
Renal Proximal Tubule Cell Cannabinoid-1 Receptor Regulates Bone Remodeling and Mass via a Kidney-to-Bone Axis

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Experimental Procedure
2.4. Biochemistry
2.5. Skeletal Phenotyping
2.6. Mechanical Testing
2.7. Cell Culture
2.8. Real-Time PCR
2.9. Western Blotting
2.10. Immunohistochemistry
2.11. Statistical Analyses
3. Results
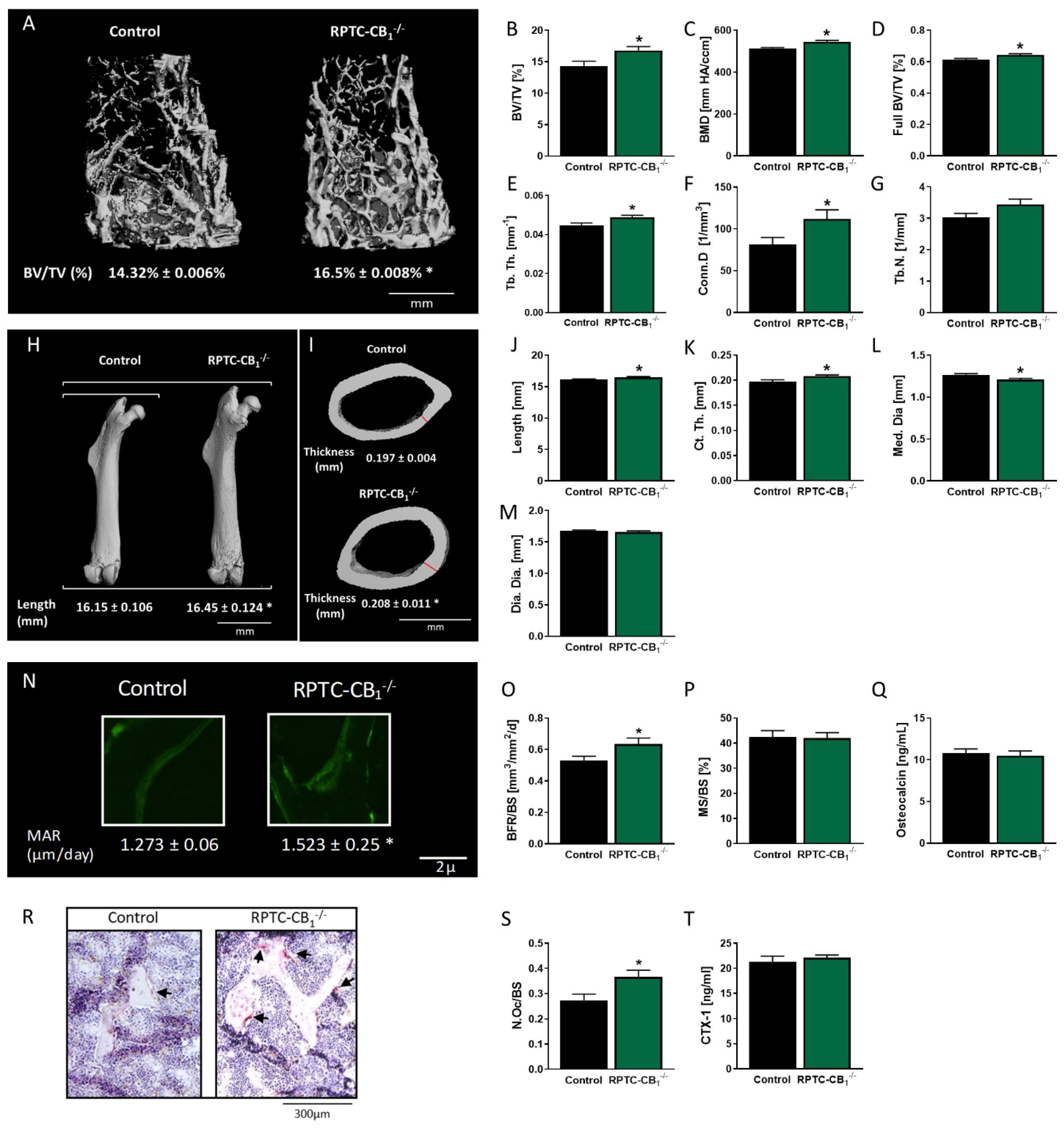
3.1. Nullification of CB1R in RPTCs Affects Bone Mass and Remodeling
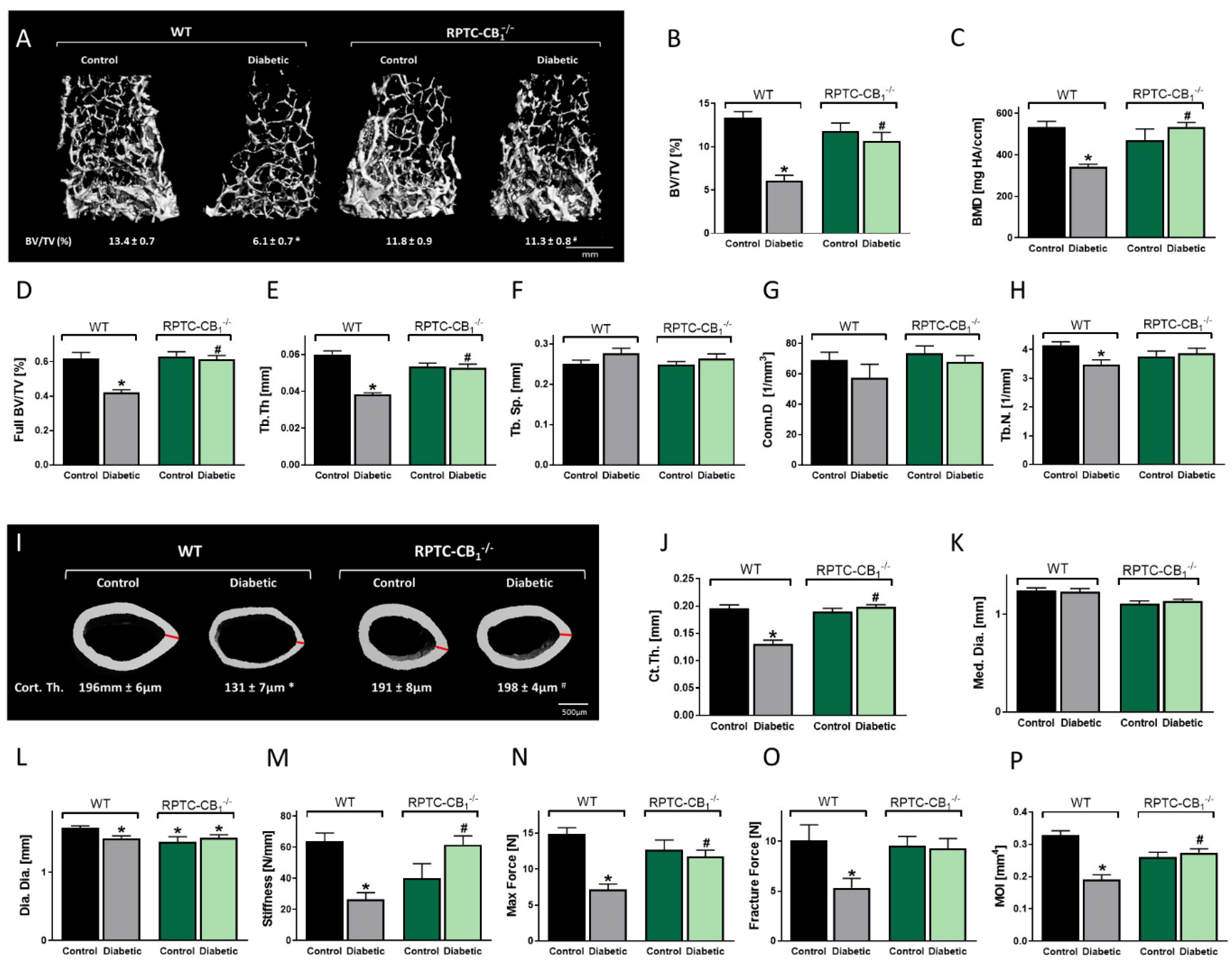
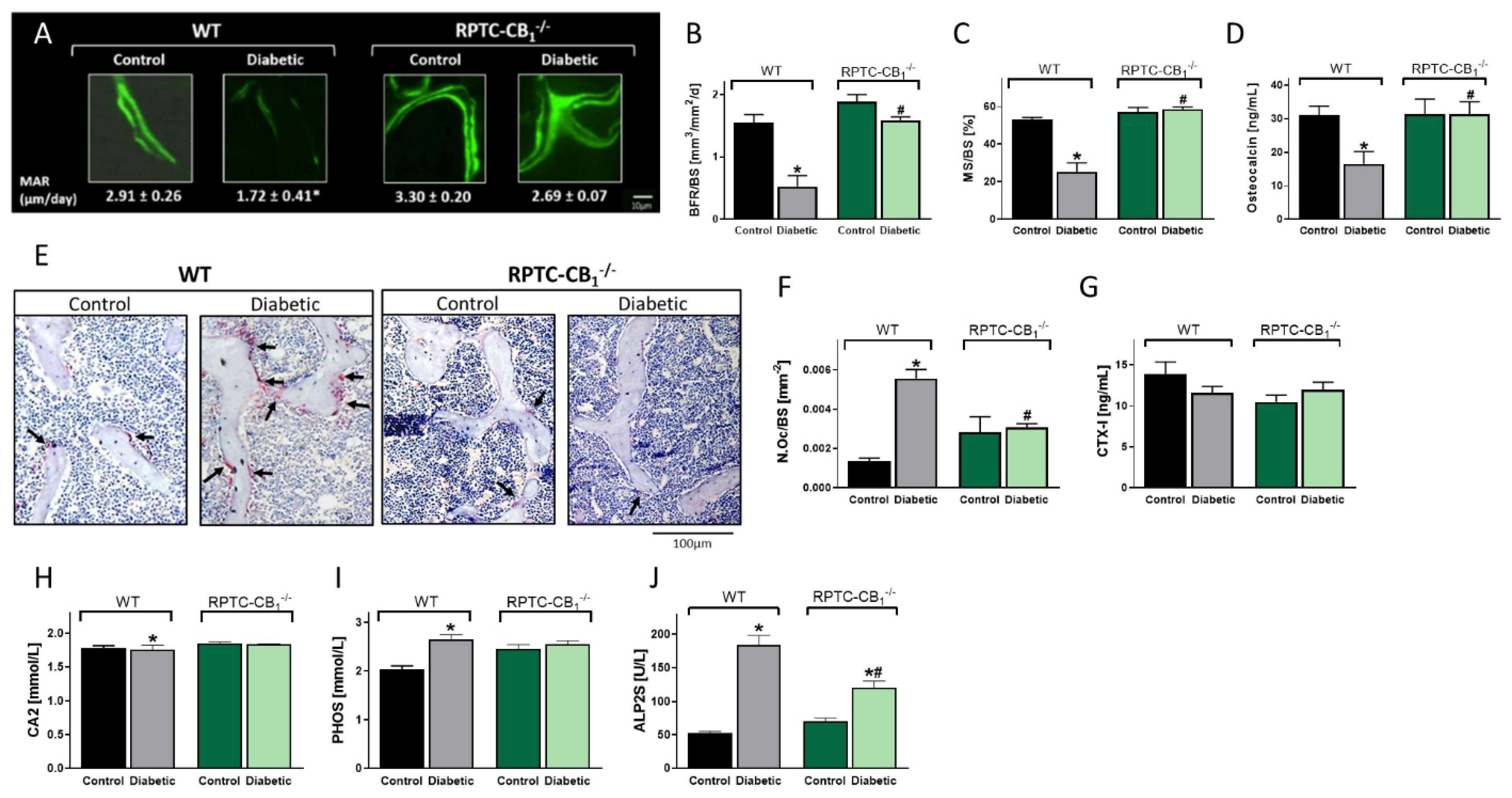
3.2. CB1R Deletion in RPTCs Protected Mice from the Development of Diabetes-Induced Bone Loss
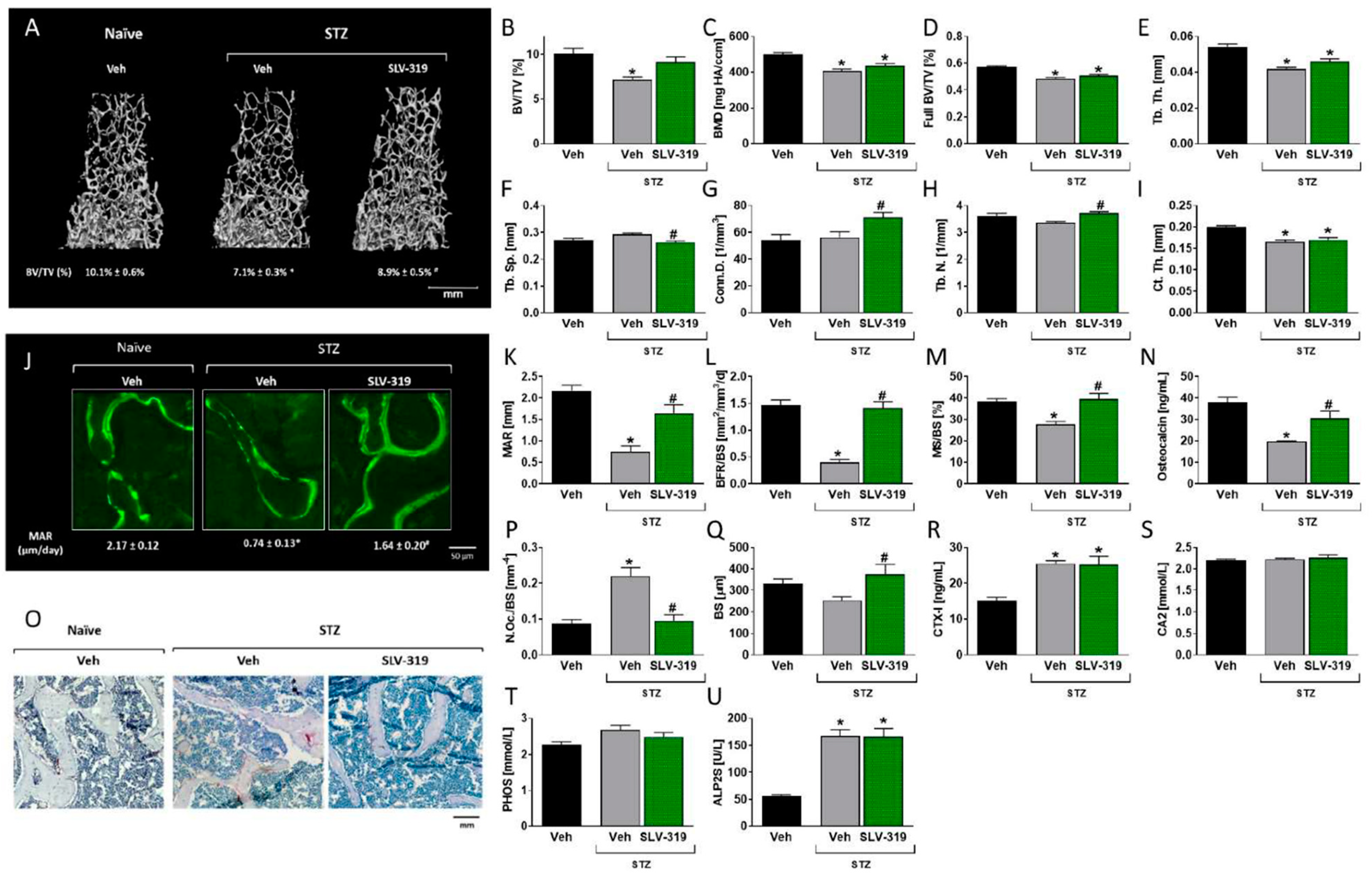
3.3. Pharmacological CB1R Blockade Prevents T1D-Induced Bone Loss
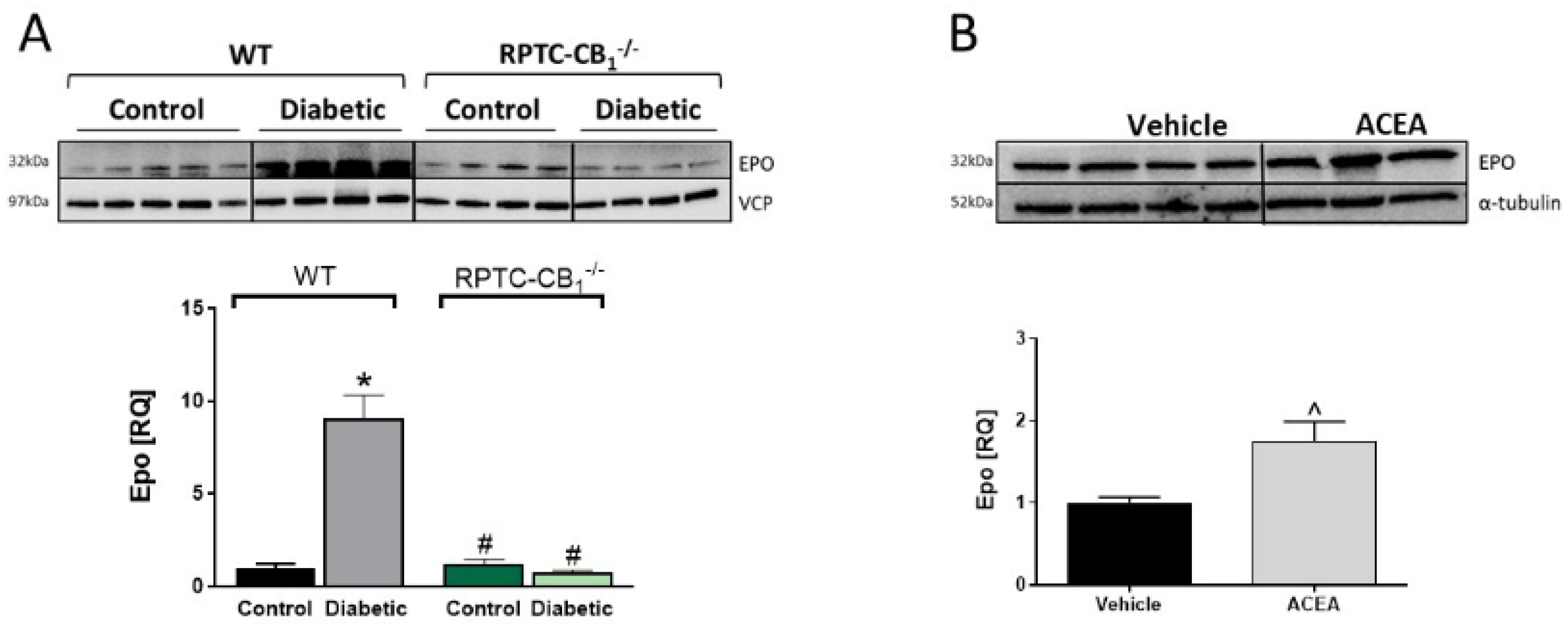
3.4. CB1R in RPTCs Regulates EPO Levels In Vivo
3.5. CB1R in RPTCs Modulates EPO Levels In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Obri, A.; Khrimian, L.; Karsenty, G.; Oury, F. Osteocalcin in the brain: From embryonic development to age-related decline in cognition. Nat. Rev. Endocrinol. 2018, 14, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; You, X.; Xing, W.; Zhang, Z.; Zou, W. Paracrine and endocrine actions of bone—the functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res. 2018, 6, 1–11. [Google Scholar] [CrossRef]
- Penido, M.G.M.G.; Alon, U.S. Phosphate homeostasis and its role in bone health. Pediatr. Nephrol. 2012, 27, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Gattineni, J.; Baum, M. Regulation of phosphate transport by fibroblast growth factor 23 (FGF23): Implications for disorders of phosphate metabolism. Pediatr. Nephrol. 2010, 25, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Pi, M.; Quarles, L.D. Novel bone endocrine networks integrating mineral and energy metabolism. Curr. Osteoporos. Rep. 2013, 11, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, H.; Border, P.; Kurokawa, K.; Nagata, N.; Ogata, E. Hormonal control of skeletal and mineral homeostasis. Am. J. Med. 1974, 56, 751–758. [Google Scholar] [CrossRef]
- Andrukhova, O.; Bayer, J.; Schüler, C.; Zeitz, U.; Murali, S.K.; Ada, S.; Alvarez-Pez, J.M.; Smorodchenko, A.; Erben, R.G. Klotho Lacks an FGF23-Independent Role in Mineral Homeostasis. J. Bone Miner. Res. 2017, 32, 2049–2061. [Google Scholar] [CrossRef] [PubMed]
- Daryadel, A.; Natale, L.; Seebeck, P.; Bettoni, C.; Schnitzbauer, U.; Gassmann, M.; Wagner, C.A. Elevated FGF23 and disordered renal mineral handling with reduced bone mineralization in chronically erythropoietin over-expressing transgenic mice. Sci. Rep. 2019, 9, 14989. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.S.; Fraser, L.-A. Type 1 Diabetes and Osteoporosis: From Molecular Pathways to Bone Phenotype. J. Osteoporos. 2015, 2015, 1–8. [Google Scholar] [CrossRef]
- Weber, D.R.; Schwartz, G. Epidemiology of Skeletal Health in Type 1 Diabetes. Curr. Osteoporos. Rep. 2016, 14, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Zhukouskaya, V.V.; Eller-Vainicher, C.; Shepelkevich, A.P.; Dydyshko, Y.; Cairoli, E.; Chiodini, I. Bone health in type 1 di-abetes: Focus on evaluation and treatment in clinical practice. J. Endocrinol. Investig. 2015, 38, 941–950. [Google Scholar] [CrossRef]
- Narres, M.; Claessen, H.; Droste, S.; Kvitkina, T.; Koch, M.; Kuss, O.; Icks, A. The Incidence of End-Stage Renal Disease in the Diabetic (Compared to the Non-Diabetic) Population: A Systematic Review. PLoS ONE 2016, 11, e0147329. [Google Scholar] [CrossRef]
- Miller, P.D. Chronic kidney disease and the skeleton. Bone Res. 2014, 2, 14044. [Google Scholar] [CrossRef]
- Tam, J.; Ofek, O.; Fride, E.; Ledent, C.; Gabet, Y.; Muller, R.; Zimmer, A.; Mackie, K.; Mechoulam, R.; Shohami, E.; et al. In-volvement of neuronal cannabinoid receptor CB1 in regulation of bone mass and bone remodeling. Mol. Pharmacol. 2006, 70, 786–792. [Google Scholar] [CrossRef]
- Tam, J.; Cinar, R.; Liu, J.; Godlewski, G.; Wesley, D.; Jourdan, T.; Szanda, G.; Mukhopadhyay, B.; Chedester, L.; Liow, J.-S.; et al. Peripheral Cannabinoid-1 Receptor Inverse Agonism Reduces Obesity by Reversing Leptin Resistance. Cell Metab. 2012, 16, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Hinden, L.; Drori, A.; Udi, S.; Azar, S.; Baraghithy, S. The therapeutic potential of targeting the peripheral endocan-nabinoid/CB1 receptor system. Eur. J. Intern. Med. 2018, 49, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.; Trembovler, V.; Di Marzo, V.; Petrosino, S.; Leo, G.; Alexandrovich, A.; Regev, E.; Casap, N.; Shteyer, A.; Ledent, C.; et al. The cannabinoid CB1 receptor regulates bone formation by modulating adrenergic signaling. FASEB J. 2007, 22, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Tam, J. The emerging role of the endocannabinoid system in the pathogenesis and treatment of kidney diseases. J. Basic Clin. Physiol. Pharm. 2016, 27, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Drori, A.; Permyakova, A.; Hadar, R.; Udi, S.; Nemirovski, A.; Tam, J. Cannabinoid-1 receptor regulates mitochondrial dy-namics and function in renal proximal tubular cells. Diabetes Obes. Metab. 2019, 21, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Hinden, L.; Udi, S.; Drori, A.; Gammal, A.; Nemirovski, A.; Hadar, R.; Baraghithy, S.; Permyakova, A.; Geron, M.; Cohen, M.; et al. Modulation of Renal GLUT2 by the Can-nabinoid-1 Receptor: Implications for the Treatment of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2018, 29, 434–448. [Google Scholar] [CrossRef]
- Udi, S.; Hinden, L.; Ahmad, M.; Drori, A.; Iyer, M.R.; Cinar, R.; Herman-Edelstein, M.; Tam, J. Dual inhibition of cannabinoid CB 1 receptor and inducible NOS attenuates obesity-induced chronic kidney disease. Br. J. Pharm. 2020, 177, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Udi, S.; Hinden, L.; Earley, B.; Drori, A.; Reuveni, N.; Hadar, R.; Cinar, R.; Nemirovski, A.; Tam, J. Proximal Tubular Canna-binoid-1 Receptor Regulates Obesity-Induced CKD. J. Am. Soc. Nephrol. 2017, 28, 3518–3532. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G.; Group NCRRGW. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br. J. Pharm. 2010, 160, 1577–1579. [Google Scholar] [CrossRef]
- Marsicano, G.; Wotjak, C.T.; Azad, S.C.; Bisogno, T.; Rammes, G.; Cascio, M.G.; Hermann, H.; Tang, J.; Hofmann, C.; Zieglgänsberger, W.; et al. The endogenous cannabinoid system controls extinction of aversive memories. Nat. Cell Biol. 2002, 418, 530–534. [Google Scholar] [CrossRef]
- Baraghithy, S.; Smoum, R.; Drori, A.; Hadar, R.; Gammal, A.; Hirsch, S.; Attar-Namdar, M.; Nemirovski, A.; Gabet, Y.; Langer, Y.; et al. Magel2 Modulates Bone Remodeling and Mass in Prader-Willi Syndrome by Affecting Oleoyl Serine Levels and Activity. J. Bone Miner. Res. 2019, 34, 93–105. [Google Scholar] [CrossRef]
- Bouxsein, M.L.; Boyd, S.K.; Christiansen, B.A.; Guldberg, R.E.; Jepsen, K.J.; Muller, R. Guidelines for assessment of bone mi-crostructure in rodents using micro-computed tomography. J. Bone Miner. Res. 2010, 25, 1468–1486. [Google Scholar] [CrossRef]
- Masarwi, M.; Gabet, Y.; Dolkart, O.; Brosh, T.; Shamir, R.; Phillip, M.; Gat-Yablonski, G. Skeletal effect of casein and whey protein intake during catch-up growth in young male Sprague–Dawley rats. Br. J. Nutr. 2016, 116, 59–69. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Alexander, J.M.; Bab, I.; Fish, S.; Müller, R.; Uchiyama, T.; Gronowicz, G.; Nahounou, M.; Zhao, Q.; White, D.W.; Chorev, M.; et al. Human Parathyroid Hormone 1-34 Reverses Bone Loss in Ovariectomized Mice. J. Bone Miner. Res. 2001, 16, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Stampa, B.; Kühn, B.; Liess, C.; Heller, M.; Glüer, C.-C. Characterization of the Integrity of Three-Dimensional Trabecular Bone Microstructure by Connectivity and Shape Analysis Using High-Resolution Magnetic Resonance Imaging In Vivo. Top. Magn. Reson. Imaging 2002, 13, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Christenson, R.H. Biochemical Markers of Bone Metabolism: An Overview. Clin. Biochem. 1997, 30, 573–593. [Google Scholar] [CrossRef]
- Maxwell, A.P.; Lappin, T.R.; Johnston, C.F.; Bridges, J.M.; McGeown, M.G. Erythropoietin production in kidney tubular cells. Br. J. Haematol. 1990, 74, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Suzuki, N.; Hirano, I.; Yamazaki, S.; Minegishi, N.; Yamamoto, M. Isolation and characterization of renal erythro-poietin-producing cells from genetically produced anemia mice. PLoS ONE 2011, 6, e25839. [Google Scholar] [CrossRef]
- Bunn, H.F. Erythropoietin. Cold Spring Harb. Perspect. Med. 2013, 3, a011619. [Google Scholar] [CrossRef] [PubMed]
- Hiram-Bab, S.; Neumann, D.; Gabet, Y. Erythropoietin in bone–Controversies and consensus. Cytokine 2017, 89, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Raphael-Mizrahi, B.; Gabet, Y. The Cannabinoids Effect on Bone Formation and Bone Healing. Curr. Osteoporos. Rep. 2020, 18, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Idris, A.I.; van ’t Hof, R.J.; Greig, I.R.; Ridge, S.A.; Baker, D.; Ross, R.A.; Ralston, S.H. Regulation of bone mass, bone loss and osteoclast activity by cannabinoid receptors. Nat. Med. 2005, 11, 774–779. [Google Scholar] [CrossRef]
- Deis, S.; Srivastava, R.K.; De Azua, I.R.; Bindila, L.; Baraghithy, S.; Lutz, B.; Bab, I.; Tam, J. Age-related regulation of bone formation by the sympathetic cannabinoid CB1 receptor. Bone 2018, 108, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, T.; Park, J.K.; Varga, Z.V.; Pálóczi, J.; Coffey, N.J.; Rosenberg, A.Z.; Godlewski, G.; Cinar, R.; Mackie, K.; Pacher, P.; et al. Cannabinoid-1 receptor deletion in podocytes mitigates both glomerular and tubular dysfunction in a mouse model of diabetic nephropathy. Diabetes Obes. Metab. 2017, 20, 698–708. [Google Scholar] [CrossRef]
- Jourdan, T.; Szanda, G.; Rosenberg, A.Z.; Tam, J.; Earley, B.J.; Godlewski, G.; Cinar, R.; Liu, Z.; Liu, J.; Ju, C.; et al. Overactive cannabinoid 1 receptor in podocytes drives type 2 diabetic nephropathy. Proc. Natl. Acad. Sci. 2014, 111, E5420–E5428. [Google Scholar] [CrossRef]
- Matias, I.; Petrosino, S.; Racioppi, A.; Capasso, R.; Izzo, A.A.; Di Marzo, V. Dysregulation of peripheral endocannabinoid levels in hyperglycemia and obesity: Effect of high fat diets. Mol. Cell. Endocrinol. 2008, 286, S66–S78. [Google Scholar] [CrossRef] [PubMed]
- Jenkin, K.A.; McAinch, A.J.; Zhang, Y.; Kelly, D.J.; Hryciw, D.H. Elevated cannabinoid receptor 1 and G protein-coupled re-ceptor 55 expression in proximal tubule cells and whole kidney exposed to diabetic conditions. Clin. Exp. Pharmacol. Physiol. 2015, 42, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Lecru, L.; Desterke, C.; Grassin-Delyle, S.; Chatziantoniou, C.; Vandermeersch, S.; Devocelle, A.; Vernochet, A.; Ivanovski, N.; Ledent, C.; Ferlicot, S.; et al. Cannabinoid receptor 1 is a major mediator of renal fibrosis. Kidney Int. 2015, 88, 72–84. [Google Scholar] [CrossRef]
- Hsu, Y.-C.; Lei, C.-C.; Shih, Y.-H.; Lin, C.-L.; Ho, C. Induction of Proteinuria by Cannabinoid Receptors 1 Signaling Activation in CB1 Transgenic Mice. Am. J. Med. Sci. 2015, 349, 162–168. [Google Scholar] [CrossRef]
- Nam, D.H.; Lee, M.H.; Kim, J.E.; Song, H.K.; Kang, Y.S.; Lee, J.E.; Kim, H.W.; Cha, J.J.; Hyun, Y.Y.; Kim, S.H.; et al. Blockade of Cannabinoid Receptor 1 Improves Insulin Resistance, Lipid Metabolism, and Diabetic Nephropathy in db/db Mice. Endocrinology 2012, 153, 1387–1396. [Google Scholar] [CrossRef]
- Lim, J.; Lim, S.; Han, H.J.; Park, S. Cannabinoid receptor 1 mediates palmitic acid-induced apoptosis via endoplasmic reticulum stress in human renal proximal tubular cells. J. Cell. Physiol. 2010, 225, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.C.; Lim, S.K.; Park, M.J.; Kim, G.Y.; Han, H.J.; Park, S.H. Cannabinoid receptor 1 mediates high glucose-induced apoptosis via endoplasmic reticulum stress in primary cultured rat mesangial cells. Am. J. Physiol. Physiol. 2011, 301, F179–F188. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Kapoor, N.; Bondu, J.D.; Thomas, N.; Paul, T.V. Bone turnover markers: Emerging tool in the management of os-teoporosis. Indian J. Endocrinol. Metab. 2016, 20, 846–852. [Google Scholar] [PubMed]
- Kuo, T.R.; Chen, C.H. Bone biomarker for the clinical assessment of osteoporosis: Recent developments and future perspec-tives. Biomark Res. 2017, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Bain, S.; Ramamurthy, N.; Impeduglia, T.; Scolman, S.; Golub, L.; Rubin, C. Tetracycline prevents cancellous bone loss and maintains near-normal rates of bone formation in streptozotocin diabetic rats. Bone 1997, 21, 147–153. [Google Scholar] [CrossRef]
- Botolin, S.; Faugere, M.C.; Malluche, H.; Orth, M.; Meyer, R.; McCabe, L.R. Increased bone adiposity and peroxisomal pro-liferator-activated receptor-gamma2 expression in type I diabetic mice. Endocrinology 2005, 146, 3622–3631. [Google Scholar] [CrossRef]
- Botolin, S.; McCabe, L.R. Bone Loss and Increased Bone Adiposity in Spontaneous and Pharmacologically Induced Diabetic Mice. Endocrinology 2007, 148, 198–205. [Google Scholar] [CrossRef] [PubMed]
- McCabe, L.; Zhang, J.; Raehtz, S. Understanding the skeletal pathology of type 1 and 2 diabetes mellitus. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 187–206. [Google Scholar] [CrossRef]
- McCabe, L.R. Understanding the pathology and mechanisms of type I diabetic bone loss. J. Cell. Biochem. 2007, 102, 1343–1357. [Google Scholar] [CrossRef]
- Dou, Z.J.; Gao, X.L.; Jia, Y.L.; Chen, J.; Yang, J.J.; Chen, Y.; Wu, S.J.; Liu, T.; Wang, M.T.; Yang, C.; et al. CB1 receptor antagonist rimonabant protects against chronic intermittent hypoxia-induced bone metabolism disorder and de-struction in rats. Sleep Breath 2020, 24, 1441–1449. [Google Scholar] [CrossRef]
- Ko, J.Y.; Wu, R.W.; Kuo, S.J.; Chen, M.W.; Yeh, D.W.; Ke, H.C.; Wu, S.L.; Wang, F.S. Cannabinoid receptor 1 mediates gluco-corticoid-induced bone loss in rats by perturbing bone mineral acquisition and marrow adipogenesis. Arthritis Rheum 2012, 64, 1204–1214. [Google Scholar] [CrossRef]
- Samir, S.M.; Malek, H.A. Effect of cannabinoid receptors 1 modulation on osteoporosis in a rat model of different ages. J. Physiol. Pharm. 2014, 65, 687–694. [Google Scholar]
- Motyl, K.J.; McCabe, L.R. Streptozotocin, Type I Diabetes Severity and Bone. Biol. Proced. Online 2009, 11, 296–315. [Google Scholar] [CrossRef]
- Coe, L.M.; Irwin, R.; Lippner, D.; McCabe, L.R. The bone marrow microenvironment contributes to type I diabetes induced osteoblast death. J. Cell. Physiol. 2010, 226, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Motyl, K.J.; Botolin, S.; Irwin, R.; Appledorn, D.M.; Kadakia, T.; Amalfitano, A.; Schwartz, R.C.; McCabe, L.R. Bone inflam-mation and altered gene expression with type I diabetes early onset. J. Cell Physiol. 2009, 218, 575–583. [Google Scholar] [CrossRef]
- Wen, B.; Zhao, L.; Zhao, H.; Wang, X. Liraglutide exerts a bone-protective effect in ovariectomized rats with streptozoto-cin-induced diabetes by inhibiting osteoclastogenesis. Exp. Ther. Med. 2018, 15, 5077–5083. [Google Scholar] [PubMed]
- Reni, C.; Mangialardi, G.; Meloni, M.; Madeddu, P. Diabetes Stimulates Osteoclastogenesis by Acidosis-Induced Activation of Transient Receptor Potential Cation Channels. Sci. Rep. 2016, 6, 30639. [Google Scholar] [CrossRef]
- Guo, C.J.; Xie, J.J.; Hong, R.H.; Pan, H.S.; Zhang, F.G.; Liang, Y.M. Puerarin alleviates streptozotocin (STZ)-induced osteo-porosis in rats through suppressing inflammation and apoptosis via HDAC1/HDAC3 signaling. Biomed. Pharm. 2019, 115, 108570. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-W.; Wu, X.-Y.; Luo, L.; Guo, L.-J.; Lei, M.-X.; Zhang, H.; Xie, H.; Peng, Y.-Q.; Wu, X.-P.; Liao, E.-Y. The relationship between bone turnover markers and BMD decreasing rates in Chinese middle-aged women. Clin. Chim. Acta 2011, 412, 1648–1657. [Google Scholar] [CrossRef] [PubMed]
- Bergman, A.; Qureshi, A.R.; Haarhaus, M.; Lindholm, B.; Barany, P.; Heimburger, O.; Stenvinkel, P.; Anderstam, B. Total and bone-specific alkaline phosphatase are associated with bone mineral density over time in end-stage renal disease patients starting dialysis. J. Nephrol. 2016, 30, 255–262. [Google Scholar] [CrossRef]
- Saha, M.K.; Agrawal, P.; Saha, S.G.; Vishwanathan, V.; Pathak, V.; Saiprasad, S.V.; Dhariwal, P.; Dave, M. Evaluation of Cor-relation between Salivary Calcium, Alkaline Phosphatase and Osteoporosis—A Prospective, Comparative and Observational Study. J. Clin. Diagn. Res. 2017, 11, ZC63–ZC66. [Google Scholar]
- Biver, E.; Chopin, F.; Coiffier, G.; Brentano, T.F.; Bouvard, B.; Garnero, P.; Cortet, B. Bone turnover markers for osteoporotic status assessment? A systematic review of their diagnosis value at baseline in osteoporosis. Jt. Bone Spine 2012, 79, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, I.; Hie, M.; Iitsuka, N.; Otsuka, T. Insulin-dependent diabetes mellitus decreases osteoblastogenesis associated with the inhibition of Wnt signaling through increased expression of Sost and Dkk1 and inhibition of Akt activation. Int. J. Mol. Med. 2011, 28, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.K.; Winocour, P.; Farrington, K. Mechanisms of Disease: The hypoxic tubular hypothesis of diabetic nephropathy. Nat. Clin. Pr. Nephrol. 2008, 4, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Basseau, F.; Tyndal, B.; Jones, R.; Deminiere, C.; Catargi, B.; Combe, C.; Moonen, C.W.; Grenier, N. Renal diffusion and BOLD MRI in experimental diabetic nephropathy. J. Magn. Reson. Imaging 2002, 17, 104–113. [Google Scholar] [CrossRef]
- Ohtomo, S.; Nangaku, M.; Izuhara, Y.; Takizawa, S.; Strihou, C.V.Y.D.; Miyata, T. Cobalt ameliorates renal injury in an obese, hypertensive type 2 diabetes rat model. Nephrol. Dial. Transpl. 2007, 23, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.; Khamaisi, M.; Abassi, Z.; Shilo, V.; Weksler-Zangen, S.; Goldfarb, M.T.; Shina, A.; Zibertrest, F.; Eckardt, K.; Rosen, S.T.; et al. Adaptation to hypoxia in the diabetic rat kidney. Kidney Int. 2008, 73, 34–42. [Google Scholar] [CrossRef]
- Norman, J.; Orphanides, C.; Garcia, P.; Fine, L. Hypoxia-Induced Changes in Extracellular Matrix Metabolism in Renal Cells. Nephron Exp. Nephrol. 1999, 7, 463–469. [Google Scholar] [CrossRef]
- Abán, C.; Martinez, N.; Carou, C.; Albamonte, I.; Toro, A.; Seyahian, A.; Franchi, A.; Leguizamón, G.; Trigubo, D.; Damiano, A.; et al. Endocannabinoids participate in placental apoptosis induced by hypoxia inducible factor-1. Apoptosis 2016, 21, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wu, S.; Dong, Y.; Huang, Y.; Chen, Y.; Qiao, Y.; Dou, Z.; Wang, B. Role of the endogenous cannabinoid receptor 1 in brain injury induced by chronic intermittent hypoxia in rats. Int. J. Neurosci. 2018, 128, 797–804. [Google Scholar] [CrossRef]
- Valk, P.; Verbakel, S.; Von Lindern, M.; Löwenberg, B.; Delwel, R. Enhancement of erythropoietin-stimulated cell proliferation by Anandamide correlates with increased activation of the mitogen-activated protein kinases ERK1 and ERK2. Hematol. J. 2000, 1, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Hiram-Bab, S.; Liron, T.; Deshet-Unger, N.; Mittelman, M.; Gassmann, M.; Rauner, M.; Franke, K.; Wielockx, B.; Neumann, D.; Gabet, Y. Erythropoietin directly stimulates osteoclast precursors and induces bone loss. FASEB J. 2015, 29, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Singbrant, S.; Russell, M.R.; Jovic, T.; Liddicoat, B.; Izon, D.J.; Purton, L.E.; Sims, N.A.; Martin, T.J.; Sankaran, V.G.; Walkley, C.R. Erythropoietin couples erythropoiesis, B-lymphopoiesis, and bone homeostasis within the bone marrow microenvironment. Blood 2011, 117, 5631–5642. [Google Scholar] [CrossRef]
- Dewamitta, S.R.; Russell, M.R.; Nandurkar, H.; Walkley, C.R. Darbepoietin-alfa has comparable erythropoietic stimulatory effects to recombinant erythropoietin whilst preserving the bone marrow microenvironment. Haematologica 2012, 98, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Redwan, E.M. Erythropoietin and co.: Intrinsic structure and functional disorder. Mol. Biosyst. 2016, 13, 56–72. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Jung, Y.; Ziegler, A.M.; Pedersen, E.A.; Wang, J.; Wang, Z.; Song, J.; Wang, J.; Lee, C.H.; Sud, S.; et al. Erythropoietin Couples Hematopoiesis with Bone Formation. PLoS ONE 2010, 5, e10853. [Google Scholar] [CrossRef]
- Guo, L.; Luo, T.; Fang, Y.; Yang, L.; Wang, L.; Liu, J.; Shi, B. Effects of erythropoietin on osteoblast proliferation and function. Clin. Exp. Med. 2012, 14, 69–76. [Google Scholar] [CrossRef]
- Garcia, P.; Speidel, V.; Scheuer, C.; Laschke, M.W.; Holstein, J.H.; Histing, T.; Pohlemann, T.; Menger, M.D. Low dose eryth-ropoietin stimulates bone healing in mice. J. Orthop. Res. 2011, 29, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Zhang, F.; He, Q.; Tsang, W.P.; Lu, L.; Li, Q.; Wu, Z.; Qiu, G.; Zhou, G.; Wan, C. EPO Promotes Bone Repair through Enhanced Cartilaginous Callus Formation and Angiogenesis. Plos One 2014, 9, e102010. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Garcia, R.; Garcia-Martin, A.; Garcia-Fontana, B.; Morales-Santana, S.; Rozas-Moreno, P.; Muñoz-Torres, M. FGF23 in Type 2 Diabetic Patients: Relationship With Bone Metabolism and Vascular Disease. Diabetes Care 2014, 37, 89–90. [Google Scholar] [CrossRef] [PubMed]
- Bortolin, R.H.; Abreu, B.J.D.G.A.; Ururahy, M.A.G.; De Souza, K.S.C.; Bezerra, J.F.; Loureiro, M.B.; Da Silva, F.S.; Marques, D.E.D.S.; Batista, A.A.D.S.; Oliveira, G.; et al. Protection against T1DM-Induced Bone Loss by Zinc Supplementation: Biomechanical, Histomorphometric, and Molecular Analyses in STZ-Induced Diabetic Rats. PLoS ONE 2015, 10, e0125349. [Google Scholar] [CrossRef]
- Gaudio, A.; Privitera, F.; Battaglia, K.; Torrisi, V.; Sidoti, M.H.; Pulvirenti, I.; Canzonieri, E.; Tringali, G.; Fiore, C.E. Sclerostin levels associated with inhibition of the Wnt/beta-catenin signaling and reduced bone turnover in type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2012, 97, 3744–3750. [Google Scholar] [CrossRef]
- Motyl, K.J.; McCauley, L.K.; McCabe, L.R. Amelioration of type I diabetes-induced osteoporosis by parathyroid hormone is associated with improved osteoblast survival. J. Cell. Physiol. 2012, 227, 1326–1334. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, L.; Lin, W.; Yin, S.; Duan, A.; Liu, Z.; Cao, W. Rhein reverses Klotho repression via promoter demethylation and protects against kidney and bone injuries in mice with chronic kidney disease. Kidney Int. 2017, 91, 144–156. [Google Scholar] [CrossRef]
- Liu, M.M.; Dong, R.; Hua, Z.; Lv, N.N.; Ma, Y.; Huang, G.C.; Cheng, J.; Xu, H.Y. Therapeutic potential of Liuwei Dihuang pill against KDM7A and Wnt/beta-catenin signaling pathway in diabetic nephropathy-related osteoporosis. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Wu, Y.-Y.; Yu, T.; Zhang, X.-H.; Liu, Y.-S.; Li, F.; Wang, Y.-Y.; Wang, Y.-Y.; Gong, P. 1,25(OH)2D3 inhibits the deleterious effects induced by high glucose on osteoblasts through undercarboxylated osteocalcin and insulin signaling. J. Steroid Biochem. Mol. Biol. 2012, 132, 112–119. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
|---|---|---|
| hRPLP0 homo sapience | AATCCCATCACCATCTTCCA | TGGACTCCACGACGTACTCA |
| hEPO homo sapience | CATGTGGATAAAGCCGTCAGT | AAGTGTCAGCAGTGATTGTTCG |
| pGAPDH sus scrofa | CACGACCATGGAGAAGGC | GAAGCAGGGATGATGTTCTGG |
| pEPO sus scrofa | GCAAGTCGAAACCTGAGCTG | ACTTGTCCCGGCCAAACTT |
| mUbc mus musculus | GCCCAGTGTTACCACCAAGA | CCCATCACACCCAAGAACA |
| mSlc34a1 mus musculus | GGCTCCAACATTGGCACTAC | ACAGTAGGATGCCCGAGATG |
| mSlc34a2 mus musculus | GCTTGACTTAGGGCAGGTGTGG | AGGGGCTCAGTTTGGCATCTC |
| mSlc34a3 mus musculus | TACCCCCTCTTCTTGGGTTC | CAGTCTCAAGACAGGCACCA |
| mSlc20a2 mus musculus | GTGCCGGCCCTGCTTAC | CAATGCCTCTGCTTTCGTTCT |
| mSlc17a1 mus musculus | ACCCGTATATGAGCAGCAGTGAGA | AAATGTCGGCGTGTATGTAACCAG |
| Protein | Reference | Supplier |
|---|---|---|
| EPO | ab224506 | Abcam |
| β-actin | ab49900 | Abcam |
| α-tubulin | DM1A | Cell signaling technology |
| VCP | ab204290 | Abcam |
| Protein | Reference | Supplier |
|---|---|---|
| Insulin | A0564 | Agilent Dako |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baraghithy, S.; Soae, Y.; Assaf, D.; Hinden, L.; Udi, S.; Drori, A.; Gabet, Y.; Tam, J. Renal Proximal Tubule Cell Cannabinoid-1 Receptor Regulates Bone Remodeling and Mass via a Kidney-to-Bone Axis. Cells 2021, 10, 414. https://doi.org/10.3390/cells10020414
Baraghithy S, Soae Y, Assaf D, Hinden L, Udi S, Drori A, Gabet Y, Tam J. Renal Proximal Tubule Cell Cannabinoid-1 Receptor Regulates Bone Remodeling and Mass via a Kidney-to-Bone Axis. Cells. 2021; 10(2):414. https://doi.org/10.3390/cells10020414
Chicago/Turabian StyleBaraghithy, Saja, Yael Soae, Dekel Assaf, Liad Hinden, Shiran Udi, Adi Drori, Yankel Gabet, and Joseph Tam. 2021. "Renal Proximal Tubule Cell Cannabinoid-1 Receptor Regulates Bone Remodeling and Mass via a Kidney-to-Bone Axis" Cells 10, no. 2: 414. https://doi.org/10.3390/cells10020414
APA StyleBaraghithy, S., Soae, Y., Assaf, D., Hinden, L., Udi, S., Drori, A., Gabet, Y., & Tam, J. (2021). Renal Proximal Tubule Cell Cannabinoid-1 Receptor Regulates Bone Remodeling and Mass via a Kidney-to-Bone Axis. Cells, 10(2), 414. https://doi.org/10.3390/cells10020414