E.; Brito, M.J.; Oramas, J. Cytokine levels (IL-6 and IFN-gamma), acute phase response and nutritional status as prognostic factors in lung cancer. Cytokine 1999, 11, 80–86. [Google Scholar] [CrossRef]
- Sirnio, P.; Tuomisto, A.; Tervahartiala, T.; Sorsa, T.; Klintrup, K.; Karhu, T.; Herzig, K.H.; Makela, J.; Karttunen, T.J.; Salo, T.; et al. High-serum MMP-8 levels are associated with decreased survival and systemic inflammation in colorectal cancer. Br. J. Cancer 2018, 119, 213–219. [Google Scholar] [CrossRef]
- Talbert, E.E.; Lewis, H.L.; Farren, M.R.; Ramsey, M.L.; Chakedis, J.M.; Rajasekera, P.; Haverick, E.; Sarna, A.; Bloomston, M.; Pawlik, T.M.; et al. Circulating monocyte chemoattractant protein-1 (MCP-1) is associated with cachexia in treatment-naive pancreatic cancer patients. J. Cachexia Sarcopenia Muscle 2018. [Google Scholar] [CrossRef] [PubMed]
- Samuels, S.E.; McLaren, T.A.; Knowles, A.L.; Stewart, S.A.; Madelmont, J.C.; Attaix, D. Liver protein synthesis stays elevated after chemotherapy in tumour-bearing mice. Cancer Lett. 2006, 239, 78–83. [Google Scholar] [CrossRef]
- Barber, M.D.; Fearon, K.C.; McMillan, D.C.; Slater, C.; Ross, J.A.; Preston, T. Liver export protein synthetic rates are increased by oral meal feeding in weight-losing cancer patients. Am. J. Physiol. Endocrinol. Metab. 2000, 279, 707. [Google Scholar] [CrossRef] [PubMed]
- Donovan, P.; Dubey, O.A.; Kallioinen, S.; Rogers, K.W.; Muehlethaler, K.; Muller, P.; Rimoldi, D.; Constam, D.B. Paracrine Activin-A Signaling Promotes Melanoma Growth and Metastasis through Immune Evasion. J. Investig. Dermatol. 2017, 137, 2578–2587. [Google Scholar] [CrossRef]
- Morianos, I.; Papadopoulou, G.; Semitekolou, M.; Xanthou, G. Activin-A in the regulation of immunity in health and disease. J. Autoimmun. 2019, 104, 102314. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, A.G.; Cuenca, A.L.; Winfield, R.D.; Joiner, D.N.; Gentile, L.; Delano, M.J.; Kelly-Scumpia, K.M.; Scumpia, P.O.; Matheny, M.K.; Scarpace, P.J.; et al. Novel role for tumor-induced expansion of myeloid-derived cells in cancer cachexia. J. Immunol. 2014, 192, 6111–6119. [Google Scholar] [CrossRef]
- Mundy-Bosse, B.L.; Lesinski, G.B.; Jaime-Ramirez, A.C.; Benninger, K.; Khan, M.; Kuppusamy, P.; Guenterberg, K.; Kondadasula, S.V.; Chaudhury, A.R.; La Perle, K.M.; et al. Myeloid-derived suppressor cell inhibition of the IFN response in tumor-bearing mice. Cancer Res. 2011, 71, 5101–5110. [Google Scholar] [CrossRef]
- Suragani, R.N.; Cawley, S.M.; Li, R.; Wallner, S.; Alexander, M.J.; Mulivor, A.W.; Gardenghi, S.; Rivella, S.; Grinberg, A.V.; Pearsall, R.S.; et al. Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine beta-thalassemia. Blood 2014, 123, 3864–3872. [Google Scholar] [CrossRef]
- Guntur, A.R.; Rosen, C.J. Bone as an endocrine organ. Endocr. Pract. 2012, 18, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.A.; Hornberger, T.A.; Robling, A.G. Bone and skeletal muscle: Key players in mechanotransduction and potential overlapping mechanisms. Bone 2015, 80, 24–36. [Google Scholar] [CrossRef]
- Brotto, M.; Bonewald, L. Bone and muscle: Interactions beyond mechanical. Bone 2015, 80, 109–114. [Google Scholar] [CrossRef]
- Puolakkainen, T.; Rummukainen, P.; Lehto, J.; Ritvos, O.; Hiltunen, A.; Saamanen, A.M.; Kiviranta, R. Soluble activin type IIB receptor improves fracture healing in a closed tibial fracture mouse model. PLoS ONE 2017, 12, e0180593. [Google Scholar] [CrossRef]
- Waning, D.L.; Mohammad, K.S.; Reiken, S.; Xie, W.; Andersson, D.C.; John, S.; Chiechi, A.; Wright, L.E.; Umanskaya, A.; Niewolna, M.; et al. Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat. Med. 2015, 21, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Huot, J.R.; Novinger, L.J.; Pin, F.; Narasimhan, A.; Zimmers, T.A.; O’Connell, T.M.; Bonetto, A. Formation of colorectal liver metastases induces musculoskeletal and metabolic abnormalities consistent with exacerbated cachexia. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.E.; Harhash, A.A.; Kozlow, W.M.; Waning, D.L.; Regan, J.N.; She, Y.; John, S.K.; Murthy, S.; Niewolna, M.; Marks, A.R.; et al. Aromatase inhibitor-induced bone loss increases the progression of estrogen receptor-negative breast cancer in bone and exacerbates muscle weakness in vivo. Oncotarget 2017, 8, 8406–8419. [Google Scholar] [CrossRef]
- Bialek, P.; Parkington, J.; Li, X.; Gavin, D.; Wallace, C.; Zhang, J.; Root, A.; Yan, G.; Warner, L.; Seeherman, H.J.; et al. A myostatin and activin decoy receptor enhances bone formation in mice. Bone 2014, 60, 162–171. [Google Scholar] [CrossRef]
- Zimmers, T.A.; Jiang, Y.; Wang, M.; Liang, T.W.; Rupert, J.E.; Au, E.D.; Marino, F.E.; Couch, M.E.; Koniaris, L.G. Exogenous GDF11 induces cardiac and skeletal muscle dysfunction and wasting. Basic Res. Cardiol. 2017, 112, 48. [Google Scholar] [CrossRef]
- David, L.; Mallet, C.; Mazerbourg, S.; Feige, J.J.; Bailly, S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007, 109, 1953–1961. [Google Scholar] [CrossRef]
- Campbell, C.; McMillan, H.J.; Mah, J.K.; Tarnopolsky, M.; Selby, K.; McClure, T.; Wilson, D.M.; Sherman, M.L.; Escolar, D.; Attie, K.M. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial. Muscle Nerve 2017, 55, 458–464. [Google Scholar] [CrossRef]
- Vaughan, D.; Mitchell, R.; Kretz, O.; Chambers, D.; Lalowski, M.; Amthor, H.; Ritvos, O.; Pasternack, A.; Matsakas, A.; Vaiyapuri, S.; et al. A muscle growth promoting treatment based on the attenuation of activin/myostatin signalling in young mice results in long-term testicular abnormalities. Dis. Models Mech. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tauer, J.T.; Rauch, F. Novel ActRIIB ligand trap increases muscle mass and improves bone geometry in a mouse model of severe osteogenesis imperfecta. Bone 2019, 128, 115036. [Google Scholar] [CrossRef] [PubMed]
- Pekkala, S.; Keskitalo, A.; Kettunen, E.; Lensu, S.; Nykanen, N.; Kuopio, T.; Ritvos, O.; Hentila, J.; Nissinen, T.A.; Hulmi, J.J. Blocking Activin Receptor Ligands Is Not Sufficient to Rescue Cancer-Associated Gut Microbiota-A Role for Gut Microbial Flagellin in Colorectal Cancer and Cachexia? Cancers 2019, 11, 1799. [Google Scholar] [CrossRef] [PubMed]
- Orell-Kotikangas, H.; Osterlund, P.; Makitie, O.; Saarilahti, K.; Ravasco, P.; Schwab, U.; Makitie, A.A. Cachexia at diagnosis is associated with poor survival in head and neck cancer patients. Acta Otolaryngol. 2017, 137, 778–785. [Google Scholar] [CrossRef]
- Wackerhage, H.; Schoenfeld, B.J.; Hamilton, D.L.; Lehti, M.; Hulmi, J.J. Stimuli and sensors that initiate skeletal muscle hypertrophy following resistance exercise. J. Appl. Physiol. 2019, 126, 30–43. [Google Scholar] [CrossRef]
- Fairman, C.M.; Zourdos, M.C.; Helms, E.R.; Focht, B.C. A Scientific Rationale to Improve Resistance Training Prescription in Exercise Oncology. Sports Med. 2017, 47, 1457–1465. [Google Scholar] [CrossRef]
- Cui, D.; Drake, J.C.; Wilson, R.J.; Shute, R.J.; Lewellen, B.; Zhang, M.; Zhao, H.; Sabik, O.L.; Onengut, S.; Berr, S.S.; et al. A novel voluntary weightlifting model in mice promotes muscle adaptation and insulin sensitivity with simultaneous enhancement of autophagy and mTOR pathway. FASEB J. 2020, 34, 7330–7344. [Google Scholar] [CrossRef]
- Murach, K.A.; McCarthy, J.J.; Peterson, C.A.; Dungan, C.M. Making Mice Mighty: Recent advances in translational models of load-induced muscle hypertrophy. J. Appl. Physiol. 2020, 129, 516–521. [Google Scholar] [CrossRef]
- Ballaro, R.; Beltra, M.; De Lucia, S.; Pin, F.; Ranjbar, K.; Hulmi, J.J.; Costelli, P.; Penna, F. Moderate exercise in mice improves cancer plus chemotherapy-induced muscle wasting and mitochondrial alterations. FASEB J. 2019, 33, 5482–5494. [Google Scholar] [CrossRef]
- Aquila, G.; Re Cecconi, A.D.; Brault, J.J.; Corli, O.; Piccirillo, R. Nutraceuticals and Exercise against Muscle Wasting during Cancer Cachexia. Cells 2020, 9, 2536. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hulmi, J.J.; Nissinen, T.A.; Penna, F.; Bonetto, A. Targeting the Activin Receptor Signaling to Counteract the Multi-Systemic Complications of Cancer and Its Treatments. Cells 2021, 10, 516. https://doi.org/10.3390/cells10030516
Hulmi JJ, Nissinen TA, Penna F, Bonetto A. Targeting the Activin Receptor Signaling to Counteract the Multi-Systemic Complications of Cancer and Its Treatments. Cells. 2021; 10(3):516. https://doi.org/10.3390/cells10030516
Chicago/Turabian StyleHulmi, Juha J., Tuuli A. Nissinen, Fabio Penna, and Andrea Bonetto. 2021. "Targeting the Activin Receptor Signaling to Counteract the Multi-Systemic Complications of Cancer and Its Treatments" Cells 10, no. 3: 516. https://doi.org/10.3390/cells10030516
APA StyleHulmi, J. J., Nissinen, T. A., Penna, F., & Bonetto, A. (2021). Targeting the Activin Receptor Signaling to Counteract the Multi-Systemic Complications of Cancer and Its Treatments. Cells, 10(3), 516. https://doi.org/10.3390/cells10030516