
Tat-Endophilin A1 Fusion Protein Protects Neurons from Ischemic Damage in the Gerbil Hippocampus: A Possible Mechanism of Lipid Peroxidation and Neuroinflammation Mitigation as Well as Synaptic Plasticity

 , , and
, , and 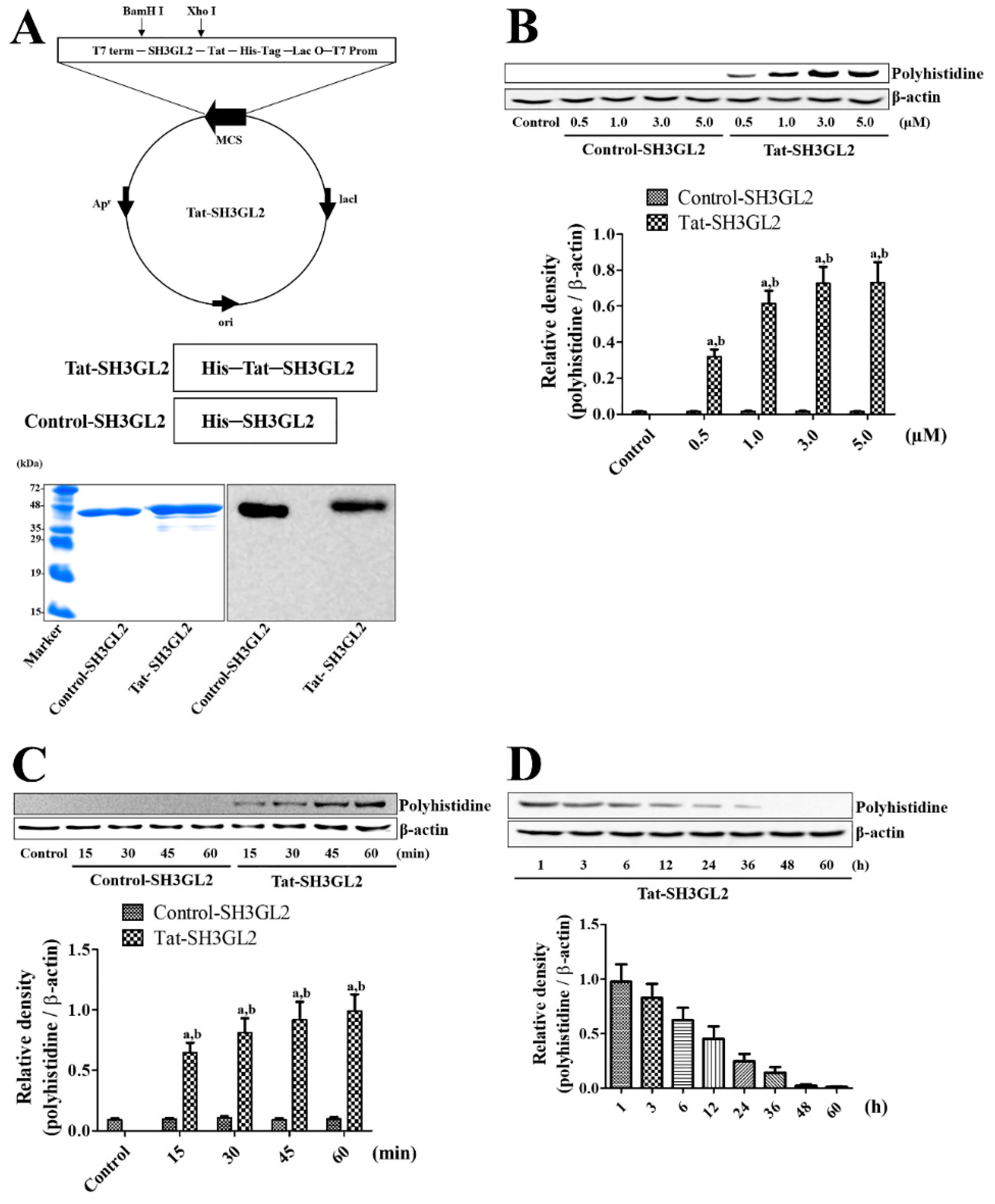{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Synthesis of Tat-SH3GL2 and Its Efficient Delivery into HT22 Cells
2.2. Confirmation of Intracellular Delivery of Tat-SH3GL2 into HT22 Cells and Gerbil Hippocampus
2.3. Effect of Tat-SH3GL2 on H2O2-Induced Oxidative Stress in HT22 Cells
2.4. Changes of SH3GL2 Immunoreactivity in the Gerbil Hippocampus after Ischemia
2.5. Effects of Tat-SH3GL2 against Brain Ischemic Damage in Gerbils
2.6. Mechanisms of Tat-SH3GL2 against Brain Ischemic Damage in Gerbils
2.7. Data Quantification and Statistical Analysis
3. Results
3.1. Construction of Tat-SH3GL2 and Control-SH3GL2 and Their Efficient Delivery to HT22 Cells
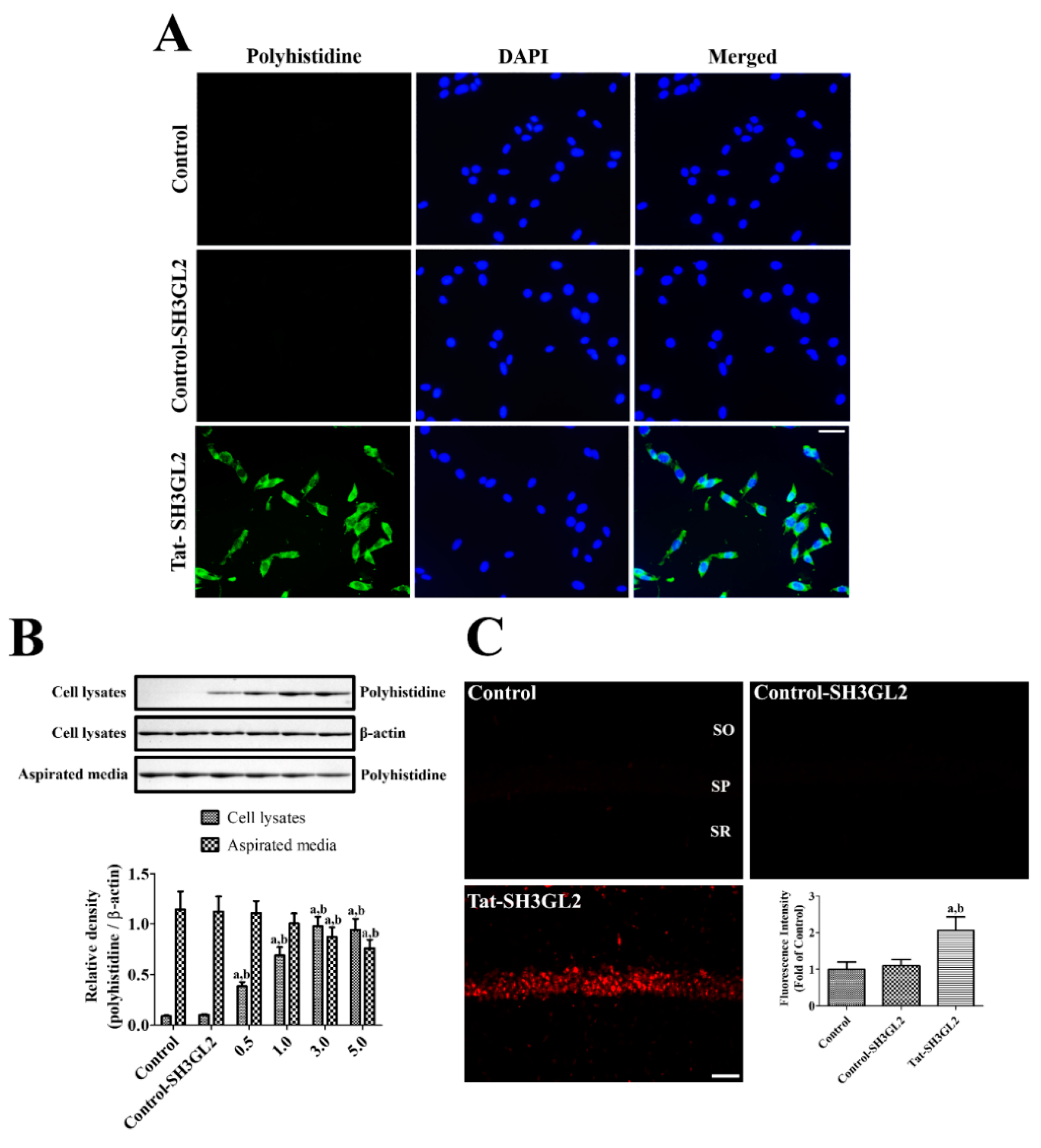
3.2. Intracellular Delivery of Tat-SH3GL2 into HT22 Cells and Gerbil Hippocampus
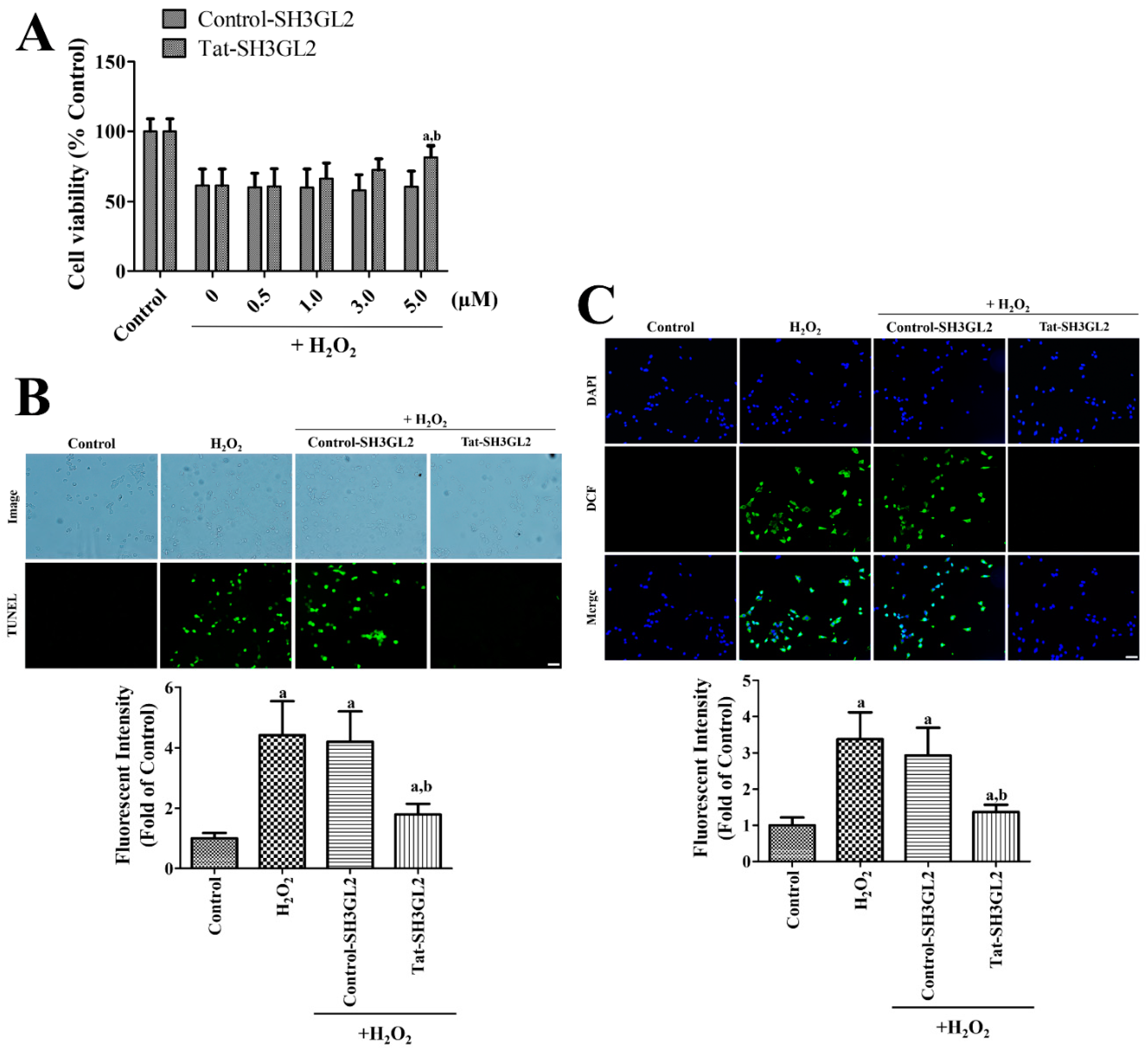
3.3. Effect of Tat-SH3GL2 and Control-SH3GL2 against H2O2-Induced Oxidative Damage in HT22 Cells
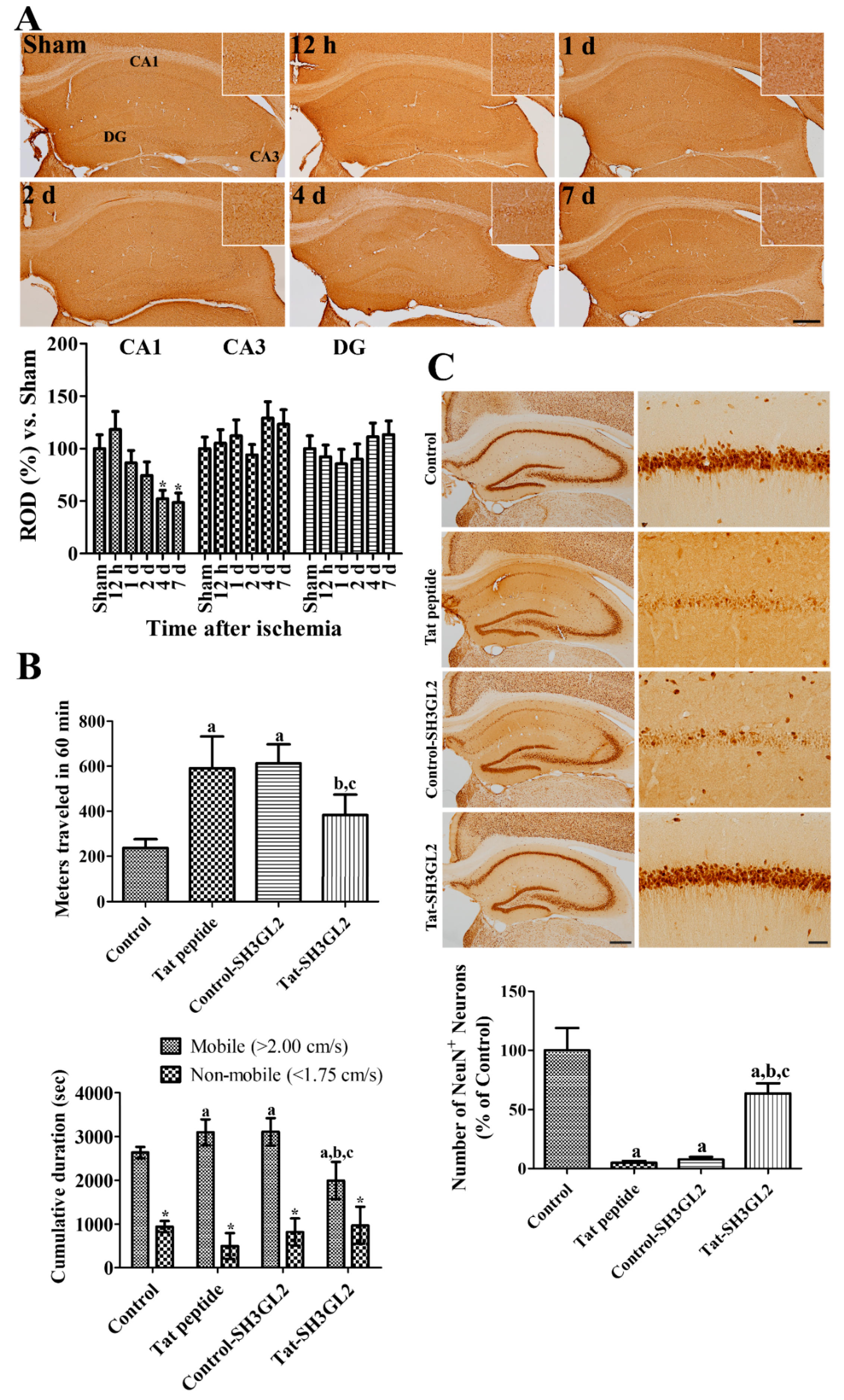
3.4. Time-Dependent Changes of Endophilin A1 Immunoreactivity in the Hippocampus after Ischemia
3.5. Effect of Tat-SH3GL2 and Control-SH3GL2 against Ischemic Injury in Gerbils
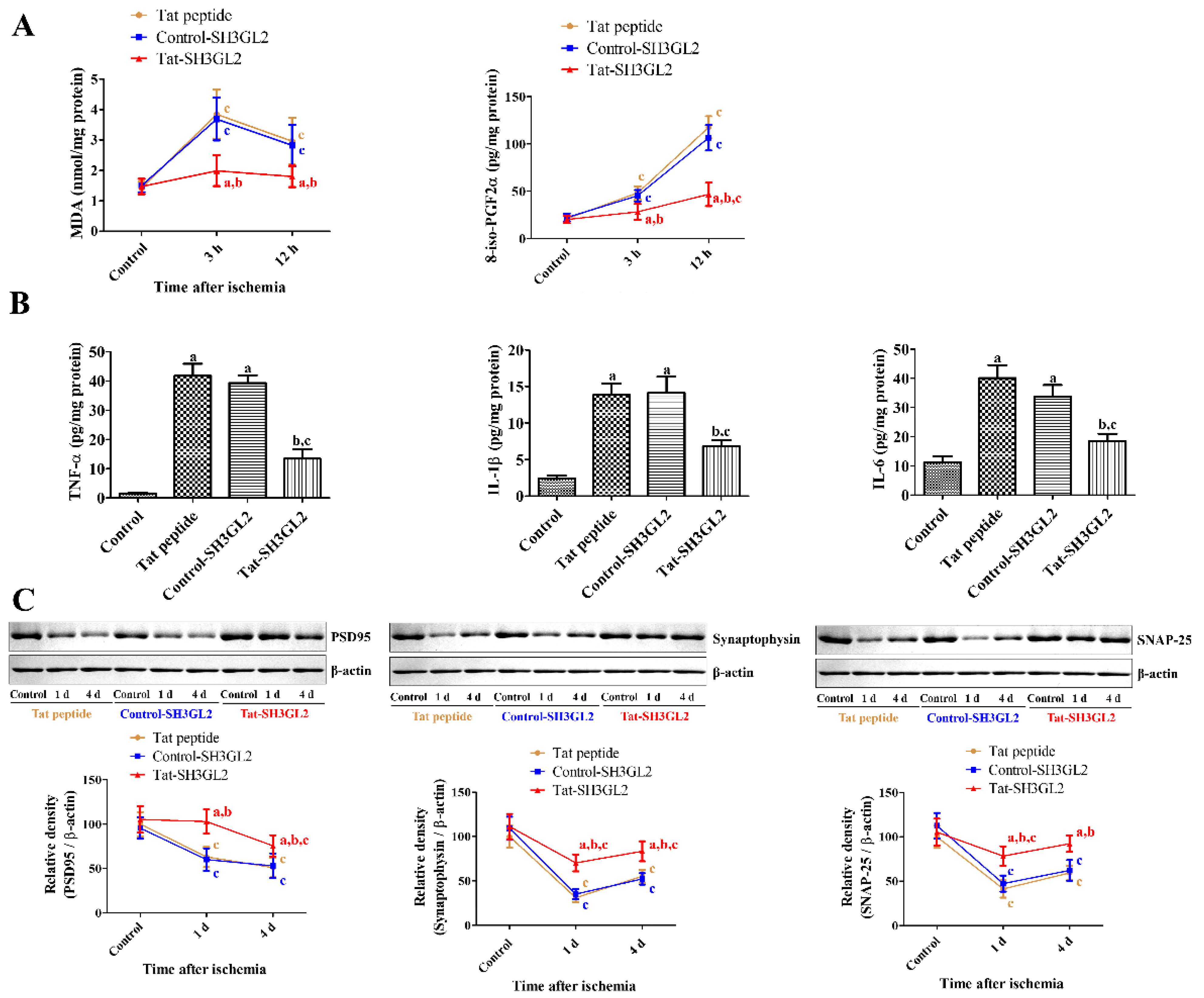
3.6. Mechanisms of Tat-SH3GL2 and Control-SH3GL2 against Ischemic Injury in Gerbils
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Du, X.Y.; Zhu, X.D.; Dong, G.; Lu, J.; Wang, Y.; Zeng, L.; Zhao, T.Y.; Ye, H.H.; Li, R.S.; Bai, J.Y.; et al. Characteristics of circle of Willis variations in the mongolian gerbil and a newly established ischemia-prone gerbil group. ILAR J. 2011, 52, E1–E7. [Google Scholar] [CrossRef]
- Lin, C.S.; Polsky, K.; Nadler, J.V.; Crain, B.J. Selective neocortical and thalamic cell death in the gerbil after transient ischemia. Neuroscience 1990, 35, 289–299. [Google Scholar] [CrossRef]
- Nitatori, T.; Sato, N.; Waguri, S.; Karasawa, Y.; Araki, H.; Shibanai, K.; Kominami, E.; Uchiyama, Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J. Neurosci. 1995, 15, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Du, X.Y.; Li, C.L.; Guo, M.; Wang, Y.; Guo, H.G.; Dai, F.W.; Sa, X.Y.; Chen, Z.W. Evaluation of an ischemic model in ischemia prone and general Mongolian gerbils by neurological symptom, injury, and sex difference. Anim. Model. Exp. Med. 2018, 1, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Roy-O’Reilly, M.; McCullough, L.D. Age and sex are critical factors in ischemic stroke pathology. Endocrinology 2018, 159, 3120–3131. [Google Scholar] [CrossRef] [PubMed]
- Kristián, T.; Siesjö, B.K. Calcium-related damage in ischemia. Life Sci. 1996, 59, 357–367. [Google Scholar] [CrossRef]
- Siesjö, B.K.; Katsura, K. Ischemic brain damage: Focus on lipids and lipid mediators. Adv. Exp. Med. Biol. 1992, 318, 41–56. [Google Scholar]
- Lee, T.K.; Kang, I.J.; Kim, B.; Sim, H.J.; Kim, D.W.; Ahn, J.H.; Lee, J.C.; Ryoo, S.; Shin, M.C.; Cho, J.H.; et al. Experimental pretreatment with chlorogenic acid prevents transient ischemia-induced cognitive decline and neuronal damage in the hippocampus through anti-oxidative and anti-inflammatory effects. Molecules 2020, 25, 3578. [Google Scholar] [CrossRef]
- Kim, W.; Hahn, K.R.; Jung, H.Y.; Kwon, H.J.; Nam, S.M.; Kim, T.H.; Kim, J.W.; Yoo, D.Y.; Kim, D.W.; Choi, J.H.; et al. Cuprizone affects hypothermia-induced neuroprotection and enhanced neuroblast differentiation in the gerbil hippocampus after ischemia. Cells 2020, 9, 1438. [Google Scholar] [CrossRef]
- Fei, F.; Su, N.; Li, X.; Fei, Z. Neuroprotection mediated by natural products and their chemical derivatives. Neural Regen. Res. 2020, 15, 2008–2015. [Google Scholar]
- González-Nieto, D.; Fernández-Serra, R.; Pérez-Rigueiro, J.; Panetsos, F.; Martinez-Murillo, R.; Guinea, G.V. Biomaterials to neuroprotect the stroke brain: A large opportunity for narrow time windows. Cells 2020, 9, 1074. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ye, A.; Ao, L.; Zhou, L.; Yan, Y.; Hu, Y.; Fang, W.; Li, Y. Protective mechanism and treatment of neurogenesis in cerebral ischemia. Neurochem. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, M.; Hällbrink, M.; Prochiantz, A.; Langel, U. Cell-penetrating peptides. Trends Pharmacol. Sci. 2000, 21, 99–103. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, A.; Akuta, T.; Okuyama, H.; Senda, T.; Yokoi, H.; Inokuchi, H.; Fujita, S.; Hayakawa, T.; Takeda, K.; Hasegawa, M.; et al. Protein transduction domain of HIV-1 Tat protein promotes efficient delivery of DNA into mammalian cells. J. Biol. Chem. 2001, 276, 26204–26210. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P.; Rammohan, R.; Weissig, V.; Levchenko, T.S. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 8786–8791. [Google Scholar] [CrossRef]
- Yoo, D.Y.; Cho, S.B.; Jung, H.Y.; Kim, W.; Lee, K.Y.; Kim, J.W.; Moon, S.M.; Won, M.H.; Choi, J.H.; Yoon, Y.S.; et al. Protein disulfide-isomerase A3 significantly reduces ischemia-induced damage by reducing oxidative and endoplasmic reticulum stress. Neurochem. Int. 2019, 122, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.Y.; Cho, S.B.; Jung, H.Y.; Kim, W.; Choi, G.M.; Won, M.H.; Kim, D.W.; Hwang, I.K.; Choi, S.Y.; Moon, S.M. Tat-protein disulfide-isomerase A3: A possible candidate for preventing ischemic damage in the spinal cord. Cell Death Dis. 2017, 8, e3075. [Google Scholar] [CrossRef] [PubMed]
- Verstreken, P.; Kjaerulff, O.; Lloyd, T.E.; Atkinson, R.; Zhou, Y.; Meinertzhagen, I.A.; Bellen, H.J. Endophilin mutations block clathrin-mediated endocytosis but not neurotransmitter release. Cell 2002, 109, 101–112. [Google Scholar] [CrossRef]
- Verstreken, P.; Koh, T.W.; Schulze, K.L.; Zhai, R.G.; Hiesinger, P.R.; Zhou, Y.; Mehta, S.Q.; Cao, Y.; Roos, J.; Bellen, H.J. Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron 2003, 40, 733–748. [Google Scholar] [CrossRef]
- Schuske, K.R.; Richmond, J.E.; Matthies, D.S.; Davis, W.S.; Runz, S.; Rube, D.A.; van der Bliek, A.M.; Jorgensen, E.M. Endophilin is required for synaptic vesicle endocytosis by localizing synaptojanin. Neuron 2003, 40, 749–762. [Google Scholar] [CrossRef]
- Milosevic, I.; Giovedi, S.; Lou, X.; Raimondi, A.; Collesi, C.; Shen, H.; Paradise, S.; O’Toole, E.; Ferguson, S.; Cremona, O.; et al. Recruitment of endophilin to clathrin-coated pit necks is required for efficient vesicle uncoating after fission. Neuron 2011, 72, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Pechstein, A.; Gerth, F.; Milosevic, I.; Jäpel, M.; Eichhorn-Grünig, M.; Vorontsova, O.; Bacetic, J.; Maritzen, T.; Shupliakov, O.; Freund, C.; et al. Vesicle uncoating regulated by SH3-SH3 domain-mediated complex formation between endophilin and intersectin at synapses. EMBO Rep. 2015, 16, 232–239. [Google Scholar] [CrossRef] [PubMed]
- de Heuvel, E.; Bell, A.W.; Ramjaun, A.R.; Wong, K.; Sossin, W.S.; McPherson, P.S. Identification of the major synaptojanin-binding proteins in brain. J. Biol. Chem. 1997, 272, 8710–8716. [Google Scholar] [CrossRef]
- Ringstad, N.; Nemoto, Y.; De Camilli, P. The SH3p4/Sh3p8/SH3p13 protein family: Binding partners for synaptojanin and dynamin via a Grb2-like Src homology 3 domain. Proc. Natl. Acad. Sci. USA 1997, 94, 8569–8674. [Google Scholar] [CrossRef]
- Ringstad, N.; Nemoto, Y.; De Camilli, P. Differential expression of endophilin 1 and 2 dimers at central nervous system synapses. J. Biol. Chem. 2001, 276, 40424–40430. [Google Scholar] [CrossRef]
- Gowrisankaran, S.; Houy, S.; Del Castillo, J.G.P.; Steubler, V.; Gelker, M.; Kroll, J.; Pinheiro, P.S.; Schwitters, D.; Halbsgut, N.; Pechstein, A.; et al. Endophilin-A coordinates priming and fusion of neurosecretory vesicles via intersectin. Nat. Commun. 2020, 11, 1266. [Google Scholar] [CrossRef]
- Chowdhury, S.; Shepherd, J.D.; Okuno, H.; Lyford, G.; Petralia, R.S.; Plath, N.; Kuhl, D.; Huganir, R.L.; Worley, P.F. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 2006, 52, 445–459. [Google Scholar] [CrossRef]
- Yang, Y.; Wei, M.; Xiong, Y.; Du, X.; Zhu, S.; Yang, L.; Zhang, C.; Liu, J.J. Endophilin A1 regulates dendritic spine morphogenesis and stability through interaction with p140Cap. Cell Res. 2015, 25, 496–516. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.D.; Rostosky, C.M.; Gowrisankaran, S.; Arora, A.S.; Soukup, S.F.; Vidal, R.; Capece, V.; Freytag, S.; Fischer, A.; Verstreken, P.; et al. Endophilin-A deficiency induces the Foxo3a-Fbxo32 network in the brain and causes dysregulation of autophagy and the ubiquitin-proteasome system. Cell Rep. 2016, 17, 1071–1086. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, J.; Guo, Z.; Deng, S.; Du, X.; Zhu, S.; Ye, C.; Shi, Y.S.; Liu, J.J. Endophilin A1 promotes actin polymerization in dendritic spines required for synaptic potentiation. Front. Mol. Neurosci. 2018, 11, 177. [Google Scholar] [CrossRef]
- Ren, Y.; Xu, H.W.; Davey, F.; Taylor, M.; Aiton, J.; Coote, P.; Fang, F.; Yao, J.; Chen, D.; Chen, J.X.; et al. Endophilin I expression is increased in the brains of Alzheimer disease patients. J. Biol. Chem. 2008, 283, 5685–5691. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Wang, Y.; Du, F.; Yan, S.; Hu, G.; Origlia, N.; Rutigliano, G.; Sun, Q.; Yu, H.; Ainge, J.; et al. Overexpression of endophilin A1 exacerbates synaptic alterations in a mouse model of Alzheimer’s disease. Nat. Commun. 2018, 9, 2968. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Yang, Y.; Xu, C.; Niu, Y.; Chen, T.; Zhou, Q.; Liu, J.J. Retrolinkin cooperates with endophilin A1 to mediate BDNF-TrkB early endocytic trafficking and signaling from early endosomes. Mol. Biol. Cell 2011, 22, 3684–3698. [Google Scholar] [CrossRef]
- Burk, K.; Murdoch, J.D.; Freytag, S.; Koenig, M.; Bharat, V.; Markworth, R.; Burkhardt, S.; Fischer, A.; Dean, C. EndophilinAs regulate endosomal sorting of BDNF-TrkB to mediate survival signaling in hippocampal neurons. Sci. Rep. 2017, 7, 2149. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Yao, Y.; Shang, C.; Xue, Y.; Ma, J.; Li, Z.; Liu, Y. MicroRNA-330 is an oncogenic factor in glioblastoma cells by regulating SH3GL2 gene. PLoS ONE 2012, 7, e46010. [Google Scholar] [CrossRef]
- Yao, Y.; Xue, Y.; Ma, J.; Shang, C.; Wang, P.; Liu, L.; Liu, W.; Li, Z.; Qu, S.; Li, Z.; et al. MiR-330-mediated regulation of SH3GL2 expression enhances malignant behaviors of glioblastoma stem cells by activating ERK and PI3K/AKT signaling pathways. PLoS ONE 2014, 9, e95060. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.; Yan, F.; Liu, Z.; Zhang, B. miR-330 regulates Drp-1 mediated mitophagy by targeting PGAM5 in a rat model of permanent focal cerebral ischemia. Eur. J. Pharmacol. 2020, 880, 173143. [Google Scholar] [CrossRef] [PubMed]
- Radtke-Schuller, S.; Schuller, G.; Angenstein, F.; Grosser, O.S.; Goldschmidt, J.; Budinger, E. Brain atlas of the Mongolian gerbil (Meriones unguiculatus) in CT/MRI-aided stereotaxic coordinates. Brain Struct. Funct. 2016, 221 (Suppl. 1), 1–272. [Google Scholar] [CrossRef]
- Kim, W.; Kwon, H.J.; Jung, H.Y.; Hahn, K.R.; Yoon, Y.S.; Hwang, I.K.; Choi, S.Y.; Kim, D.W. P27 protects neurons from ischemic damage by suppressing oxidative stress and increasing autophagy in the hippocampus. Int. J. Mol. Sci. 2020, 21, 9496. [Google Scholar] [CrossRef]
- Jung, H.Y.; Kim, W.; Hahn, K.R.; Kang, M.S.; Kim, T.H.; Kwon, H.J.; Nam, S.M.; Chung, J.Y.; Choi, J.H.; Yoon, Y.S.; et al. Pyridoxine deficiency exacerbates neuronal damage after ischemia by increasing oxidative stress and reduces proliferating cells and neuroblasts in the gerbil hippocampus. Int. J. Mol. Sci. 2020, 21, 5551. [Google Scholar] [CrossRef]
- Babcock, A.M.; Baker, D.A.; Lovec, R. Locomotor activity in the ischemic gerbil. Brain Res. 1993, 625, 351–354. [Google Scholar] [CrossRef]
- Jung, H.Y.; Cho, S.B.; Kim, W.; Yoo, D.Y.; Won, M.H.; Choi, G.M.; Cho, T.G.; Kim, D.W.; Hwang, I.K.; Choi, S.Y.; et al. Phosphatidylethanolamine-binding protein 1 protects CA1 neurons against ischemic damage via ERK-CREB signaling in Mongolian gerbils. Neurochem. Int. 2018, 118, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.D.; Hill, K.E.; Burk, R.F.; Nammour, T.M.; Badr, K.F.; Roberts, L.J., II. A series of prostaglandin F2-like compounds are produced in vitro in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc. Natl. Acad. Sci. USA 1990, 87, 9383–9387. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Jung, H.Y.; Kwon, H.J.; Kim, W.; Hahn, K.R.; Moon, S.M.; Yoon, Y.S.; Kim, D.W.; Hwang, I.K. Phosphoglycerate mutase 1 prevents neuronal death from ischemic damage by reducing neuroinflammation in the rabbit spinal cord. Int. J. Mol. Sci. 2020, 21, 7425. [Google Scholar] [CrossRef]
- Micheva, K.D.; Kay, B.K.; McPherson, P.S. Synaptojanin forms two separate complexes in the nerve terminal. Interactions with endophilin and amphiphysin. J. Biol. Chem. 1997, 272, 27239–27245. [Google Scholar] [CrossRef]
- Ambroso, M.R.; Hegde, B.G.; Langen, R. Endophilin A1 induces different membrane shapes using a conformational switch that is regulated by phosphorylation. Proc. Natl. Acad. Sci. USA 2014, 111, 6982–6987. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Keo, A.; Mahfouz, A.; Ingrassia, A.M.T.; Meneboo, J.P.; Villenet, C.; Mutez, E.; Comptdaer, T.; Lelieveldt, B.P.F.; Figeac, M.; Chartier-Harlin, M.C.; et al. Transcriptomic signatures of brain regional vulnerability to Parkinson’s disease. Commun. Biol. 2020, 3, 101. [Google Scholar] [CrossRef]
- Karasawa, Y.; Araki, H.; Otomo, S. Changes in locomotor activity and passive avoidance task performance induced by cerebral ischemia in Mongolian gerbils. Stroke 1994, 25, 645–650. [Google Scholar] [CrossRef]
- Tappel, A.L. Lipid peroxidation damage to cell components. Fed. Proc. 1973, 32, 1870–1874. [Google Scholar] [PubMed]
- Mead, J.F. Free radical mechanisms of lipid damage and consequences for cellular membranes. In Free Radicals in Biology; Pryor, W.A., Ed.; Academic Press: New York, NY, USA, 1976; pp. 51–68. [Google Scholar]
- Qu, Y.; Zhang, H.L.; Zhang, X.P.; Jiang, H.L. Arachidonic acid attenuates brain damage in a rat model of ischemia/reperfusion by inhibiting inflammatory response and oxidative stress. Hum. Exp. Toxicol. 2018, 37, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Q.; Li, L.; Liu, Y.; Diao, H. Arachidonic acid attenuates learning and memory dysfunction induced by repeated isoflurane anesthesia in rats. Int. J. Clin. Exp. Med. 2015, 8, 12365–12373. [Google Scholar] [PubMed]
- Hooper, L.; Al-Khudairy, L.; Abdelhamid, A.S.; Rees, K.; Brainard, J.S.; Brown, T.J.; Ajabnoor, S.M.; O’Brien, A.T.; Winstanley, L.E.; Donaldson, D.H.; et al. Omega-6 fats for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2018, 7, CD011094. [Google Scholar]
- Saito, K.; Suyama, K.; Nishida, K.; Sei, Y.; Basile, A.S. Early increases in TNF-alpha, IL-6 and IL-1 beta levels following transient cerebral ischemia in gerbil brain. Neurosci. Lett. 1996, 206, 149–152. [Google Scholar] [CrossRef]
- Suzuki, S.; Tanaka, K.; Nagata, E.; Ito, D.; Dembo, T.; Fukuuchi, Y. Cerebral neurons express interleukin-6 after transient forebrain ischemia in gerbils. Neurosci. Lett. 1999, 262, 117–120. [Google Scholar] [CrossRef]
- Ishimaru, H.; Casamenti, F.; Uéda, K.; Maruyama, Y.; Pepeu, G. Changes in presynaptic proteins, SNAP-25 and synaptophysin, in the hippocampal CA1 area in ischemic gerbils. Brain Res. 2001, 903, 94–101. [Google Scholar] [CrossRef]
- Yan, B.C.; Park, J.H.; Ahn, J.H.; Lee, J.C.; Won, M.H.; Kang, I.J. Postsynaptic density protein (PSD)-95 expression is markedly decreased in the hippocampal CA1 region after experimental ischemia-reperfusion injury. J. Neurol. Sci. 2013, 330, 111–116. [Google Scholar] [CrossRef]
- Zhang, M.; Zhai, Y.; Sun, Y.; Zhang, W.; Li, Q.; Brann, D.; Wang, R. Swimming improves cognitive reserve in ovariectomized rats and enhances neuroprotection after global cerebral ischemia. Brain Res. 2018, 1692, 110–117. [Google Scholar] [CrossRef]
- Fernandes, J.; Vieira, M.; Carreto, L.; Santos, M.A.; Duarte, C.B.; Carvalho, A.L.; Santos, A.E. In vitro ischemia triggers a transcriptional response to down-regulate synaptic proteins in hippocampal neurons. PLoS ONE 2014, 9, e99958. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, H.Y.; Kwon, H.J.; Kim, W.; Hwang, I.K.; Choi, G.-M.; Chang, I.B.; Kim, D.W.; Moon, S.M. Tat-Endophilin A1 Fusion Protein Protects Neurons from Ischemic Damage in the Gerbil Hippocampus: A Possible Mechanism of Lipid Peroxidation and Neuroinflammation Mitigation as Well as Synaptic Plasticity. Cells 2021, 10, 357. https://doi.org/10.3390/cells10020357
Jung HY, Kwon HJ, Kim W, Hwang IK, Choi G-M, Chang IB, Kim DW, Moon SM. Tat-Endophilin A1 Fusion Protein Protects Neurons from Ischemic Damage in the Gerbil Hippocampus: A Possible Mechanism of Lipid Peroxidation and Neuroinflammation Mitigation as Well as Synaptic Plasticity. Cells. 2021; 10(2):357. https://doi.org/10.3390/cells10020357
Chicago/Turabian StyleJung, Hyo Young, Hyun Jung Kwon, Woosuk Kim, In Koo Hwang, Goang-Min Choi, In Bok Chang, Dae Won Kim, and Seung Myung Moon. 2021. "Tat-Endophilin A1 Fusion Protein Protects Neurons from Ischemic Damage in the Gerbil Hippocampus: A Possible Mechanism of Lipid Peroxidation and Neuroinflammation Mitigation as Well as Synaptic Plasticity" Cells 10, no. 2: 357. https://doi.org/10.3390/cells10020357
APA StyleJung, H. Y., Kwon, H. J., Kim, W., Hwang, I. K., Choi, G.-M., Chang, I. B., Kim, D. W., & Moon, S. M. (2021). Tat-Endophilin A1 Fusion Protein Protects Neurons from Ischemic Damage in the Gerbil Hippocampus: A Possible Mechanism of Lipid Peroxidation and Neuroinflammation Mitigation as Well as Synaptic Plasticity. Cells, 10(2), 357. https://doi.org/10.3390/cells10020357