
Ablation of Aquaporin-9 Ameliorates the Systemic Inflammatory Response of LPS-Induced Endotoxic Shock in Mouse



Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. LPS-Induced Endotoxic Shock
2.3. Cell Culture
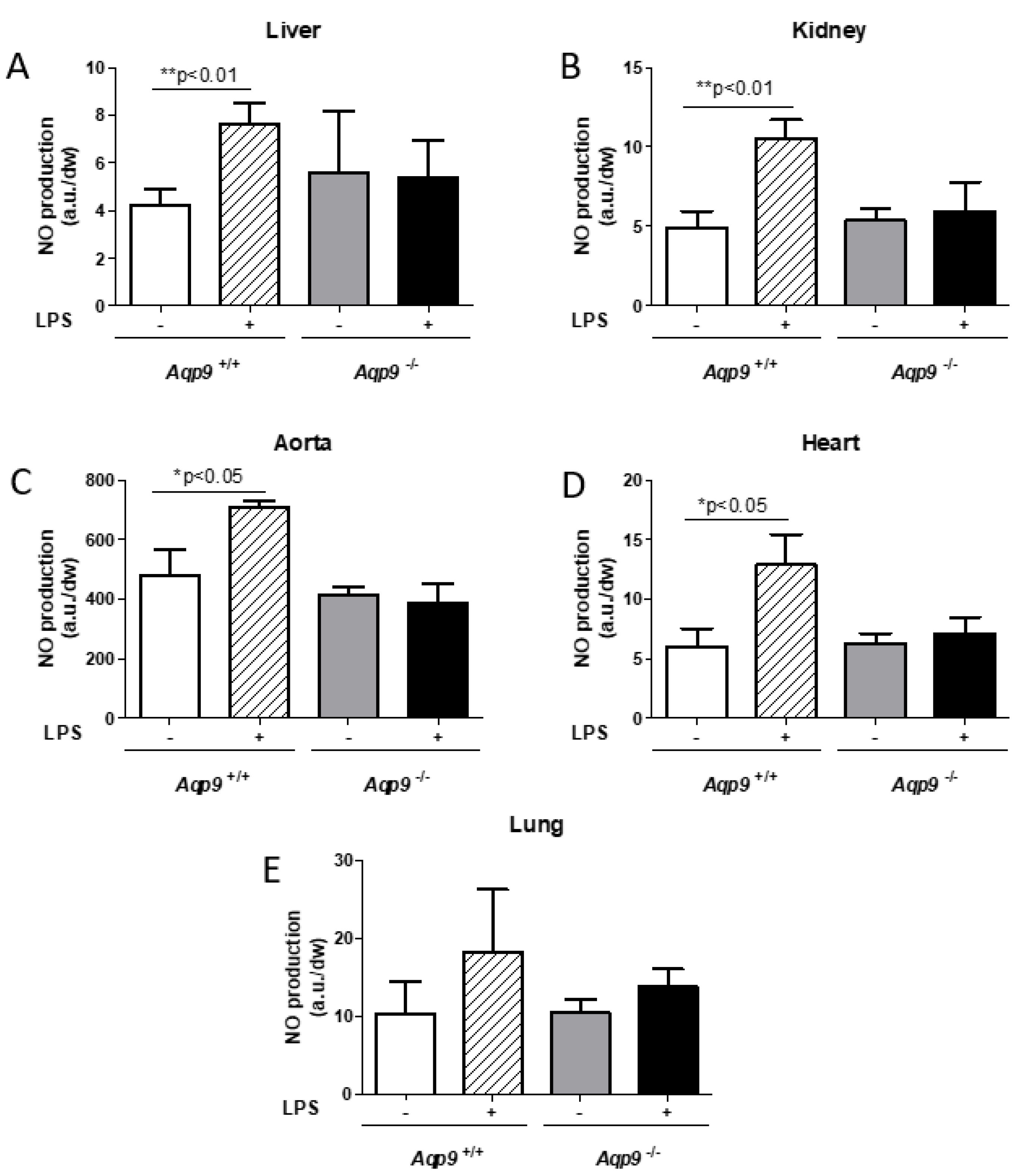
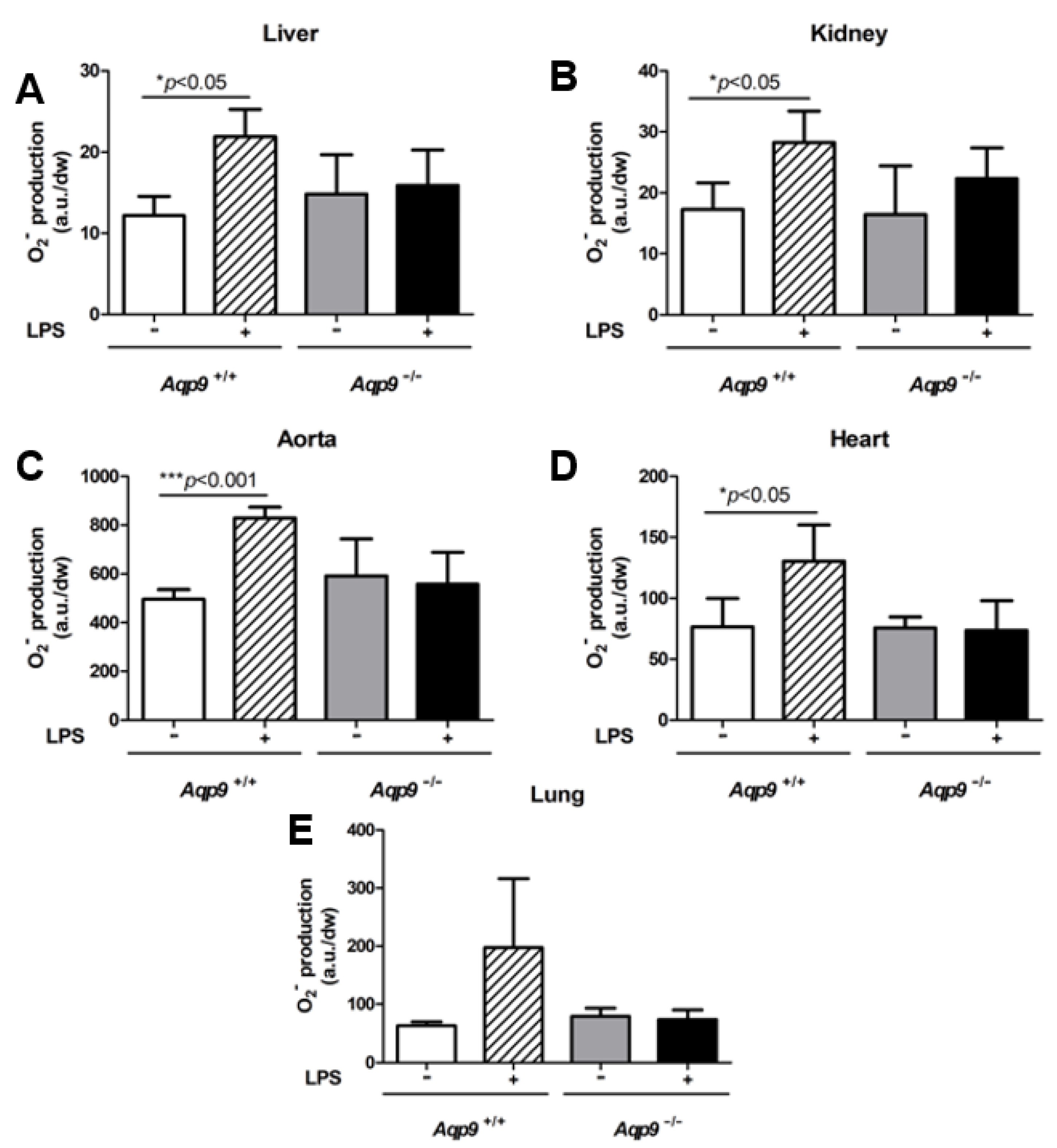
2.4. Electronic Paramagnetic Resonance (EPR) Studies for NO and O2− Measurements
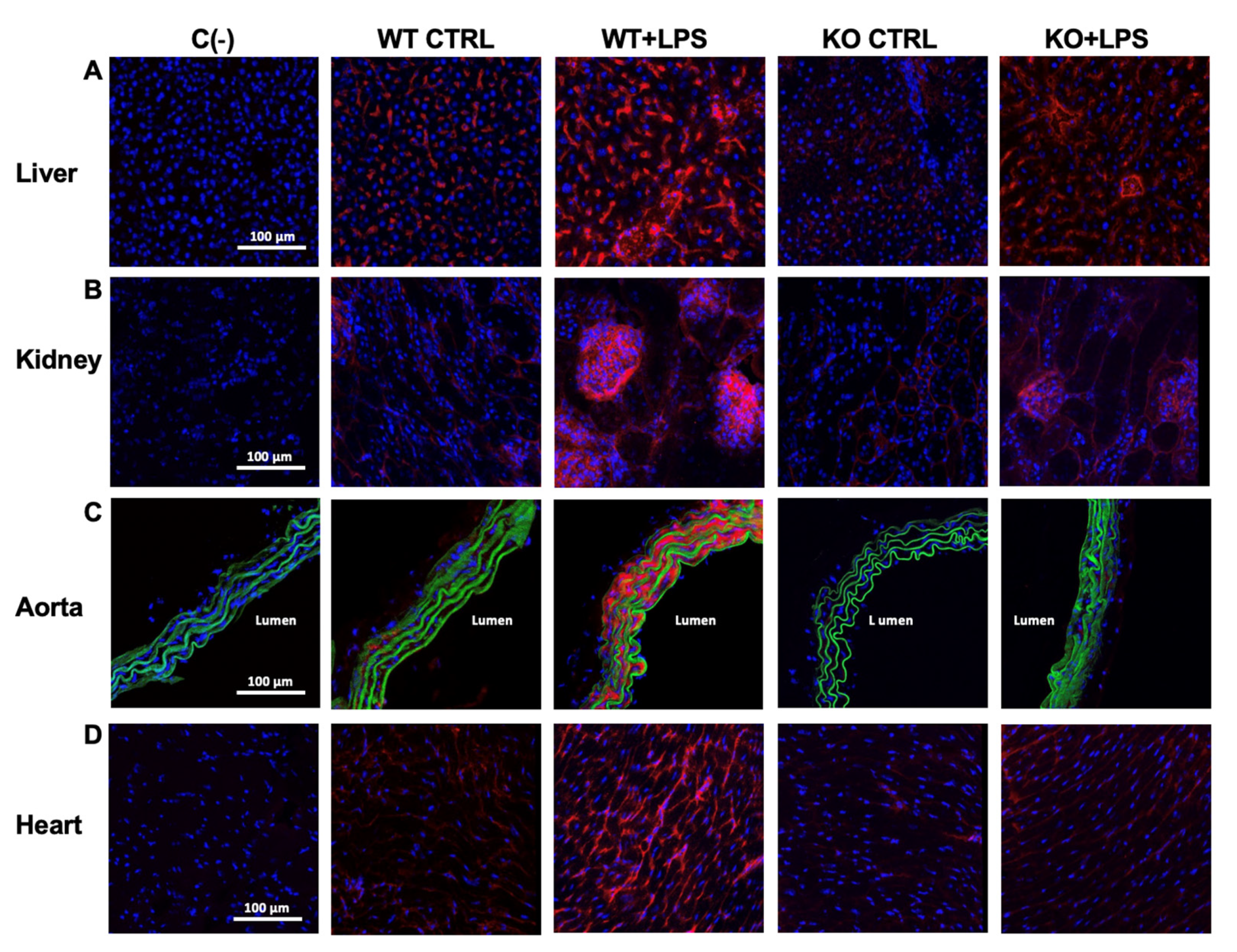
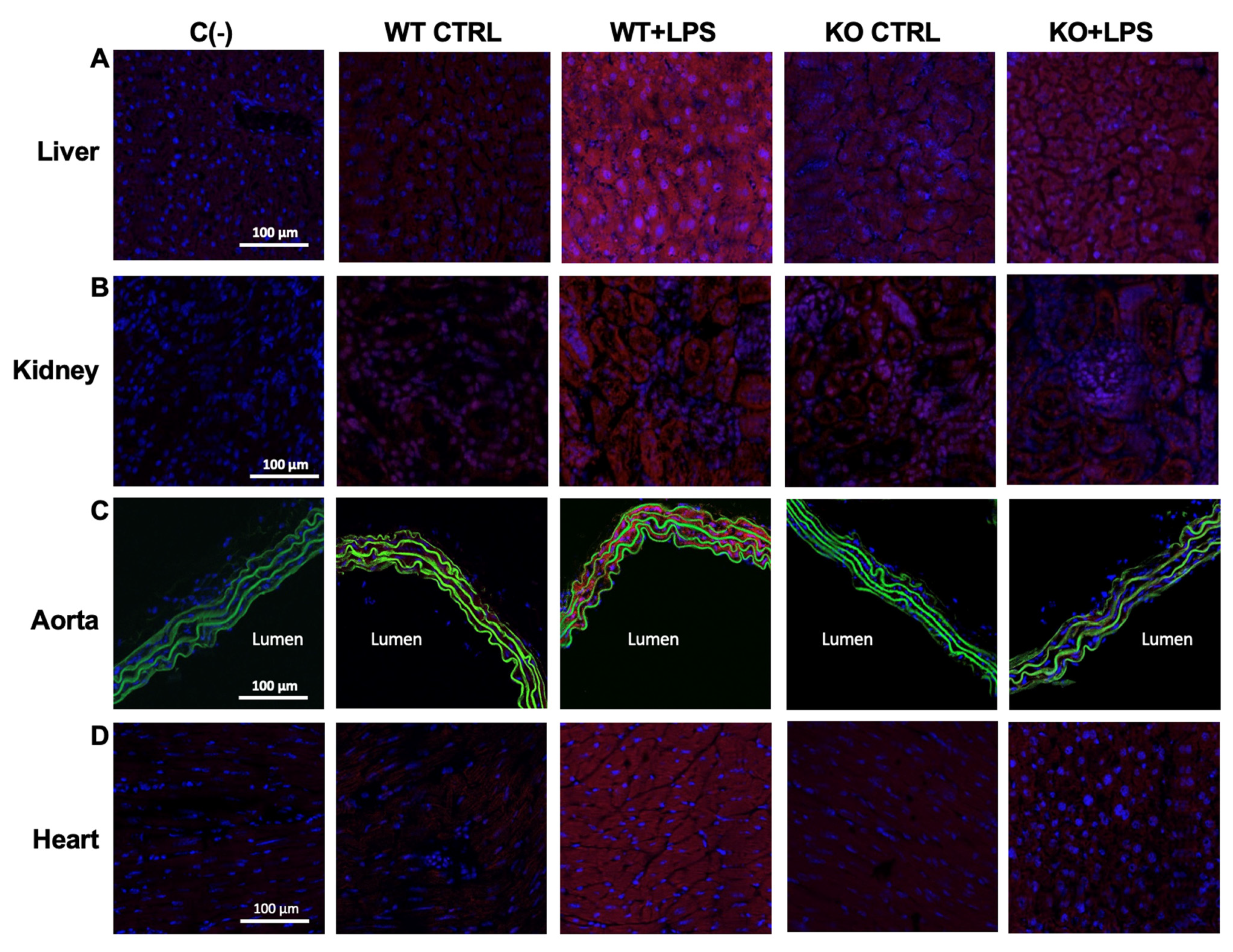
2.5. Staining and Confocal Microscopy Imaging
2.6. Data Analysis
3. Results
3.1. Aqp9 Deletion Improves Survival of Mice after LPS Exposure
3.2. Deletion of AQP9 Protein Protects Against LPS Induced Inflammation
3.3. Aqp9 Deletion Reduces NF-κB p65 Elevation by LPS
3.4. Lack of AQP9 Protects Against LPS Induced Oxidative Stress
3.5. Inhibition of AQP9 in Rat Hepatoma Cells Reduces the LPS-Induced NO and Superoxide anion Production
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Mayr, F.B.; Yende, S.; Angus, D.C. Epidemiology of severe sepsis. Virulence 2014, 5, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Schorr, C.A.; Zanotti, S.; Dellinger, R.P. Severe sepsis and septic shock: Management and performance improvement. Virulence 2014, 5, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Erridge, C.; Bennett-Guerrero, E.; Poxton, I.R. Structure and function of lipopolysaccharides. Microbes Infect. 2002, 4, 837–851. [Google Scholar] [CrossRef]
- Van Amersfoot, E.S.; Van Berkel, T.J.C.; Kuiper, J. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clin. Microbiol. Rev. 2003, 16, 379–414. [Google Scholar] [CrossRef] [PubMed]
- Mariajoseph-Antony, L.F.; Kannan, A.; Panneerselvam, A.; Loganathan, C.; Shankar, E.M.; Anbarasu, K.; Prahalathan, C. Role of Aquaporins in Inflammation—A scientific curation. Inflammation 2020, 43, 1599–1610. [Google Scholar] [CrossRef]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The Role of Hydrogen Peroxide in Redox-dependent signaling: Homeostatic and pathological responses in mammalian cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef]
- Tamma, G.; Valenti, G.; Grossini, E.; Donnini, S.; Marino, A.; Marinelli, R.A.; Calamita, G. Aquaporin Membrane Channels in Oxidative Stress, Cell Signaling, and Aging: Recent Advances and Research Trends. Oxid. Med. Cell. Long. 2018, 2018, 1501847. [Google Scholar] [CrossRef]
- Rump, K.; Adamzik, M. Function of aquaporins in sepsis: A systematic review. Cell Biosci. 2018, 8, 10. [Google Scholar] [CrossRef]
- Jahn, T.P.; Møller, A.L.; Zeuthen, T.; Holm, L.M.; Klaerke, D.A.; Mohsin, B.; Kühlbrandt, W.; Schjoerring, J.K. Aquaporin homologues in plants and mammals transport ammonia. FEBS Lett. 2004, 574, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Bestetti, S.; Galli, M.; Sorrentino, I.; Pinton, P.; Rimessi, A.; Sitia, R.; Medraño-Fernandez, I. Human aquaporin-11 guarantees efficient transport of H2O2 across the endoplasmic reticulum membrane. Redox Biol. 2020, 28, 101326. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Møller, A.L.B.; Kristiansen, K.A.; Schulz, A.; Møller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, R.A.; Marchissio, M.J. Mitochondrial aquaporin-8: A functional peroxiporin? Antioxid. Redox Signal. 2013, 19, 896. [Google Scholar] [CrossRef]
- Bertolotti, M.; Bestetti, S.; García-Manteiga, J.M.; Medraño-Fernandez, I.; Dal Mas, A.; Malosio, M.L.; Sitia, R. Tyrosine kinase signal modulation: A matter of H2O2 membrane permeability? Antioxid. Redox Signal. 2013, 19, 1447–1451. [Google Scholar] [CrossRef]
- Sies, H. Role of metabolic H2O2 generation: Redox signaling and oxidative stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [PubMed]
- Tsukaguchi, H.; Weremowicz, S.; Morton, C.C.; Hediger, M.A. Functional and molecular characterization of the human neutral solute channel aquaporin-9. Am. J. Physiol. 1999, 277, F685–F696. [Google Scholar] [CrossRef] [PubMed]
- Nakhoul, N.L.; Davis, B.A.; Romero, M.F.; Boron, W.F. Effect of expressing the water channel aquaporin-1 on the CO2 permeability of Xenopus oocytes. Am. J. Physiol. 1998, 274, C543–C548. [Google Scholar] [CrossRef]
- Herrera, M.; Hong, N.J.; Garvin, J.L. Aquaporin-1 Transports NO Across Cell Membranes. Hypertension 2006, 48, 157–164. [Google Scholar] [CrossRef]
- Wang, Y.; Tajkhorshid, E. Molecular Mechanisms of Conduction and Selectivity in Aquaporin Water Channels. J. Nutr. 2007, 137, 1509–1515. [Google Scholar] [CrossRef] [PubMed]
- Soveral, G.; Casini, A. Aquaporin modulators: A patent review (2010-2015). Expert Opin. Ther. Pat. 2017, 27, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Calamita, G.; Perret, J.; Delporte, C. Aquaglyceroporins: Drug Targets for Metabolic Diseases? Front. Physiol. 2018, 9, 851. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. More than just water channels: Unexpected cellular roles of aquaporins. J. Cell Sci. 2005, 118, 3225–3232. [Google Scholar] [CrossRef] [PubMed]
- Agre, P. Aquaporin Water Channels (Nobel Lecture). Angew Chem. Int. Ed. Engl. 2004, 43, 4278–4290. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Chikuma, S.; Sugiyama, Y.; Kabashima, K.; Verkman, A.S.; Inoue, S.; Miyachi, Y. Chemokine-dependent T cell migration requires aquaporin-3-mediated hydrogen peroxide uptake. J. Exp. Med. 2012, 209, 1743–1752. [Google Scholar] [CrossRef]
- Cui, G.; Staron, M.M.; Gray, S.M.; Ho, P.C.; Amezquita, R.A.; Wu, J.; Kaech, S.M. IL-7-Induced Glycerol Transport and TAG Synthesis Promotes Memory CD8+ T Cell Longevity. Cell 2015, 161, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Holm, A.; Karlsson, T.; Vikström, E. Pseudomonas aeruginosa lasI/rhlI quorum sensing genes promote phagocytosis and aquaporin 9 redistribution to the leading and trailing regions in macrophages. Front. Microbiol. 2015, 6, 915. [Google Scholar] [CrossRef]
- Moniaga, C.S.; Watanabe, S.; Honda, T.; Nielsen, S.; Hara-Chikuma, M. Aquaporin-9-expressing neutrophils are required for the establishment of contact hypersensitivity. Sci. Rep. 2015, 5, 15319. [Google Scholar] [CrossRef]
- Rump, K.; Unterberg, M.; Bergmann, L.; Bankfalvi, A.; Menon, A.; Schäfer, S.; Scherag, A.; Bazzi, Z.; Siffert, W.; Peters, J.; et al. AQP5-1364A/C polymorphism and the AQP5 expression influence sepsis survival and immune cell migration: A prospective laboratory and patient study. J. Transl. Med. 2016, 14, 321. [Google Scholar] [CrossRef]
- Meli, R.; Pirozzi, C.; Pelagalli, A. New Perspectives on the Potential Role of Aquaporins (AQPs) in the Physiology of Inflammation. Front. Physiol. 2018, 9, 101. [Google Scholar] [CrossRef]
- Rabolli, V.; Wallemme, L.; Lo Re, S.; Uwambayinema, F.; Palmai-Pallag, M.; Thomassen, L.; Tyteca, D.; Octave, J.N.; Marbaix, E.; Lison, D.; et al. Critical role of aquaporins in interleukin 1β (IL-1β)–induced inflammation. J. Biol. Chem. 2014, 289, 13937–13947. [Google Scholar] [CrossRef]
- da Silva, I.V.; Cardoso, C.; Martìnez-Banaclocha, H.; Casini, A.; Pelegrìn, P.; Soveral, G. Aquaporin-3 is involved in NLRP3-inflammasome activation contributing to the setting of inflammatory response. Cell Mol. Life Sci. 2020. Online ahead of print. [Google Scholar] [CrossRef]
- Esquerdo, K.F.; Sharma, N.K.; Brunialti, M.K.; Baggio-Zappia, G.L.; Assunção, M.; Azevedo, L.C.P.; Bafi, A.T.; Salomao, R. Inflammasome gene profile is modulated in septic patients, with a greater magnitude in non-survivors. Clin. Exp. Immunol. 2017, 189, 232–240. [Google Scholar] [CrossRef]
- Rodríguez, A.; Marinelli, R.A.; Tesse, A.; Frühbeck, G.; Calamita, G. Sexual dimorphism of adipose and hepatic aquaglyceroporins in health and metabolic disorders. Front. Endocrinol. (Lausanne) 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Jelen, S.; Wacker, S.; Aponte-Santamaría, C.; Skott, M.; Rojek, A.; Johanson, U.; Kjellbom, P.; Nielsen, S.; de Groot, B.L.; Rützler, M. Aquaporin-9 protein is the primary route of hepatocyte glycerol uptake for glycerol gluconeogenesis in mice. J. Biol. Chem. 2011, 286, 44319–44325. [Google Scholar] [CrossRef]
- Calamita, G.; Gena, P.; Ferri, D.; Rosito, A.; Rojek, A.; Nielsen, S.; Marinelli, R.A.; Frühbeck, G.; Svelto, M. Biophysical assessment of aquaporin-9 as principal facilitative pathway in mouse liver import of glucogenetic glycerol. Biol. Cell 2012, 104, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Gena, P.; Del Buono, N.; D’Abbicco, M.; Mastrodonato, M.; Berardi, M.; Svelto, M.; Lopez, L.; Calamita, G. Dynamical modeling of liver Aquaporin-9 expression and glycerol permeability in hepatic glucose metabolism. Eur. J. Cell Biol. 2017, 96, 61–69. [Google Scholar] [CrossRef] [PubMed]
- De Santis, S.; Serino, G.; Fiorentino, M.R.; Galleggiante, V.; Gena, P.; Verna, G.; Liso, M.; Massaro, M.; Lan, J.; Troisi, J.; et al. Aquaporin 9 contributes to the maturation process and inflammatory cytokine secretion of murine dendritic cells. Front. Immunol. 2018, 9, 2355. [Google Scholar] [CrossRef] [PubMed]
- Rojek, A.M.; Skowronski, M.T.; Füchtbauer, E.M.; Füchtbauer, A.C.; Fenton, R.A.; Agre, P.; Frøkiaer, J.; Nielsen, S. Defective glycerol metabolism in aquaporin 9 (AQP9) knockout mice. Proc. Natl. Acad. Sci. USA 2007, 104, 3609–3614. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.F.; Weiss, M.; Darnell, J.E., Jr. Liver-specific RNA metabolism in hepatoma cells: Variations in transcription rates and mRNA levels. Mol. Cell Biol. 1985, 5, 2633–2641. [Google Scholar] [CrossRef] [PubMed]
- Coué, M.; Tesse, A.; Falewée, J.; Aguesse, A.; Croyal, M.; Fizanne, L.; Chaigneau, J.; Boursier, J.; Ouguerram, K. Spirulina liquid extract protects against fibrosis related to non-alcoholic steatohepatitis and increases ursodeoxycholic acid. Nutrients 2019, 11, 194. [Google Scholar] [CrossRef]
- Tesse, A.; Martínez, M.C.; Hugel, B.; Chalupsky, K.; Muller, C.D.; Meziani, F.; Mitolo-Chieppa, D.; Freyssinet, J.M.; Andriantsitohaina, R. Upregulation of proinflammatory proteins through NF-kappaB pathway by shed membrane microparticles results in vascular hyporeactivity. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2522–2527. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, S.; Reynoso, M.; Geddis, A.V.; Mitrophanov, A.Y.; Matheny, R.W., Jr. LPS-stimulated NF-κB p65 dynamic response marks the initiation of TNF expression and transition to IL-10 expression in RAW 264.7 macrophages. Physiol. Rep. 2018, 6, e13914. [Google Scholar] [CrossRef]
- Baldini, F.; Portincasa, P.; Grasselli, E.; Damonte, G.; Salis, A.; Bonomo, M.; Florio, M.; Serale, N.; Voci, A.; Gena, P.; et al. Aquaporin-9 is involved in the lipid-lowering activity of the nutraceutical silybin on hepatocytes through modulation of autophagy and lipid droplets composition. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2020, 1865, 158586. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Gaynor, R.B. IkappaB kinases: Key regulators of the NF-kappaB pathway. Trends Biochem. Sci. 2004, 29, 72–79. [Google Scholar] [CrossRef]
- Moon, D.O.; Choi, Y.H.; Kim, N.D.; Park, Y.M.; Kim, G.Y. Anti-inflammatory effects of beta-lapachone in lipopolysaccharide-stimulated BV2 microglia. Int. Immunopharmacol. 2007, 7, 506–514. [Google Scholar] [CrossRef]
- Lindskog, C.; Asplund, A.; Catrina, A.; Nielsen, S.; Rützler, M. A Systematic Characterization of Aquaporin-9 Expression in Human Normal and Pathological Tissues. J. Histochem. Cytochem. 2016, 64, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Khemlani, L.S.; Shapiro, R.A.; Johnson, M.L.; Liu, K.; Geller, D.A.; Watkins, S.C.; Goyert, S.M.; Billiar, T.R. Expression of CD14 by hepatocytes: Upregulation by cytokines during endotoxemia. Infect. Immun. 1998, 66, 5089–5098. [Google Scholar] [CrossRef]
- Vodovotz, Y.; Liu, S.; McCloskey, C.; Shapiro, R.; Green, A.; Billiar, T.R. The hepatocyte as a microbial product-responsive cell. J. Endotoxin. Res. 2001, 7, 365–373. [Google Scholar] [CrossRef]
- Liu, S.; Gallo, D.J.; Green, A.M.; Williams, D.L.; Gong, X.; Shapiro, R.A.; Gambotto, A.A.; Humphris, E.L.; Vodovotz, Y.; Billiar, T.R. Role of toll-like receptors in changes in gene expression and NF-kappa B activation in mouse hepatocytes stimulated with lipopolysaccharide. Infect. Immun. 2002, 70, 3433–3442. [Google Scholar] [CrossRef] [PubMed]
- Geller, D.A.; Nussler, A.K.; Di Silvio, M.; Lowenstein, C.J.; Shapiro, R.A.; Wang, S.C.; Simmons, R.L.; Billiar, T.R. Cytokines, endotoxin, and glucocorticoids regulate the expression of inducible nitric oxide synthase in hepatocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 522–546. [Google Scholar] [CrossRef]
- Wright, S.D.; Ramos, R.A.; Tobias, P.S.; Ulevitch, R.J.; Mathison, J.C. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 1990, 249, 1431–1433. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, S.T.; Larivière, L.; Leveque, G.; Clermont, S.; Moore, K.J.; Gros, P.; Malo, D. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). J. Exp. Med. 1999, 189, 615–625. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef]
- Jiang, J.X.; Török, N.J. NADPH Oxidases in Chronic Liver Diseases. Adv. Hepatol. 2014, 2014, 742931. [Google Scholar] [CrossRef]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef]
- Watanabe, S.; Moniaga, C.S.; Nielsen, S.; Hara-Chikuma, M. Aquaporin-9 facilitates membrane transport of hydrogen peroxide in mammalian cells. Biochem. Biophys. Res. Commun. 2016, 471, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Satooka, H.; Watanabe, S.; Honda, T.; Miyachi, Y.; Watanabe, T.; Verkman, A.S. Aquaporin-3-mediated hydrogen peroxide transport is required for NF-κB signalling in keratinocytes and development of psoriasis. Nat. Commun. 2015, 6, 7454. [Google Scholar] [CrossRef]
- Hara-Chikuma, M.; Tanaka, M.; Verkman, A.S.; Yasui, M. Inhibition of aquaporin-3 in macrophages by a monoclonal antibody as potential therapy for liver injury. Nat. Commun. 2020, 11, 5666. [Google Scholar] [CrossRef]
- Bertolotti, M.; Farinelli, G.; Galli, M.; Aiuti, A.; Sitia, R. AQP8 transports NOX2-generated H2O2 across the plasma membrane to promote signaling in B cells. J. Leukoc. Biol. 2016, 100, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Abram, C.L.; Lowell, C.A. Shp1 function in myeloid cells. J. Leukoc. Biol. 2017, 102, 657–675. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide–production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef]
- Miller, E.W.; Dickinson, B.C.; Chang, C.J. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 15681–15686. [Google Scholar] [CrossRef]
- Vieceli Dalla Sega, F.; Zambonin, L.; Fiorentini, D.; Rizzo, B.; Caliceti, C.; Landi, L.; Hrelia, S.; Prata, C. Specific aquaporins facilitate Nox-produced hydrogen peroxide transport through plasma membrane in leukaemia cells. BBA Mol. Cell Res. 2014, 1843, 806–814. [Google Scholar] [CrossRef]
- Takashi, Y.; Tomita, K.; Kuwahara, Y.; Roudkenar, M.H.; Roushandeh, A.M.; Igarashi, K.; Nagasawa, T.; Nishitani, Y.; Sato, T. Mitochondrial dysfunction promotes aquaporin expression that controls hydrogen peroxide permeability and ferroptosis. Free. Radic. Biol. Med. 2020, 161, 60–70. [Google Scholar] [CrossRef]
- Medraño-Fernandez, I.; Sitia, R. Chapter 11–Aquaporins: Gatekeepers in the borders of oxidative stress and redox signaling. In Oxidative Stress. Eustress and Distress; Sies, H., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 1, pp. 167–171. [Google Scholar]
- Huang, X.; Yu, X.; Li, H.; Han, L.; Yang, X. Regulation mechanism of aquaporin 9 gene on inflammatory response and cardiac function in rats with myocardial infarction through extracellular signal-regulated kinase1/2 pathway. Heart Vess. 2019, 34, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, A.; Ogura, H.; Koh, T.; Shimazu, T.; Sugimoto, H. Enhanced expression of aquaporin 9 in activated polymorphonuclear leukocytes in patients with systemic inflammatory response syndrome. Shock 2014, 42, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Filep, J.G. Impaired neutrophil migration during sepsis: Paying the toll. Crit. Care Med. 2012, 40, 2712–2722. [Google Scholar] [CrossRef] [PubMed]
- Zonneveld, R.; Molema, G.; Plötz, F.B. Analyzing neutrophil morphology, mechanics, and motility in sepsis: Options and challenges for novel bedside technologies. Crit. Care Med. 2016, 44, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Loitto, V.-M.; Forslund, T.; Sundqvist, T.; Magnusson, K.-E.; Gustafsson, K. Neutrophil leukocytes motility requires directed water influx. J. Leukoc. Biol. 2002, 71, 212–222. [Google Scholar] [PubMed]
- Alves-Filho, J.C.; de Freitas, A.; Spiller, F.; Souto, F.O.; Cunha, F.Q. The role of neutrophils in severe sepsis. Shock 2008, 30, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Craciun, F.L.; Schuller, E.R.; Remick, D.G. Early enhanced local neutrophil recruitment in peritonitis-induced sepsis improves bacterial clearance and survival. J. Immunol. 2010, 185, 6930–6938. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT CTRL | WT + LPS | KO CTRL | KO + LPS | ||
|---|---|---|---|---|---|
| iNOS (a.u.) | liver | 4.48 ± 1.23 | 26.97 ± 5.23 *** | 3.32 ± 0.32 | 7.69 ± 0.96 ###, ++ |
| kidney | 3.07 ± 1.03 | 50.56 ± 16.39 *** | 1.79 ± 0.33 | 9.63 ± 1.49 ## | |
| aorta | 1.16 ± 0.50 | 69.35 ± 14.02 *** | 1.43 ± 0.27 | 1.26 ± 0.35 ### | |
| heart | 2.42 ± 0.40 | 20.37 ± 2.80 *** | 1.98 ± 0.42 | 4.53 ± 1.17 ###, + | |
| COX-2 staining (a.u.) | liver | 0.17 ± 0.07 | 8.37 ± 1.88 *** | 0.82 ± 0.80 | 0.44 ± 0.18 ### |
| kidney | 0.014 ± 0.01 | 27.31 ± 6.00 *** | 0.02 ± 0.01 | 0.02 ± 0.004 ### | |
| aorta | 0.29 ± 0.19 | 15.80 ± 2.84 *** | 0.50 ± 0.17 | 3.37 ± 0.44 ### | |
| heart | 0.10 ± 0.11 | 18.49 ± 6.62 *** | 0.95 ± 0.27 | 1.68 ± 1.34 ### | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tesse, A.; Gena, P.; Rützler, M.; Calamita, G. Ablation of Aquaporin-9 Ameliorates the Systemic Inflammatory Response of LPS-Induced Endotoxic Shock in Mouse. Cells 2021, 10, 435. https://doi.org/10.3390/cells10020435
Tesse A, Gena P, Rützler M, Calamita G. Ablation of Aquaporin-9 Ameliorates the Systemic Inflammatory Response of LPS-Induced Endotoxic Shock in Mouse. Cells. 2021; 10(2):435. https://doi.org/10.3390/cells10020435
Chicago/Turabian StyleTesse, Angela, Patrizia Gena, Michael Rützler, and Giuseppe Calamita. 2021. "Ablation of Aquaporin-9 Ameliorates the Systemic Inflammatory Response of LPS-Induced Endotoxic Shock in Mouse" Cells 10, no. 2: 435. https://doi.org/10.3390/cells10020435
APA StyleTesse, A., Gena, P., Rützler, M., & Calamita, G. (2021). Ablation of Aquaporin-9 Ameliorates the Systemic Inflammatory Response of LPS-Induced Endotoxic Shock in Mouse. Cells, 10(2), 435. https://doi.org/10.3390/cells10020435