
Regulation of Mitochondrial Homeostasis by sAC-Derived cAMP Pool: Basic and Translational Aspects



Abstract
1. Introduction
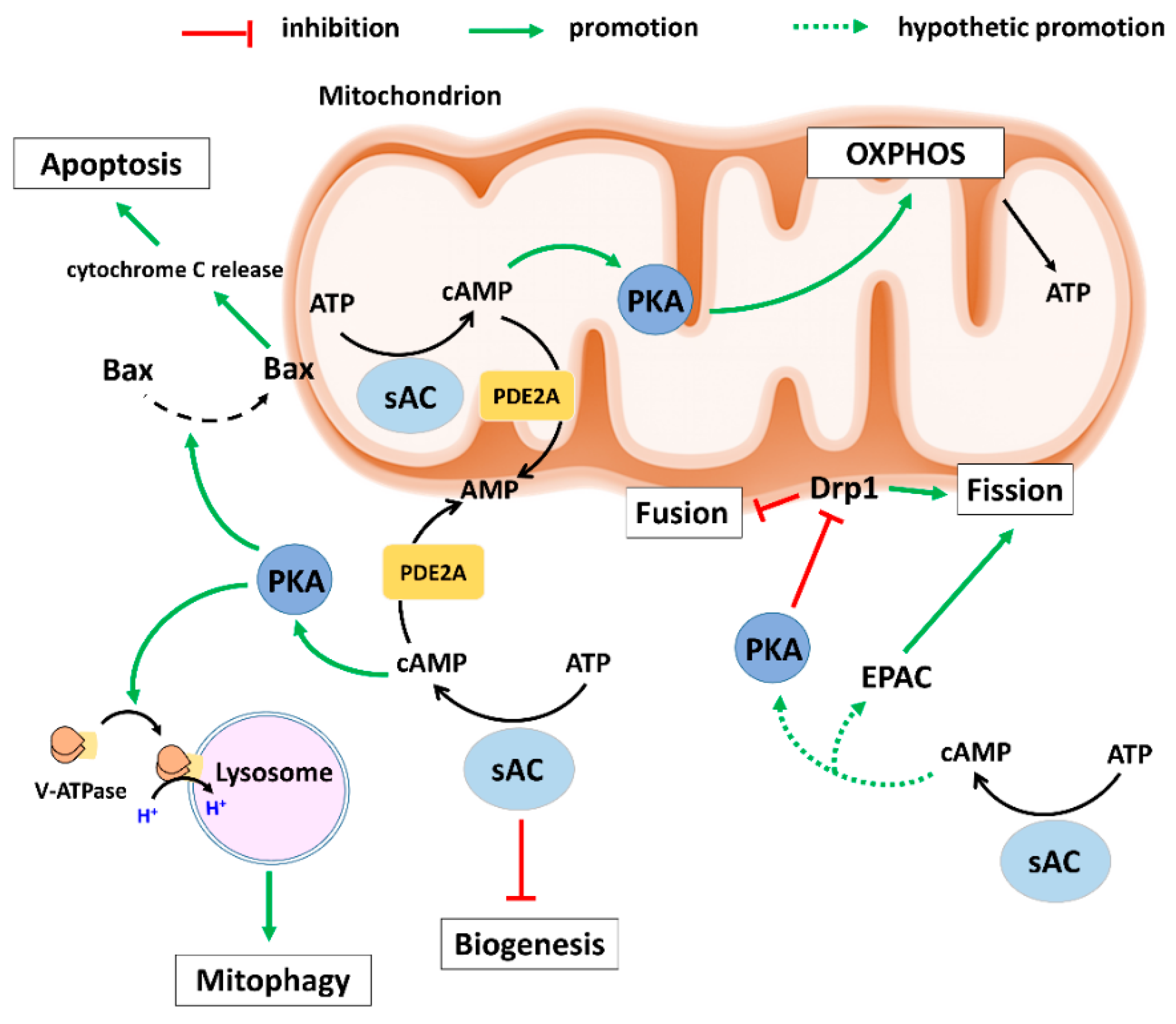
2. Role of the sAC-Signalling in Mitochondrial Biology
2.1. sAC and OXPHOS Activity
2.2. sAC and Mitochondrial Biogenesis
2.3. sAC and Mitochondrial Dynamics
2.4. sAC and Mitophagy
2.5. sAC and Apoptosis
3. Role of Dysregulation of Mitochondrial sAC Signalling in Pathologies
3.1. Ischaemia/Reperfusion
3.2. Preconditioning to I/R Injury
3.3. Heart Failure
3.4. Septic Cardiomyopathy
3.5. Amnesia
3.6. Cancer
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Corbin, J.; Sugden, P.; West, L.; Flockhart, D.; Lincoln, T.; McCarthy, D. Studies on the properties and mode of action of the purified regulatory subunit of bovine heart adenosine 3‘:5‘-monophosphate-dependent protein kinase. J. Biol. Chem. 1978, 253, 3997–4003. [Google Scholar] [CrossRef]
- Montminy, M. Transcriptional Regulation by Cyclic AMP. Annu. Rev. Biochem. 1997, 66, 807–822. [Google Scholar] [CrossRef]
- De Rooij, J.; Zwartkruis, F.J.T.; Verheijen, M.H.G.; Cool, R.H.; Nijman, S.M.B.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nat. Cell Biol. 1998, 396, 474–477. [Google Scholar] [CrossRef]
- Bos, J.L. Epac proteins: Multi-purpose cAMP targets. Trends Biochem. Sci. 2006, 31, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Zerio, A.; Lobo, M.J. Subcellular Organization of the cAMP Signaling Pathway. Pharmacol. Rev. 2021, 73, 278–309. [Google Scholar] [CrossRef]
- Wiggins, S.V.; Steegborn, C.; Levin, L.R.; Buck, J. Pharmacological modulation of the CO2/HCO3(-)/pH-, calcium-, and ATP-sensing soluble adenylyl cyclase. Pharmacol. Ther. 2018, 190, 173–186. [Google Scholar] [CrossRef]
- Zippin, J.H.; Chen, Y.; Straub, S.G.; Hess, K.C.; Diaz, A.; Lee, D.; Tso, P.; Holz, G.G.; Sharp, G.W.G.; Levin, L.R.; et al. CO2/HCO3−- and Calcium-regulated Soluble Adenylyl Cyclase as a Physiological ATP Sensor. J. Biol. Chem. 2013, 288, 33283–33291. [Google Scholar] [CrossRef]
- Zippin, J.H.; Chen, Y.; Nahirney, P.; Kamenetsky, M.; Wuttke, M.S.; Fischman, D.A.; Levin, L.R.; Buck, J. Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J. 2002, 17, 82–84. [Google Scholar] [CrossRef]
- Mika, D.; Leroy, J.; Vandecasteele, G.; Fischmeister, R. PDEs create local domains of cAMP signaling. J. Mol. Cell. Cardiol. 2012, 52, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bouadjel, K.; Manoury, B.; Vandecasteele, G.; Fischmeister, R.; Leblais, V. Cyclic nucleotide signalling compartmentation by PDEs in cultured vascular smooth muscle cells. Br. J. Pharmacol. 2019, 176, 1780–1792. [Google Scholar] [CrossRef] [PubMed]
- Beene, D.L.; Scott, J.D. A-kinase anchoring proteins take shape. Curr. Opin. Cell Biol. 2007, 19, 192–198. [Google Scholar] [CrossRef]
- Skroblin, P.; Grossmann, S.; Schäfer, G.; Rosenthal, W.; Klussmann, E. Mechanisms of Protein Kinase A Anchoring. Int. Rev. Cell Mol. Biol. 2010, 283, 235–330. [Google Scholar] [CrossRef]
- Ercu, M.; Klussmann, E. Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2018, 5, 14. [Google Scholar] [CrossRef]
- Godbole, A.; Lyga, S.; Lohse, M.J.; Calebiro, D. Internalized TSH receptors en route to the TGN induce local Gs-protein signaling and gene transcription. Nat. Commun. 2017, 8, 443. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP Produced inside Mitochondria Regulates Oxidative Phosphorylation. Cell Metab. 2009, 9, 265–276. [Google Scholar] [CrossRef]
- Pozdniakova, S.; Guitart-Mampel, M.; Garrabou, G.; Di Benedetto, G.; Ladilov, Y.; Regitz-Zagrosek, V. 17beta-Estradiol reduces mitochondrial cAMP content and cytochrome oxidase activity in a phosphodiesterase 2-dependent manner. Br. J. Pharmacol. 2018, 175, 3876–3890. [Google Scholar] [CrossRef]
- Zippin, J.H.; Chadwick, P.A.; Levin, L.R.; Buck, J.; Magro, C.M. Soluble Adenylyl Cyclase Defines a Nuclear cAMP Microdomain in Keratinocyte Hyperproliferative Skin Diseases. J. Investig. Dermatol. 2010, 130, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.L.; Buck, J.; Levin, L.R.; Winger, R.C.; Wang, J.; Arase, H.; Muller, W.A. Endothelial CD99 signals through soluble adenylyl cyclase and PKA to regulate leukocyte transendothelial migration. J. Exp. Med. 2015, 212, 1021–1041. [Google Scholar] [CrossRef]
- Ladilov, Y.; Appukuttan, A. Role of soluble adenylyl cyclase in cell death and growth. Biochim. Biophys. Acta 2014, 1842, 2646–2655. [Google Scholar] [CrossRef]
- Schmitz, B.; Nedele, J.; Guske, K.; Maase, M.; Lenders, M.; Schelleckes, M.; Kusche-Vihrog, K.; Brand, S.M.; Brand, E. Soluble adenylyl cyclase in vascular endothelium: Gene expression control of epithelial sodium channel-alpha, Na+/K+-ATPase-alpha/beta, and mineralocorticoid receptor. Hypertension 2014, 63, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Salazar, E.; Brosel, S.; Yang, H.; Schon, E.A.; Manfredi, G. Modulation of mitochondrial protein phosphorylation by soluble adenylyl cyclase ameliorates cytochrome oxidase defects. EMBO Mol. Med. 2009, 1, 392–406. [Google Scholar] [CrossRef]
- Pozdniakova, S.; Ladilov, Y. Functional Significance of the Adcy10-Dependent Intracellular cAMP Compartments. J. Cardiovasc. Dev. Dis. 2018, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Russwurm, M.; Günnewig, K.; Gertz, M.; Zoidl, G.; Ramos, L.; Buck, J.; Levin, L.R.; Rassow, J.; Manfredi, G.; et al. A Phosphodiesterase 2A Isoform Localized to Mitochondria Regulates Respiration. J. Biol. Chem. 2011, 286, 30423–30432. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Gerbino, A.; Lefkimmiatis, K. Shaping mitochondrial dynamics: The role of cAMP signalling. Biochem. Biophys. Res. Commun. 2018, 500, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Monterisi, S.; Zaccolo, M. Components of the mitochondrial cAMP signalosome. Biochem. Soc. Trans. 2017, 45, 269–274. [Google Scholar] [CrossRef]
- Grimsrud, P.A.; Carson, J.J.; Hebert, A.S.; Hubler, S.L.; Niemi, N.M.; Bailey, D.J.; Jochem, A.; Stapleton, D.S.; Keller, M.P.; Westphall, M.S.; et al. A Quantitative Map of the Liver Mitochondrial Phosphoproteome Reveals Posttranslational Control of Ketogenesis. Cell Metab. 2012, 16, 672–683. [Google Scholar] [CrossRef]
- Amer, Y.O.; Hebert-Chatelain, E. Insight into the Interactome of Intramitochondrial PKA Using Biotinylation-Proximity Labeling. Int. J. Mol. Sci. 2020, 21, 8283. [Google Scholar] [CrossRef]
- Jakobsen, E.; Lange, S.C.; Bak, L.K. Soluble adenylyl cyclase-mediated cAMP signaling and the putative role of PKA and EPAC in cerebral mitochondrial function. J. Neurosci. Res. 2019, 97, 1018–1038. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, D.; Varin, A.; Nicolas, V.; Courilleau, D.; Mateo, P.; Caubere, C.; Rouet, P.; Gomez, A.-M.; Vandecasteele, G.; et al. A cardiac mitochondrial cAMP signaling pathway regulates calcium accumulation, permeability transition and cell death. Cell Death Dis. 2016, 7, e2198. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Gatti, D.L.; Bai, Y.; Manfredi, G. Protein Phosphorylation and Prevention of Cytochrome Oxidase Inhibition by ATP: Coupled Mechanisms of Energy Metabolism Regulation. Cell Metab. 2011, 13, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Valsecchi, F.; Konrad, C.; D’Aurelio, M.; Ramos-Espiritu, L.S.; Stepanova, A.; Burstein, S.R.; Galkin, A.; Magrane, J.; Starkov, A.; Buck, J.; et al. Distinct intracellular sAC-cAMP domains regulate ER Ca(2+) signaling and OXPHOS function. J. Cell Sci. 2017, 130, 3713–3727. [Google Scholar] [CrossRef]
- Hess, K.C.; Liu, J.; Manfredi, G.; Muhlschlegel, F.A.; Buck, J.; Levin, L.R.; Barrientos, A. A mitochondrial CO2-adenylyl cyclase-cAMP signalosome controls yeast normoxic cytochrome c oxidase activity. FASEB J. 2014, 28, 4369–4380. [Google Scholar] [CrossRef]
- De Rasmo, D.; Signorile, A.; Santeramo, A.; Larizza, M.; Lattanzio, P.; Capitanio, G.; Papa, S. Intramitochondrial adenylyl cyclase controls the turnover of nuclear-encoded subunits and activity of mammalian complex I of the respiratory chain. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1853, 183–191. [Google Scholar] [CrossRef]
- De Rasmo, D.; Micelli, L.; Santeramo, A.; Signorile, A.; Lattanzio, P.; Papa, S. cAMP regulates the functional activity, coupling efficiency and structural organization of mammalian FOF1 ATP synthase. Biochim. Biophys. Acta 2016, 1857, 350–358. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Scalzotto, E.; Mongillo, M.; Pozzan, T. Mitochondrial Ca2+ Uptake Induces Cyclic AMP Generation in the Matrix and Modulates Organelle ATP Levels. Cell Metab. 2013, 17, 965–975. [Google Scholar] [CrossRef]
- Hebert-Chatelain, E.; Desprez, T.; Serrat, R.; Bellocchio, L.; Soria-Gomez, E.; Busquets-Garcia, A.; Zottola, A.C.P.; Delamarre, A.; Cannich, A.; Vincent, P.; et al. A cannabinoid link between mitochondria and memory. Nature 2016, 539, 555–559. [Google Scholar] [CrossRef]
- Pavlaki, N.; Nikolaev, V.O. Imaging of PDE2- and PDE3-Mediated cGMP-to-cAMP Cross-Talk in Cardiomyocytes. J. Cardiovasc. Dev. Dis. 2018, 5, 4. [Google Scholar] [CrossRef]
- Monterisi, S.; Lobo, M.J.; Livie, C.; Castle, J.C.; Weinberger, M.; Baillie, G.S.; Surdo, N.C.; Musheshe, N.; Stangherlin, A.; Gottlieb, E.; et al. PDE2A2 regulates mitochondria morphology and apoptotic cell death via local modulation of cAMP/PKA signalling. eLife 2017, 6, e21374. [Google Scholar] [CrossRef] [PubMed]
- Pattengale, P.K.; Holloszy, J.O. Augmentation of skeletal muscle myoglobin by a program of treadmill running. Am. J. Physiol. Content 1967, 213, 783–785. [Google Scholar] [CrossRef]
- Gopalakrishnan, L.; Scarpulla, R. Differential regulation of respiratory chain subunits by a CREB-dependent signal transduction pathway. Role of cyclic AMP in cytochrome c and COXIV gene expression. J. Biol. Chem. 1994, 269, 105–113. [Google Scholar] [CrossRef]
- Bouchez, C.; Devin, A. Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (ROS): A Complex Relationship Regulated by the cAMP/PKA Signaling Pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef]
- Ding, H.; Bai, F.; Cao, H.; Xu, J.; Fang, L.; Wu, J.; Yuan, Q.; Zhou, Y.; Sun, Q.; He, W.; et al. PDE/cAMP/Epac/C/EBP-beta Signaling Cascade Regulates Mitochondria Biogenesis of Tubular Epithelial Cells in Renal Fibrosis. Antioxid. Redox Signal. 2018, 29, 637–652. [Google Scholar] [CrossRef]
- Yoboue, E.D.; Augier, E.; Galinier, A.; Blancard, C.; Pinson, B.; Casteilla, L.; Rigoulet, M.; Devin, A. cAMP-induced mitochondrial compartment biogenesis: Role of glutathione redox state. J. Biol. Chem. 2012, 287, 14569–14578. [Google Scholar] [CrossRef]
- Bogacka, I.; Ukropcova, B.; McNeil, M.; Gimble, J.M.; Smith, S.R. Structural and functional consequences of mitochondrial biogenesis in human adipocytes in vitro. J. Clin. Endocrinol. Metab. 2005, 90, 6650–6656. [Google Scholar] [CrossRef]
- Wang, Y.; Kang, Y.; Qi, C.; Zhang, T.; Zhao, H.; Ji, X.; Yan, W.; Huang, Y.; Cui, R.; Zhang, G.; et al. Pentoxifylline enhances antioxidative capability and promotes mitochondrial biogenesis for improving age-related behavioral deficits. Aging 2020, 12, 25487–25504. [Google Scholar] [CrossRef]
- Gak, I.A.; Radovic, S.M.; Dukic, A.R.; Janjic, M.M.; Stojkov-Mimic, N.J.; Kostic, T.S.; Andric, S.A. Stress triggers mitochondrial biogenesis to preserve steroidogenesis in Leydig cells. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1853, 2217–2227. [Google Scholar] [CrossRef]
- Park, S.-J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol Ameliorates Aging-Related Metabolic Phenotypes by Inhibiting cAMP Phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef]
- Lee, J.; Kim, C.-H.; Simon, D.K.; Aminova, L.R.; Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Lonze, B.E.; Kim, K.-S.; Ginty, D.D.; et al. Mitochondrial Cyclic AMP Response Element-binding Protein (CREB) Mediates Mitochondrial Gene Expression and Neuronal Survival. J. Biol. Chem. 2005, 280, 40398–40401. [Google Scholar] [CrossRef]
- Sepuri, N.B.V.; Tammineni, P.; Mohammed, F.; Paripati, A. Nuclear Transcription Factors in the Mitochondria: A New Paradigm in Fine-Tuning Mitochondrial Metabolism. Handb. Exp. Pharmacol. 2016, 240, 3–20. [Google Scholar] [CrossRef]
- Archer, S.L. Mitochondrial Dynamics—Mitochondrial Fission and Fusion in Human Diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [PubMed]
- Mewes, M.; Lenders, M.; Stappers, F.; Scharnetzki, D.; Nedele, J.; Fels, J.; Wedlich-Söldner, R.; Brand, S.-M.; Schmitz, B.; Brand, E. Soluble adenylyl cyclase (sAC) regulates calcium signaling in the vascular endothelium. FASEB J. 2019, 33, 13762–13774. [Google Scholar] [CrossRef]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, K.-H.; Cao, W.; Zeng, J.; Liao, H.; Zhao, L.; Guo, X. Mutation of the protein kinase A phosphorylation site influences the anti-proliferative activity of mitofusin 2. Atherosclerosis 2010, 211, 216–223. [Google Scholar] [CrossRef]
- Wang, H.; Robichaux, W.G.; Wang, Z.; Mei, F.C.; Cai, M.; Du, G.; Chen, J.; Cheng, X. Inhibition of Epac1 suppresses mitochondrial fission and reduces neointima formation induced by vascular injury. Sci. Rep. 2016, 6, 36552. [Google Scholar] [CrossRef] [PubMed]
- Means, C.K.; Lygren, B.; Langeberg, L.K.; Jain, A.; Dixon, R.E.; Vega, A.L.; Gold, M.G.; Petrosyan, S.; Taylor, S.S.; Murphy, A.N.; et al. An entirely specific type I A-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, E1227–E1235. [Google Scholar] [CrossRef]
- Kovanich, D.; van der Heyden, M.A.; Aye, T.T.; van Veen, T.A.; Heck, A.J.; Scholten, A. Sphingosine kinase interacting protein is an A-kinase anchoring protein specific for type I cAMP-dependent protein kinase. Chembiochem 2010, 11, 963–971. [Google Scholar] [CrossRef]
- Kozjak-Pavlovic, V. The MICOS complex of human mitochondria. Cell Tissue Res. 2016, 367, 83–93. [Google Scholar] [CrossRef]
- Scholten, A.; Poh, M.K.; Van Veen, T.A.B.; Van Breukelen, B.; Vos, M.A.; Heck, A.J.R. Analysis of the cGMP/cAMP Interactome Using a Chemical Proteomics Approach in Mammalian Heart Tissue Validates Sphingosine Kinase Type 1-interacting Protein as a Genuine and Highly Abundant AKAP. J. Proteome Res. 2006, 5, 1435–1447. [Google Scholar] [CrossRef]
- Darshi, M.; Mendiola, V.L.; Mackey, M.R.; Murphy, A.N.; Koller, A.; Perkins, G.A.; Ellisman, M.H.; Taylor, S.S. ChChd3, an Inner Mitochondrial Membrane Protein, Is Essential for Maintaining Crista Integrity and Mitochondrial Function*. J. Biol. Chem. 2011, 286, 2918–2932. [Google Scholar] [CrossRef]
- Lefkimmiatis, K.; Leronni, D.; Hofer, A.M. The inner and outer compartments of mitochondria are sites of distinct cAMP/PKA signaling dynamics. J. Cell Biol. 2013, 202, 453–462. [Google Scholar] [CrossRef]
- Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R.; et al. Mitophagy in Cardiovascular Diseases. J. Clin. Med. 2020, 9, 892. [Google Scholar] [CrossRef]
- Sekine, S. PINK1 import regulation at a crossroad of mitochondrial fate: The molecular mechanisms of PINK1 import. J. Biochem. 2020, 167, 217–224. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Lobo, M.J.; Reverte-Salisa, L.; Chao, Y.-C.; Koschinski, A.; Gesellchen, F.; Subramaniam, G.; Jiang, H.; Pace, S.; Larcom, N.; Paolocci, E.; et al. Phosphodiesterase 2A2 regulates mitochondria clearance through Parkin-dependent mitophagy. Commun. Biol. 2020, 3, 1–16. [Google Scholar] [CrossRef]
- Akabane, S.; Uno, M.; Tani, N.; Shimazaki, S.; Ebara, N.; Kato, H.; Kosako, H.; Oka, T. PKA Regulates PINK1 Stability and Parkin Recruitment to Damaged Mitochondria through Phosphorylation of MIC60. Mol. Cell 2016, 62, 371–384. [Google Scholar] [CrossRef]
- Jayarajan, V.; Appukuttan, A.; Aslam, M.; Reusch, P.; Regitz-Zagrosek, V.; Ladilov, Y. Regulation of AMPK activity by type 10 adenylyl cyclase: Contribution to the mitochondrial biology, cellular redox and energy homeostasis. Cell. Mol. Life Sci. 2019, 76, 4945–4959. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.; Ramos-Espiritu, L.; Milner, T.A.; Buck, J.; Levin, L.R. Soluble adenylyl cyclase is essential for proper lysosomal acidification. J. Gen. Physiol. 2016, 148, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Chagtoo, M.; George, N.; Pathak, N.; Tiwari, S.; Godbole, M.M.; Ladilov, Y. Inhibition of Intracellular Type 10 Adenylyl Cyclase Protects Cortical Neurons Against Reperfusion-Induced Mitochondrial Injury and Apoptosis. Mol. Neurobiol. 2018, 55, 2471–2482. [Google Scholar] [CrossRef] [PubMed]
- Appukuttan, A.; Kasseckert, S.A.; Micoogullari, M.; Flacke, J.-P.; Kumar, S.; Woste, A.; Abdallah, Y.; Pott, L.; Reusch, H.P.; Ladilov, Y. Type 10 adenylyl cyclase mediates mitochondrial Bax translocation and apoptosis of adult rat cardiomyocytes under simulated ischaemia/reperfusion. Cardiovasc. Res. 2011, 93, 340–349. [Google Scholar] [CrossRef]
- Kumar, S.; Kostin, S.; Flacke, J.-P.; Reusch, H.P.; Ladilov, Y. Soluble Adenylyl Cyclase Controls Mitochondria-dependent Apoptosis in Coronary Endothelial Cells. J. Biol. Chem. 2009, 284, 14760–14768. [Google Scholar] [CrossRef]
- Appukuttan, A.; Kasseckert, S.A.; Kumar, S.; Reusch, H.P.; Ladilov, Y. Oxysterol-induced apoptosis of smooth muscle cells is under the control of a soluble adenylyl cyclase. Cardiovasc. Res. 2013, 99, 734–742. [Google Scholar] [CrossRef]
- Chang, J.-C.; Go, S.; De Waart, D.R.; Munoz-Garrido, P.; Beuers, U.; Paulusma, C.C.; Elferink, R.O. Soluble Adenylyl Cyclase Regulates Bile Salt-Induced Apoptosis in Human Cholangiocytes. Hepatology 2016, 64, 522–534. [Google Scholar] [CrossRef]
- Kumar, S.; Appukuttan, A.; Maghnouj, A.; Hahn, S.; Reusch, H.P.; Ladilov, Y. Suppression of soluble adenylyl cyclase protects smooth muscle cells against oxidative stress-induced apoptosis. Apoptosis 2014, 19, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Signorile, A.; Santeramo, A.; Tamma, G.; Pellegrino, T.; D’Oria, S.; Lattanzio, P.; De Rasmo, D. Mitochondrial cAMP prevents apoptosis modulating Sirt3 protein level and OPA1 processing in cardiac myoblast cells. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1864, 355–366. [Google Scholar] [CrossRef]
- Abdallah, Y.; Kasseckert, S.A.; Iraqi, W.; Said, M.; Shahzad, T.; Erdogan, A.; Neuhof, C.; Gündüz, D.; Schlüter, K.-D.; Tillmanns, H.; et al. Interplay between Ca2+ cycling and mitochondrial permeability transition pores promotes reperfusion-induced injury of cardiac myocytes. J. Cell. Mol. Med. 2011, 15, 2478–2485. [Google Scholar] [CrossRef]
- Rinaldi, L.; Pozdniakova, S.; Jayarajan, V.; Troidl, C.; Abdallah, Y.; Aslam, M.; Ladilov, Y. Protective role of soluble adenylyl cyclase against reperfusion-induced injury of cardiac cells. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Soares, L.M.; Meyer, E.; Milani, H.; Steinbusch, H.W.M.; Prickaerts, J.; De Oliveira, R.M.W. The phosphodiesterase type 2 inhibitor BAY 60-7550 reverses functional impairments induced by brain ischemia by decreasing hippocampal neurodegeneration and enhancing hippocampal neuronal plasticity. Eur. J. Neurosci. 2017, 45, 510–520. [Google Scholar] [CrossRef]
- Fazal, L.; Laudette, M.; Paula-Gomes, S.; Pons, S.; Conte, C.; Tortosa, F.; Sicard, P.; Sainte-Marie, Y.; Bisserier, M.; Lairez, O.; et al. Multifunctional Mitochondrial Epac1 Controls Myocardial Cell Death. Circ. Res. 2017, 120, 645–657. [Google Scholar] [CrossRef]
- Jakobsen, E.; Lange, S.C.; Andersen, J.V.; Desler, C.; Kihl, H.F.; Hohnholt, M.C.; Stridh, M.H.; Rasmussen, L.J.; Waagepetersen, H.S.; Bak, L.K. The inhibitors of soluble adenylate cyclase 2-OHE, KH7, and bithionol compromise mitochondrial ATP production by distinct mechanisms. Biochem. Pharmacol. 2018, 155, 92–101. [Google Scholar] [CrossRef]
- Siraj, M.A.; Mundil, D.; Beca, S.; Momen, A.; Shikatani, E.A.; Afroze, T.; Sun, X.; Liu, Y.; Ghaffari, S.; Lee, W.; et al. Cardioprotective GLP-1 metabolite prevents ischemic cardiac injury by inhibiting mitochondrial trifunctional protein-α. J. Clin. Investig. 2020, 130, 1392–1404. [Google Scholar] [CrossRef]
- Teodoro, J.S.; Amorim, J.A.; Machado, I.F.; Castela, A.C.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P.; Palmeira, C.M. The Soluble Adenylyl Cyclase Inhibitor LRE1 Prevents Hepatic Ischemia/Reperfusion Damage Through Improvement of Mitochondrial Function. Int. J. Mol. Sci. 2020, 21, 4896. [Google Scholar] [CrossRef]
- Court, O.; Kumar, A.; Parrillo, J.E. Clinical review: Myocardial depression in sepsis and septic shock. Crit. Care 2002, 6, 500. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-C.; Yu, M.-M.; Shou, S.-T.; Chai, Y.-F. Sepsis-Induced Cardiomyopathy: Mechanisms and Treatments. Front. Immunol. 2017, 8, 1021. [Google Scholar] [CrossRef] [PubMed]
- Fenton, K.E.; Parker, M.M. Cardiac Function and Dysfunction in Sepsis. Clin. Chest Med. 2016, 37, 289–298. [Google Scholar] [CrossRef]
- Martin, L.; Derwall, M.; Al Zoubi, S.; Zechendorf, E.; Reuter, D.A.; Thiemermann, C.; Schuerholz, T. The Septic Heart: Current Understanding of Molecular Mechanisms and Clinical Implications. Chest 2019, 155, 427–437. [Google Scholar] [CrossRef]
- Takasu, O.; Gaut, J.P.; Watanabe, E.; To, K.; Fagley, R.E.; Sato, B.; Jarman, S.; Efimov, I.R.; Janks, D.L.; Srivastava, A.; et al. Mechanisms of Cardiac and Renal Dysfunction in Patients Dying of Sepsis. Am. J. Respir. Crit. Care Med. 2013, 187, 509–517. [Google Scholar] [CrossRef]
- Vanasco, V.; Saez, T.; Magnani, N.D.; Pereyra, L.; Marchini, T.; Corach, A.; Vaccaro, M.I.; Corach, D.; Evelson, P.; Alvarez, S. Cardiac mitochondrial biogenesis in endotoxemia is not accompanied by mitochondrial function recovery. Free. Radic. Biol. Med. 2014, 77, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zang, Q.S.; Wolf, S.E.; Minei, J.P. Sepsis-induced Cardiac Mitochondrial Damage and Potential Therapeutic Interventions in the Elderly. Aging Dis. 2014, 5, 137–149. [Google Scholar] [CrossRef]
- Suliman, H.B.; Welty-Wolf, K.E.; Carraway, M.; Tatro, L.; Piantadosi, C.A. Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc. Res. 2004, 64, 279–288. [Google Scholar] [CrossRef]
- Sánchez-Villamil, J.P.; D’Annunzio, V.; Finocchietto, P.; Holod, S.; Rebagliati, I.; Pérez, H.; Peralta, J.G.; Gelpi, R.J.; Poderoso, J.J.; Carreras, M.C. Cardiac-specific overexpression of thioredoxin 1 attenuates mitochondrial and myocardial dysfunction in septic mice. Int. J. Biochem. Cell Biol. 2016, 81, 323–334. [Google Scholar] [CrossRef]
- Gonzalez, A.S.; Elguero, M.E.; Finocchietto, P.; Holod, S.; Romorini, L.; Miriuka, S.G.; Peralta, J.G.; Poderoso, J.J.; Carreras, M.C. Abnormal mitochondrial fusion–fission balance contributes to the progression of experimental sepsis. Free. Radic. Res. 2014, 48, 769–783. [Google Scholar] [CrossRef]
- Wu, Y.; Yao, Y.-M.; Lu, Z.-Q. Mitochondrial quality control mechanisms as potential therapeutic targets in sepsis-induced multiple organ failure. J. Mol. Med. 2019, 97, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Neviere, R.; Delguste, F.; Durand, A.; Inamo, J.; Boulanger, E.; Preau, S. Abnormal Mitochondrial cAMP/PKA Signaling Is Involved in Sepsis-Induced Mitochondrial and Myocardial Dysfunction. Int. J. Mol. Sci. 2016, 17, 2075. [Google Scholar] [CrossRef]
- Didier, S.; Sauvé, F.; Domise, M.; Buée, L.; Marinangeli, C.; Vingtdeux, V. AMP-activated Protein Kinase Controls Immediate Early Genes Expression Following Synaptic Activation Through the PKA/CREB Pathway. Int. J. Mol. Sci. 2018, 19, 3716. [Google Scholar] [CrossRef] [PubMed]
- Flacke, J.-P.; Flacke, H.; Appukuttan, A.; Palisaar, R.-J.; Noldus, J.; Robinson, B.D.; Reusch, H.P.; Zippin, J.H.; Ladilov, Y. Type 10 Soluble Adenylyl Cyclase Is Overexpressed in Prostate Carcinoma and Controls Proliferation of Prostate Cancer Cells. J. Biol. Chem. 2013, 288, 3126–3135. [Google Scholar] [CrossRef]
- Appukuttan, A.; Flacke, J.-P.; Flacke, H.; Posadowsky, A.; Reusch, H.P.; Ladilov, Y. Inhibition of soluble adenylyl cyclase increases the radiosensitivity of prostate cancer cells. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 2656–2663. [Google Scholar] [CrossRef][Green Version]
- Ramos-Espiritu, L.; Diaz, A.; Nardin, C.; Saviola, A.J.; Shaw, F.; Plitt, T.; Yang, X.; Wolchok, J.; Pirog, E.C.; Desman, G.; et al. The metabolic/pH sensor soluble adenylyl cyclase is a tumor suppressor protein. Oncotarget 2016, 7, 45597–45607. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Xing, F.; Luan, Y.; Cai, J.; Wu, S.; Mai, J.; Gu, J.; Zhang, H.; Li, K.; Lin, Y.; Xiao, X.; et al. The Anti-Warburg Effect Elicited by the cAMP-PGC1alpha Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep. 2017, 18, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-C.; Go, S.; Gilglioni, E.H.; Duijst, S.; Panneman, D.M.; Rodenburg, R.J.; Li, H.L.; Huang, H.-L.; Levin, L.R.; Buck, J.; et al. Soluble adenylyl cyclase regulates the cytosolic NADH/NAD+ redox state and the bioenergetic switch between glycolysis and oxidative phosphorylation. Biochim. Biophys. Acta (BBA) Bioenerg. 2021, 1862, 148367. [Google Scholar] [CrossRef]
- Choi, H.B.; Gordon, G.R.; Zhou, N.; Tai, C.; Rungta, R.L.; Martinez, J.; Milner, T.A.; Ryu, J.K.; McLarnon, J.G.; Tresguerres, M.; et al. Metabolic Communication between Astrocytes and Neurons via Bicarbonate-Responsive Soluble Adenylyl Cyclase. Neuron 2012, 75, 1094–1104. [Google Scholar] [CrossRef]


{kind=link}
| Pathologies | Species | sAC Alterations | Mitochondrial Effects | References |
|---|---|---|---|---|
| Amnesia | mice | Inhibition of activity | Reduced respiration | [36] |
| Ischemia/reperfusion | rats | Not studied | Mitochondrial apoptosis | [69,70,76] |
| Heart failure | rats | Reduced expression | Reduced mitochondrial Ca2+-resistance | [29] |
| Septic cardiomyopathy | mice | Not studied | Impaired ADP-stimulated respiratory rate | [93] |
| Prostate cancer | human | Upregulated | Not studied | [95,97] |
| Anaplastic oligoastrocytoma | human | Upregulated | Not studied | [97] |
| Several haematologic cancers | human | Downregulated | Not studied | [97] |
| Several solid cancers | human | Downregulated | Not studied | [97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aslam, M.; Ladilov, Y. Regulation of Mitochondrial Homeostasis by sAC-Derived cAMP Pool: Basic and Translational Aspects. Cells 2021, 10, 473. https://doi.org/10.3390/cells10020473
Aslam M, Ladilov Y. Regulation of Mitochondrial Homeostasis by sAC-Derived cAMP Pool: Basic and Translational Aspects. Cells. 2021; 10(2):473. https://doi.org/10.3390/cells10020473
Chicago/Turabian StyleAslam, Muhammad, and Yury Ladilov. 2021. "Regulation of Mitochondrial Homeostasis by sAC-Derived cAMP Pool: Basic and Translational Aspects" Cells 10, no. 2: 473. https://doi.org/10.3390/cells10020473
APA StyleAslam, M., & Ladilov, Y. (2021). Regulation of Mitochondrial Homeostasis by sAC-Derived cAMP Pool: Basic and Translational Aspects. Cells, 10(2), 473. https://doi.org/10.3390/cells10020473