
Gel-Free 3D Tumoroids with Stem Cell Properties Modeling Drug Resistance to Cisplatin and Imatinib in Metastatic Colorectal Cancer

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Gel-Free 3D Culture
2.2. Chemicals and Drugs
2.3. Drug Treatment
2.4. 3D Tumoroid-Based Multiplex Reporter Assay
2.5. Cell Viability Assay
2.6. Side Population Analysis
2.7. Microarray and Bioinformatics
2.8. RT-qPCR
2.9. Statistical Analysis
3. Results
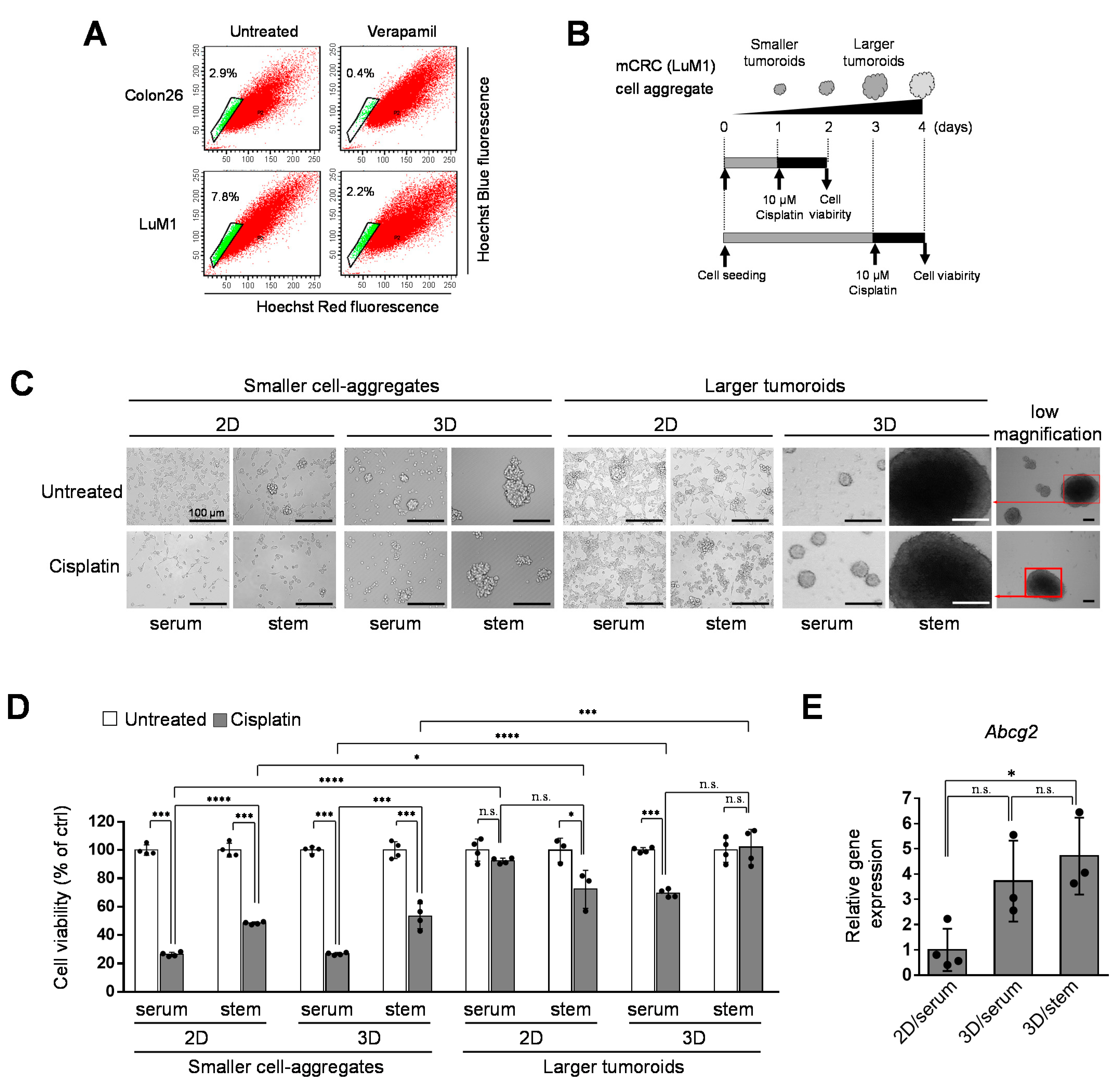
3.1. Stem Cell Properties of the Aggregative, Metastatic CRC Cell Line LuM1
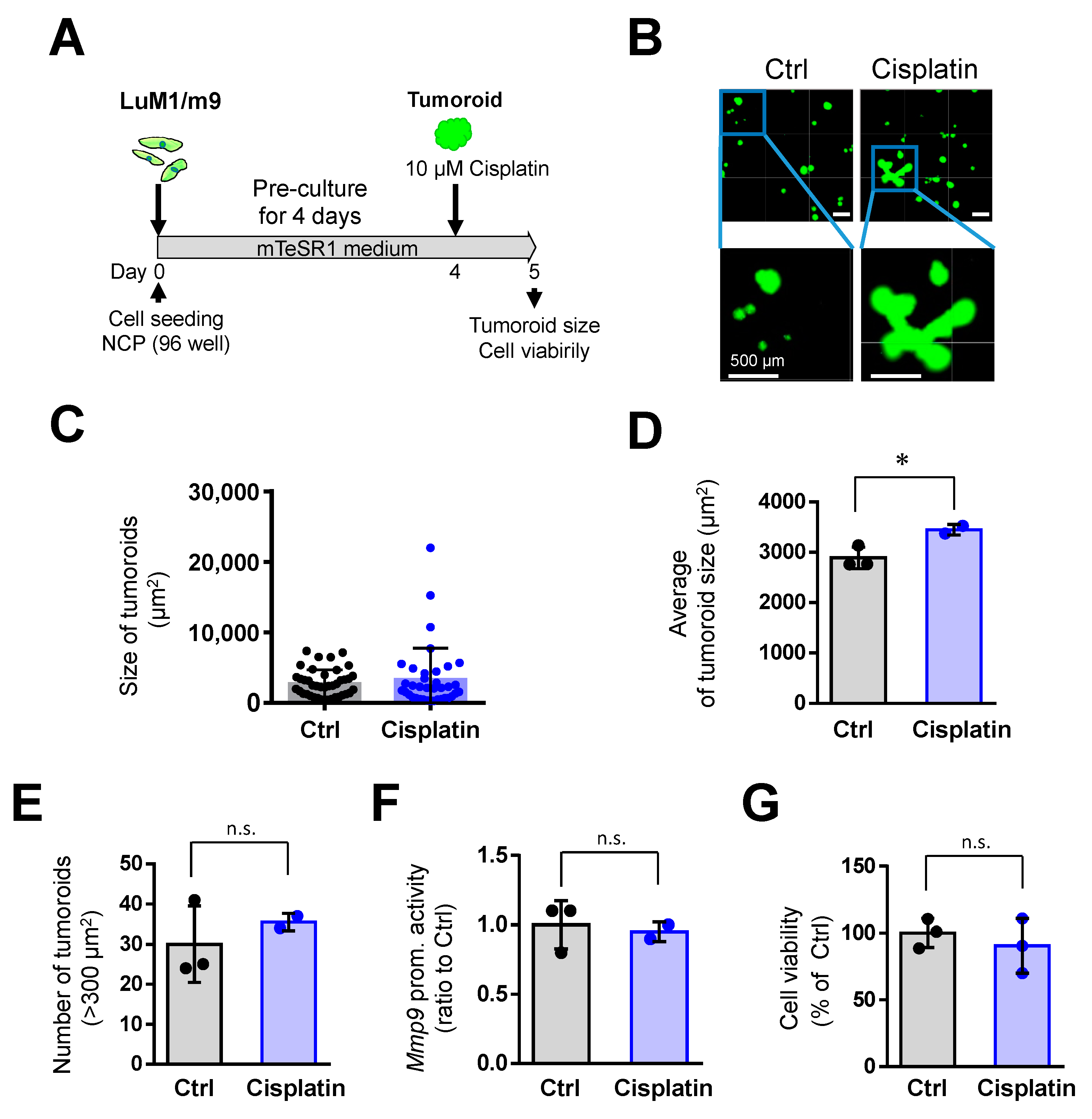
3.2. Tumoroids Acquired Platinum-Resistance with ABCG2 Expression in Metastatic Colorectal Cancer Cells
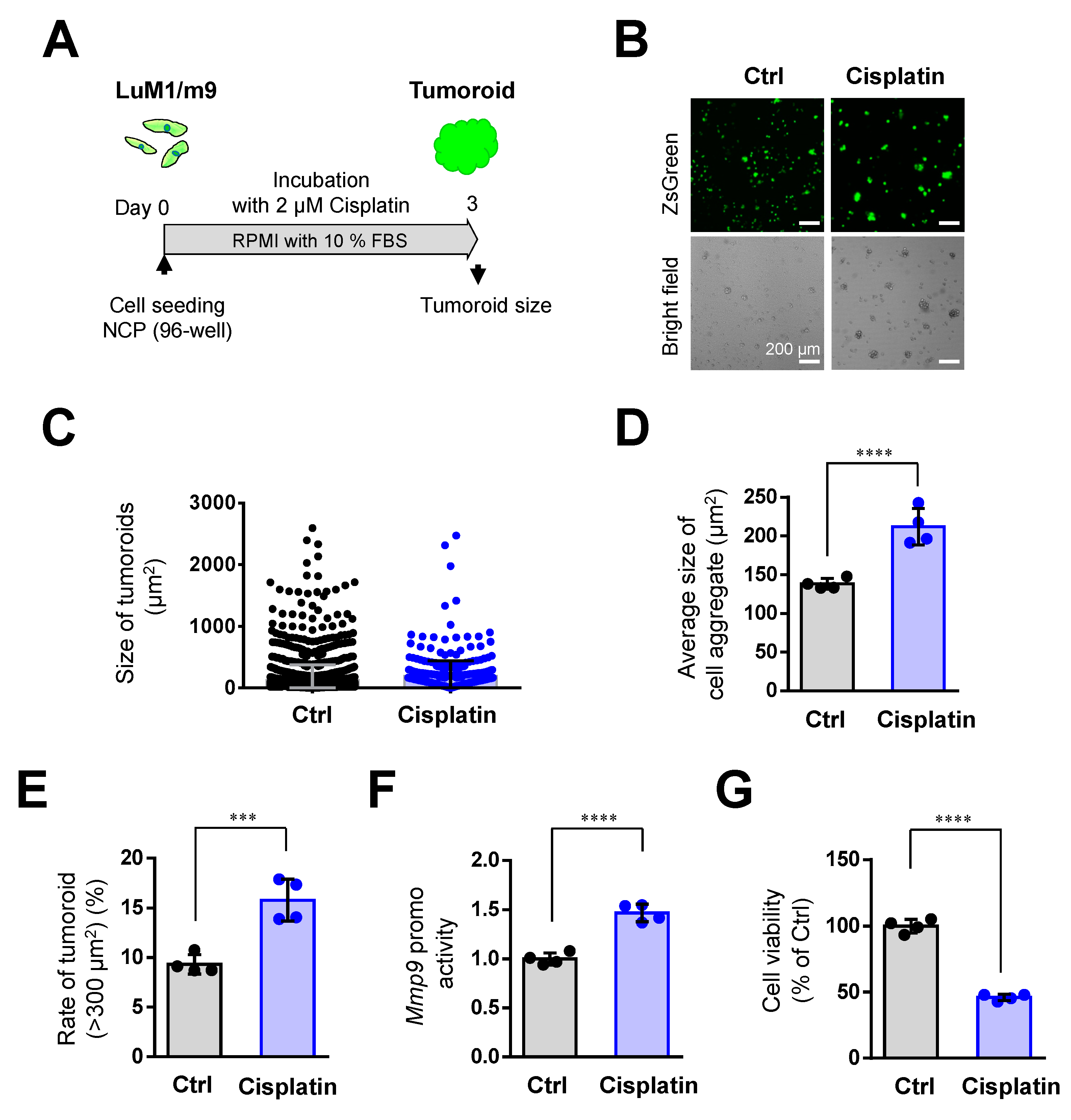
3.3. Cisplatin Promoted Tumoroid Formation of Metastatic Colorectal Cancer
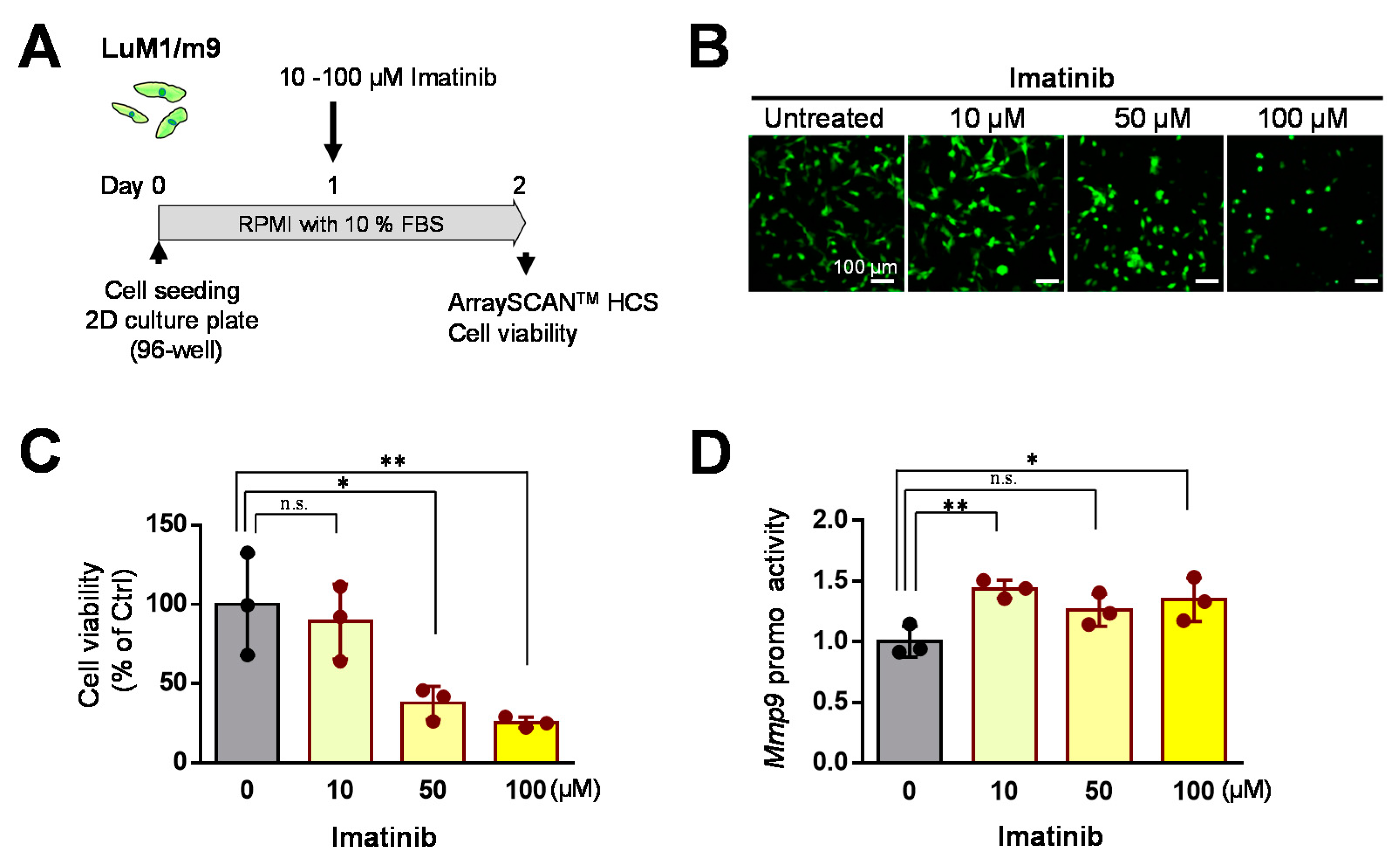
3.4. Concentration-Dependent Effects of Imatinib on mCRC Cells in 2D Culture
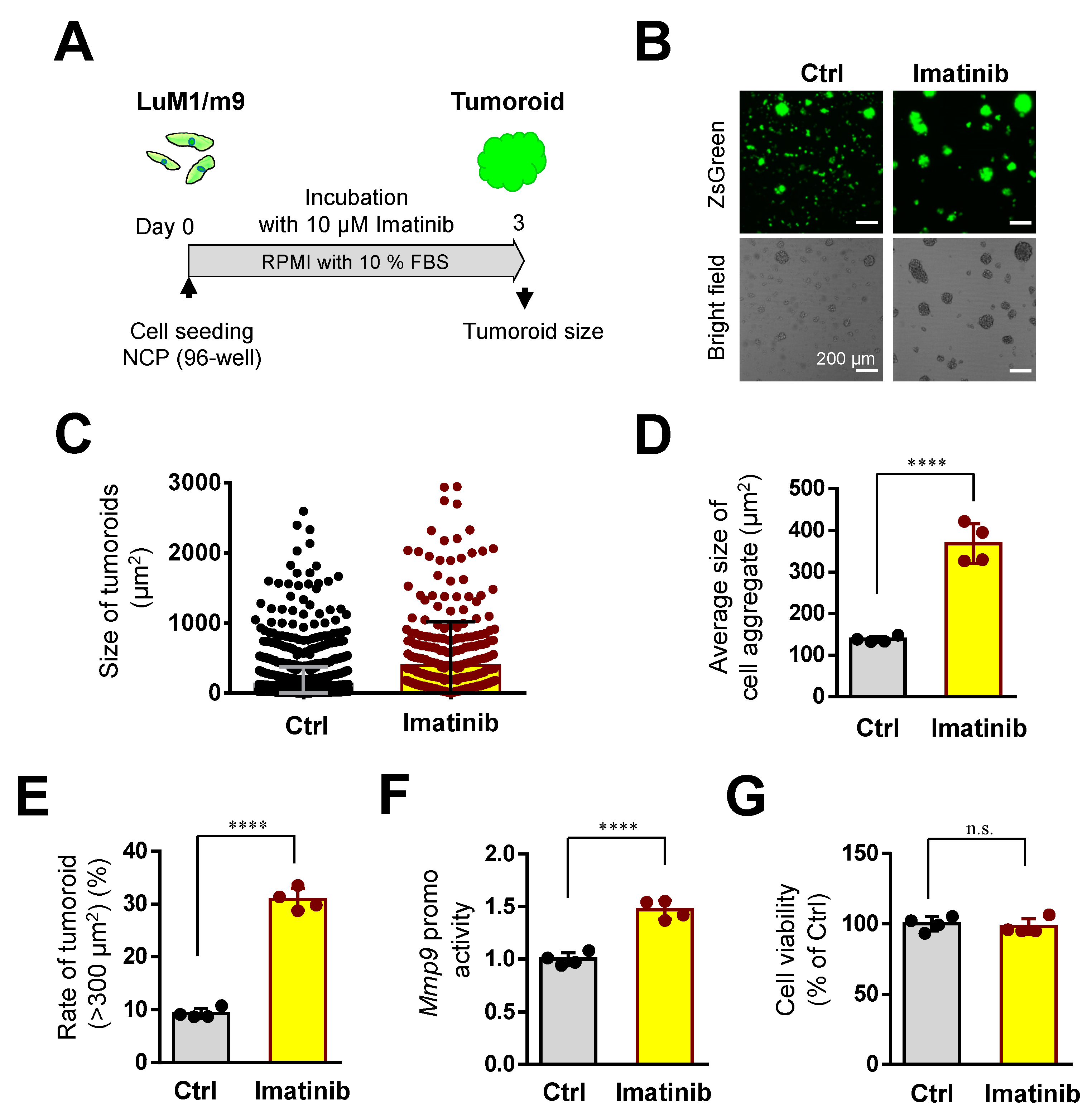
3.5. Imatinib Promoted Tumoroid Growth of mCRC
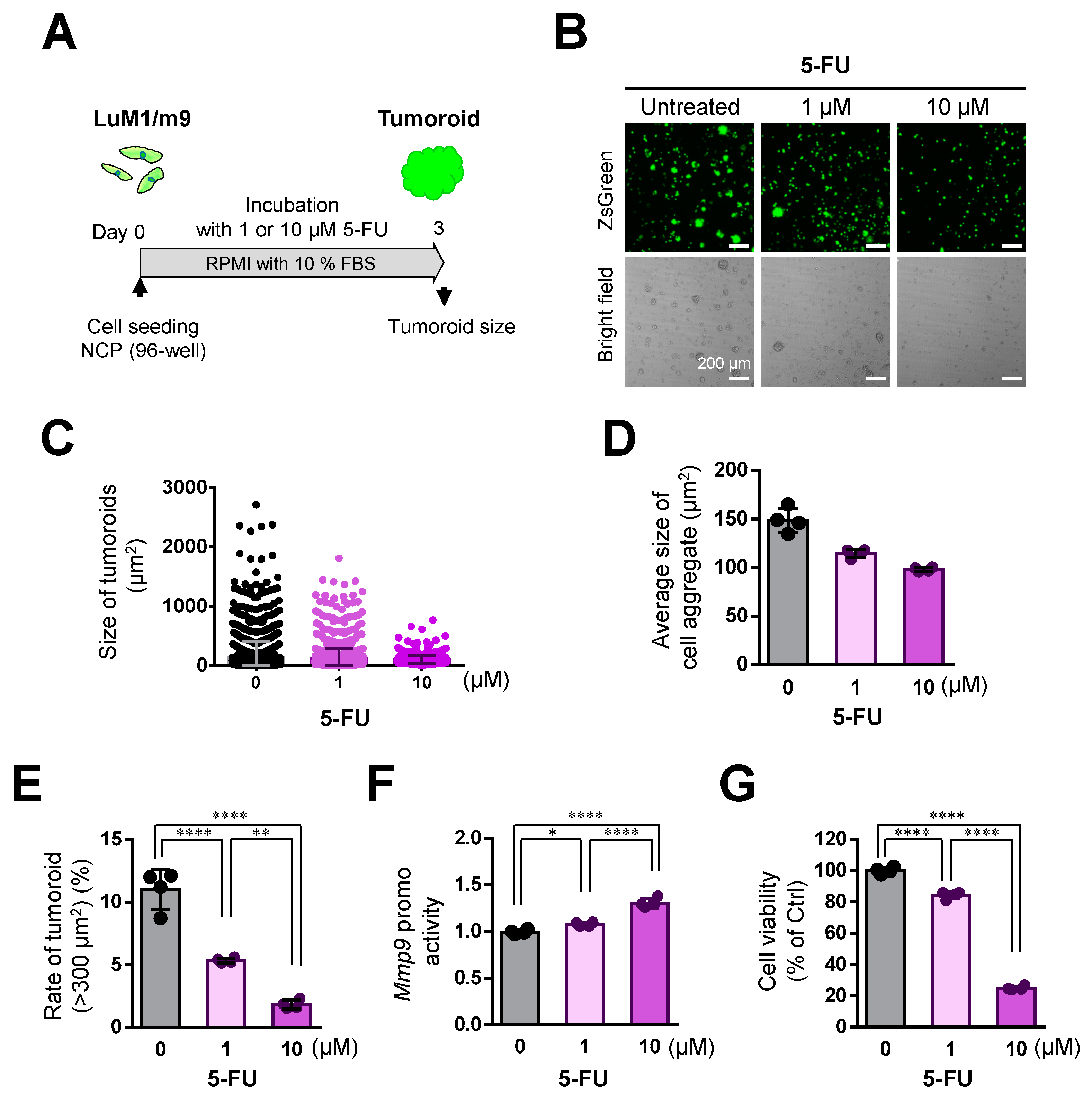
3.6. 5-FU Inhibited Tumoroid Growth of mCRC, Although Not Completely
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chou, S.D.; Murshid, A.; Eguchi, T.; Gong, J.; Calderwood, S.K. HSF1 regulation of beta-catenin in mammary cancer cells through control of HuR/elavL1 expression. Oncogene 2015, 34, 2178–2188. [Google Scholar] [CrossRef]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef]
- Arai, K.; Eguchi, T.; Rahman, M.M.; Sakamoto, R.; Masuda, N.; Nakatsura, T.; Calderwood, S.K.; Kozaki, K.; Itoh, M. A Novel High-Throughput 3D Screening System for EMT Inhibitors: A Pilot Screening Discovered the EMT Inhibitory Activity of CDK2 Inhibitor SU9516. PLoS ONE 2016, 11, e0162394. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef]
- Takai, A.; Fako, V.; Dang, H.; Forgues, M.; Yu, Z.; Budhu, A.; Wang, X.W. Three-dimensional Organotypic Culture Models of Human Hepatocellular Carcinoma. Sci. Rep. 2016, 6, 21174. [Google Scholar] [CrossRef]
- Taha, E.A.; Sogawa, C.; Okusha, Y.; Kawai, H.; Oo, M.W.; Elseoudi, A.; Lu, Y.; Nagatsuka, H.; Kubota, S.; Satoh, A.; et al. Knockout of MMP3 Weakens Solid Tumor Organoids and Cancer Extracellular Vesicles. Cancers (Basel) 2020, 12, 1260. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Sogawa, C.; Okusha, Y.; Uchibe, K.; Iinuma, R.; Ono, K.; Nakano, K.; Murakami, J.; Itoh, M.; Arai, K.; et al. Organoids with Cancer Stem Cell-like Properties Secrete Exosomes and HSP90 in a 3D NanoEnvironment. PLoS ONE 2018, 13, e0191109. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Rossi, G.; Ferrari, P.; Carpi, A. Minimal residual disease in advanced or metastatic solid cancers: The G0-G1 state and immunotherapy are key to unwinding cancer complexity. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; de Prati, A.C.; Boriero, D.; Mariotto, S. Tumor Dormancy and Interplay with Hypoxic Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 4305. [Google Scholar] [CrossRef]
- Walker, N.D.; Patel, J.; Munoz, J.L.; Hu, M.; Guiro, K.; Sinha, G.; Rameshwar, P. The bone marrow niche in support of breast cancer dormancy. Cancer Lett. 2016, 380, 263–271. [Google Scholar] [CrossRef]
- Senkowski, W.; Zhang, X.; Olofsson, M.H.; Isacson, R.; Hoglund, U.; Gustafsson, M.; Nygren, P.; Linder, S.; Larsson, R.; Fryknas, M. Three-Dimensional Cell Culture-Based Screening Identifies the Anthelmintic Drug Nitazoxanide as a Candidate for Treatment of Colorectal Cancer. Mol. Cancer Ther. 2015, 14, 1504–1516. [Google Scholar] [CrossRef]
- Sogawa, C.; Eguchi, T.; Tran, M.T.; Ishige, M.; Trin, K.; Okusha, Y.; Taha, E.A.; Lu, Y.; Kawai, H.; Sogawa, N.; et al. Antiparkinson Drug Benztropine Suppresses Tumor Growth, Circulating Tumor Cells, and Metastasis by Acting on SLC6A3/DAT and Reducing STAT3. Cancers (Basel) 2020, 12, 523. [Google Scholar] [CrossRef]
- Sogawa, C.; Eguchi, T.; Okusha, Y.; Ono, K.; Ohyama, K.; Iizuka, M.; Kawasaki, R.; Hamada, Y.; Takigawa, M.; Sogawa, N.; et al. A reporter system evaluates tumorigenesis, metastasis, beta-catenin/MMP regulation, and druggability. Tissue Eng. Part A 2019, 25, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Sogawa, C.; Kawai, H.; Tran, M.T.; Taha, E.A.; Lu, Y.; Oo, M.W.; Okusha, Y.; Okamura, H.; Ibaragi, S.; et al. Triple knockdown of CDC37, HSP90-alpha and HSP90-beta diminishes extracellular vesicles-driven malignancy events and macrophage M2 polarization in oral cancer. J. Extracell. Vesicles 2020, 9, 1769373. [Google Scholar] [CrossRef] [PubMed]
- Namba, Y.; Sogawa, C.; Okusha, Y.; Kawai, H.; Itagaki, M.; Ono, K.; Murakami, J.; Aoyama, E.; Ohyama, K.; Asaumi, J.; et al. Depletion of Lipid Efflux Pump ABCG1 Triggers the Intracellular Accumulation of Extracellular Vesicles and Reduces Aggregation and Tumorigenesis of Metastatic Cancer Cells. Front. Oncol. 2018, 8, 376. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.L.; Joshi, R.; Thakuri, P.S.; Tavana, H. Liquid-based three-dimensional tumor models for cancer research and drug discovery. Exp. Biol. Med. (Maywood) 2016, 241, 939–954. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Translational applications of adult stem cell-derived organoids. Development 2017, 144, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Karthaus, W.R.; Iaquinta, P.J.; Drost, J.; Gracanin, A.; van Boxtel, R.; Wongvipat, J.; Dowling, C.M.; Gao, D.; Begthel, H.; Sachs, N.; et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014, 159, 163–175. [Google Scholar] [CrossRef]
- Palikuqi, B.; Nguyen, D.T.; Li, G.; Schreiner, R.; Pellegata, A.F.; Liu, Y.; Redmond, D.; Geng, F.; Lin, Y.; Gomez-Salinero, J.M.; et al. Adaptable haemodynamic endothelial cells for organogenesis and tumorigenesis. Nature 2020, 585, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Bhagwandin, V.J.; Bishop, J.M.; Wright, W.E.; Shay, J.W. The Metastatic Potential and Chemoresistance of Human Pancreatic Cancer Stem Cells. PLoS ONE 2016, 11, e0148807. [Google Scholar] [CrossRef]
- Wang, Y.; Teng, J.S. Increased multi-drug resistance and reduced apoptosis in osteosarcoma side population cells are crucial factors for tumor recurrence. Exp. Ther. Med. 2016, 12, 81–86. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Ross, D.D.; Nakanishi, T.; Bailey-Dell, K.; Zhou, S.; Mercer, K.E.; Sarkadi, B.; Sorrentino, B.P.; Schuetz, J.D. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J. Biol. Chem. 2004, 279, 24218–24225. [Google Scholar] [CrossRef]
- Huang, Y.; Sadee, W. Membrane transporters and channels in chemoresistance and -sensitivity of tumor cells. Cancer Lett. 2006, 239, 168–182. [Google Scholar] [CrossRef]
- van Herwaarden, A.E.; Schinkel, A.H. The function of breast cancer resistance protein in epithelial barriers, stem cells and milk secretion of drugs and xenotoxins. Trends Pharmacol. Sci. 2006, 27, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Katayama, K.; Sugimoto, Y. Human ABC transporter ABCG2/BCRP expression in chemoresistance: Basic and clinical perspectives for molecular cancer therapeutics. Pharm. Pers. Med. 2014, 7, 53–64. [Google Scholar] [CrossRef]
- Cabana, R.; Frolova, E.G.; Kapoor, V.; Thomas, R.A.; Krishan, A.; Telford, W.G. The minimal instrumentation requirements for Hoechst side population analysis: Stem cell analysis on low-cost flow cytometry platforms. Stem Cells 2006, 24, 2573–2581. [Google Scholar] [CrossRef][Green Version]
- Liu, P.P.; Liao, J.; Tang, Z.J.; Wu, W.J.; Yang, J.; Zeng, Z.L.; Hu, Y.; Wang, P.; Ju, H.Q.; Xu, R.H.; et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. 2014, 21, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.Y.; Wang, F.F.; Xiao, Z.H.; Liu, S.F.; Tang, S.L.; Lai, Y.L. Overexpressing microRNA-34a overcomes ABCG2-mediated drug resistance to 5-FU in side population cells from colon cancer via suppressing DLL1. J. Biochem. 2020, 167, 557–564. [Google Scholar] [CrossRef]
- Mori, T.; Nishizawa, S.; Hirohashi, Y.; Torigoe, T.; Tamura, Y.; Takahashi, A.; Kochin, V.; Fujii, R.; Kondo, T.; Greene, M.I.; et al. Efficiency of G2/M-related tumor-associated antigen-targeting cancer immunotherapy depends on antigen expression in the cancer stem-like population. Exp. Mol. Pathol. 2012, 92, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Hara, E.S.; Ono, M.; Eguchi, T.; Kubota, S.; Pham, H.T.; Sonoyama, W.; Tajima, S.; Takigawa, M.; Calderwood, S.K.; Kuboki, T. miRNA-720 controls stem cell phenotype, proliferation and differentiation of human dental pulp cells. PLoS ONE 2013, 8, e83545. [Google Scholar] [CrossRef]
- Guan, G.F.; Zhang, D.J.; Zheng, Y.; Wen, L.J.; Yu, D.J.; Lu, Y.Q.; Zhao, Y. Abnormal Wnt signaling and overexpression of ABCG2 contributes to drug efflux properties of side population cells in nasopharyngeal carcinoma. Mol. Med. Rep. 2015, 12, 4352–4357. [Google Scholar] [CrossRef] [PubMed]
- Guan, G.F.; Zhang, D.J.; Zheng, Y.; Wen, L.J.; Yu, D.J.; Lu, Y.Q.; Zhao, Y. Significance of ATP-binding cassette transporter proteins in multidrug resistance of head and neck squamous cell carcinoma. Oncol. Lett. 2015, 10, 631–636. [Google Scholar] [CrossRef]
- Eguchi, T.; Taha, E.A.; Calderwood, S.K.; Ono, K. A Novel Model of Cancer Drug Resistance: Oncosomal Release of Cytotoxic and Antibody-Based Drugs. Biology (Basel) 2020, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Sakata, K.; Kozaki, K.; Iida, K.; Tanaka, R.; Yamagata, S.; Utsumi, K.R.; Saga, S.; Shimizu, S.; Matsuyama, M. Establishment and characterization of high- and low-lung-metastatic cell lines derived from murine colon adenocarcinoma 26 tumor line. Jpn. J. Cancer Res. 1996, 87, 78–85. [Google Scholar] [CrossRef]
- Okusha, Y.; Eguchi, T.; Sogawa, C.; Okui, T.; Nakano, K.; Okamoto, K.; Kozaki, K. The intranuclear PEX domain of MMP involves proliferation, migration, and metastasis of aggressive adenocarcinoma cells. J. Cell Biochem. 2018, 119, 7363–7376. [Google Scholar] [CrossRef]
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol. Cancer 2005, 4, 1595–1604. [Google Scholar] [CrossRef]
- Yin, J.; Yan, X.; Yao, X.; Zhang, Y.; Shan, Y.; Mao, N.; Yang, Y.; Pan, L. Secretion of annexin A3 from ovarian cancer cells and its association with platinum resistance in ovarian cancer patients. J. Cell Mol. Med. 2012, 16, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Okusha, Y.; Eguchi, T.; Tran, M.T.; Sogawa, C.; Yoshida, K.; Itagaki, M.; Taha, E.A.; Ono, K.; Aoyama, E.; Okamura, H.; et al. Extracellular Vesicles Enriched with Moonlighting Metalloproteinase Are Highly Transmissive, Pro-Tumorigenic, and Trans-Activates Cellular Communication Network Factor (CCN2/CTGF): CRISPR against Cancer. Cancers (Basel) 2020, 12, 881. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Xiao, X.; Xu, H.; Lu, W.; Wang, Y. Efficacy of atovaquone on EpCAM(+)CD44(+) HCT-116 human colon cancer stem cells under hypoxia. Exp. Ther. Med. 2020, 20, 286. [Google Scholar] [CrossRef]
- Atkinson, C.J.; Kawamata, F.; Liu, C.; Ham, S.; Gyorffy, B.; Munn, A.L.; Wei, M.Q.; Moller, A.; Whitehall, V.; Wiegmans, A.P. EGFR and Prion protein promote signaling via FOXO3a-KLF5 resulting in clinical resistance to platinum agents in colorectal cancer. Mol. Oncol. 2019, 13, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Taha, E.A. Extracellular Vesicle-Associated Moonlighting Proteins: Heat Shock Proteins and Metalloproteinases. In Heat Shock Proteins; Asea, A.A.A., Kaur, P., Eds.; Springer: Dordrecht, The Netherlands, 2020; pp. 1–18. [Google Scholar]
- Eguchi, T.; Calderwood, S.K.; Takigawa, M.; Kubota, S.; Kozaki, K. Intracellular MMP3 Promotes HSP Gene Expression in Collaboration With Chromobox Proteins. J. Cell Biochem. 2017, 118, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, T.; Kubota, S.; Kawata, K.; Mukudai, Y.; Uehara, J.; Ohgawara, T.; Ibaragi, S.; Sasaki, A.; Kuboki, T.; Takigawa, M. Novel Transcriptional Regulation of CCN2/CTGF by Nuclear Translocation of MMP3. In CCN Proteins in Health and Disease; Perbal, A., Takigawa, M., Perbal, B., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 255–264. [Google Scholar]
- Eguchi, T.; Kubota, S.; Kawata, K.; Mukudai, Y.; Uehara, J.; Ohgawara, T.; Ibaragi, S.; Sasaki, A.; Kuboki, T.; Takigawa, M. Novel transcription-factor-like function of human matrix metalloproteinase 3 regulating the CTGF/CCN2 gene. Mol. Cell Biol. 2008, 28, 2391–2413. [Google Scholar] [CrossRef] [PubMed]
- Chau, W.K.; Ip, C.K.; Mak, A.S.; Lai, H.C.; Wong, A.S. c-Kit mediates chemoresistance and tumor-initiating capacity of ovarian cancer cells through activation of Wnt/beta-catenin-ATP-binding cassette G2 signaling. Oncogene 2013, 32, 2767–2781. [Google Scholar] [CrossRef]
- Rossi, F.; Yozgat, Y.; de Stanchina, E.; Veach, D.; Clarkson, B.; Manova, K.; Giancotti, F.G.; Antonescu, C.R.; Besmer, P. Imatinib upregulates compensatory integrin signaling in a mouse model of gastrointestinal stromal tumor and is more effective when combined with dasatinib. Mol. Cancer Res. 2010, 8, 1271–1283. [Google Scholar] [CrossRef][Green Version]
- Pene-Dumitrescu, T.; Smithgall, T.E. Expression of a Src family kinase in chronic myelogenous leukemia cells induces resistance to imatinib in a kinase-dependent manner. J. Biol Chem. 2010, 285, 21446–21457. [Google Scholar] [CrossRef]
- Larue, L.; Bellacosa, A. Epithelial-mesenchymal transition in development and cancer: Role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 2005, 24, 7443–7454. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Liu, A.; Wang, J.; Li, L.; Chen, X.; Gao, X.; Xue, Y.; Zhang, X.; Liu, Y. Distinct Dasatinib-Induced Mechanisms of Apoptotic Response and Exosome Release in Imatinib-Resistant Human Chronic Myeloid Leukemia Cells. Int. J. Mol. Sci. 2016, 17, 531. [Google Scholar] [CrossRef]
- Fujiwara, T.; Eguchi, T.; Sogawa, C.; Ono, K.; Murakami, J.; Ibaragi, S.; Asaumi, J.-I.; Calderwood, S.K.; Okamoto, K.; Kozaki, K.-I. Carcinogenic epithelial-mesenchymal transition initiated by oral cancer exosomes is inhibited by anti-EGFR antibody cetuximab. Oral Oncol. 2018, 86, 25–257. [Google Scholar] [CrossRef]
- Perone, Y.; Farrugia, A.J.; Rodriguez-Meira, A.; Gyorffy, B.; Ion, C.; Uggetti, A.; Chronopoulos, A.; Marrazzo, P.; Faronato, M.; Shousha, S.; et al. SREBP1 drives Keratin-80-dependent cytoskeletal changes and invasive behavior in endocrine-resistant ERalpha breast cancer. Nat. Commun. 2019, 10, 2115. [Google Scholar] [CrossRef]
- Braig, F.; Kriegs, M.; Voigtlaender, M.; Habel, B.; Grob, T.; Biskup, K.; Blanchard, V.; Sack, M.; Thalhammer, A.; Batalla, I.B.; et al. Cetuximab Resistance in Head and Neck Cancer Is Mediated by EGFR-K521 Polymorphism. Cancer Res. 2017, 77, 1188–1199. [Google Scholar] [CrossRef] [PubMed]
- Stangl, S.; Gehrmann, M.; Riegger, J.; Kuhs, K.; Riederer, I.; Sievert, W.; Hube, K.; Mocikat, R.; Dressel, R.; Kremmer, E.; et al. Targeting membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70.1 antibody. Proc. Natl. Acad. Sci. USA 2011, 108, 733–738. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sogawa, C.; Eguchi, T.; Namba, Y.; Okusha, Y.; Aoyama, E.; Ohyama, K.; Okamoto, K. Gel-Free 3D Tumoroids with Stem Cell Properties Modeling Drug Resistance to Cisplatin and Imatinib in Metastatic Colorectal Cancer. Cells 2021, 10, 344. https://doi.org/10.3390/cells10020344
Sogawa C, Eguchi T, Namba Y, Okusha Y, Aoyama E, Ohyama K, Okamoto K. Gel-Free 3D Tumoroids with Stem Cell Properties Modeling Drug Resistance to Cisplatin and Imatinib in Metastatic Colorectal Cancer. Cells. 2021; 10(2):344. https://doi.org/10.3390/cells10020344
Chicago/Turabian StyleSogawa, Chiharu, Takanori Eguchi, Yuri Namba, Yuka Okusha, Eriko Aoyama, Kazumi Ohyama, and Kuniaki Okamoto. 2021. "Gel-Free 3D Tumoroids with Stem Cell Properties Modeling Drug Resistance to Cisplatin and Imatinib in Metastatic Colorectal Cancer" Cells 10, no. 2: 344. https://doi.org/10.3390/cells10020344
APA StyleSogawa, C., Eguchi, T., Namba, Y., Okusha, Y., Aoyama, E., Ohyama, K., & Okamoto, K. (2021). Gel-Free 3D Tumoroids with Stem Cell Properties Modeling Drug Resistance to Cisplatin and Imatinib in Metastatic Colorectal Cancer. Cells, 10(2), 344. https://doi.org/10.3390/cells10020344