
Genetic Abnormalities in Multiple Myeloma: Prognostic and Therapeutic Implications



Abstract
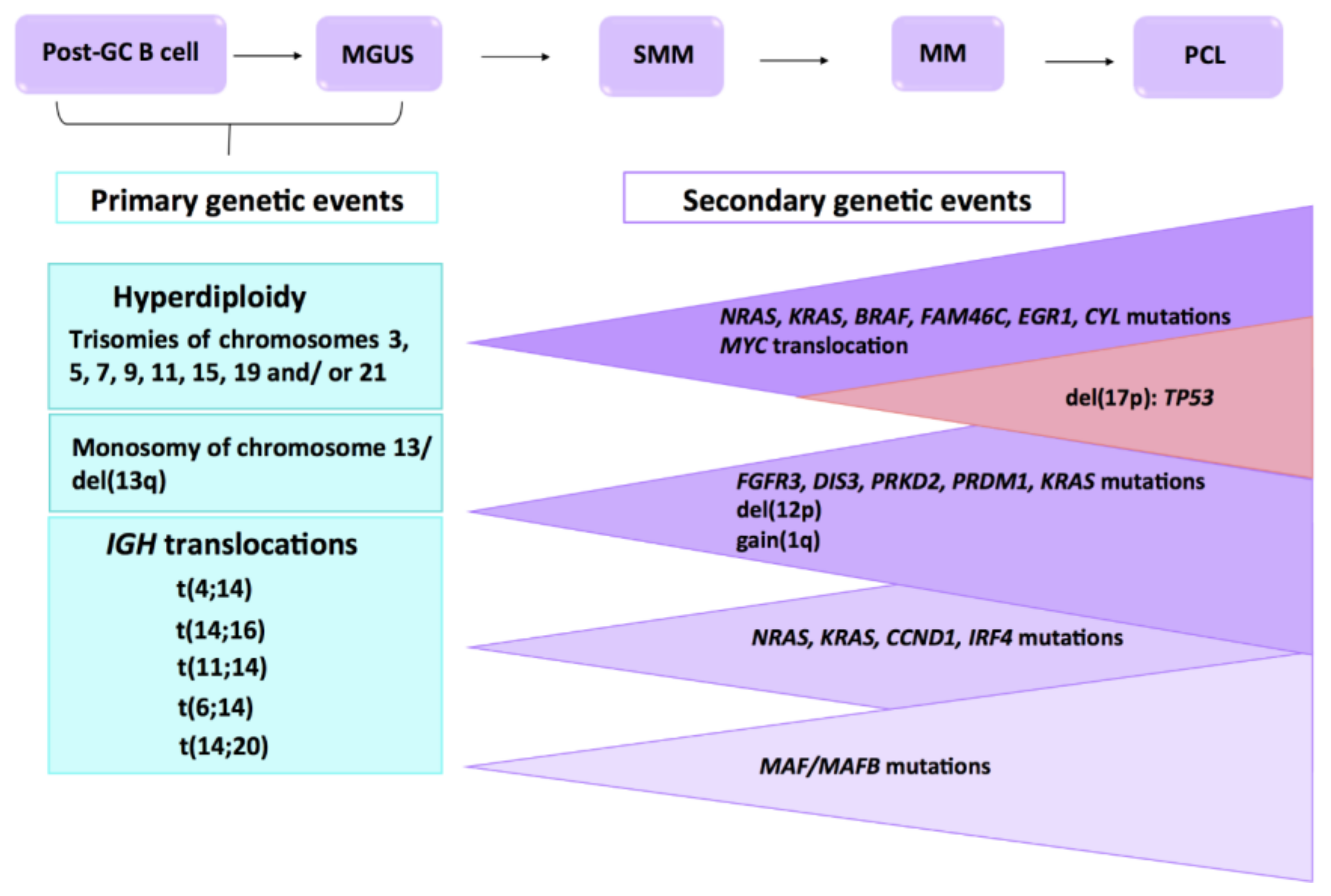
1. Introduction
2. Genetic Abnormalities
2.1. Chromosomal Translocations
2.1.1. Translocations t(11;14) and t(6;14)
Prognosis
Therapeutic Implications
2.1.2. Translocation t(4;14)
Prognosis
Therapeutic Implications
2.1.3. Translocations t(14;16) and t(14;20)
Prognosis
Therapeutic Implications
2.1.4. Translocation of MYC Oncogene
Prognosis
Therapeutic Implications
2.2. Copy Number Abnormalities
2.2.1. Hyperdiploid/Hypodiploid
Prognosis
Therapeutic Implications
2.2.2. Deletion of 1p
Prognosis
Therapeutic Implications
2.2.3. Gain of 1q
Prognosis
Therapeutic Implications
2.2.4. Deletion of 13q
Prognosis
Therapeutic Implications
2.2.5. Deletion of 17p
Prognosis
Therapeutic Implications
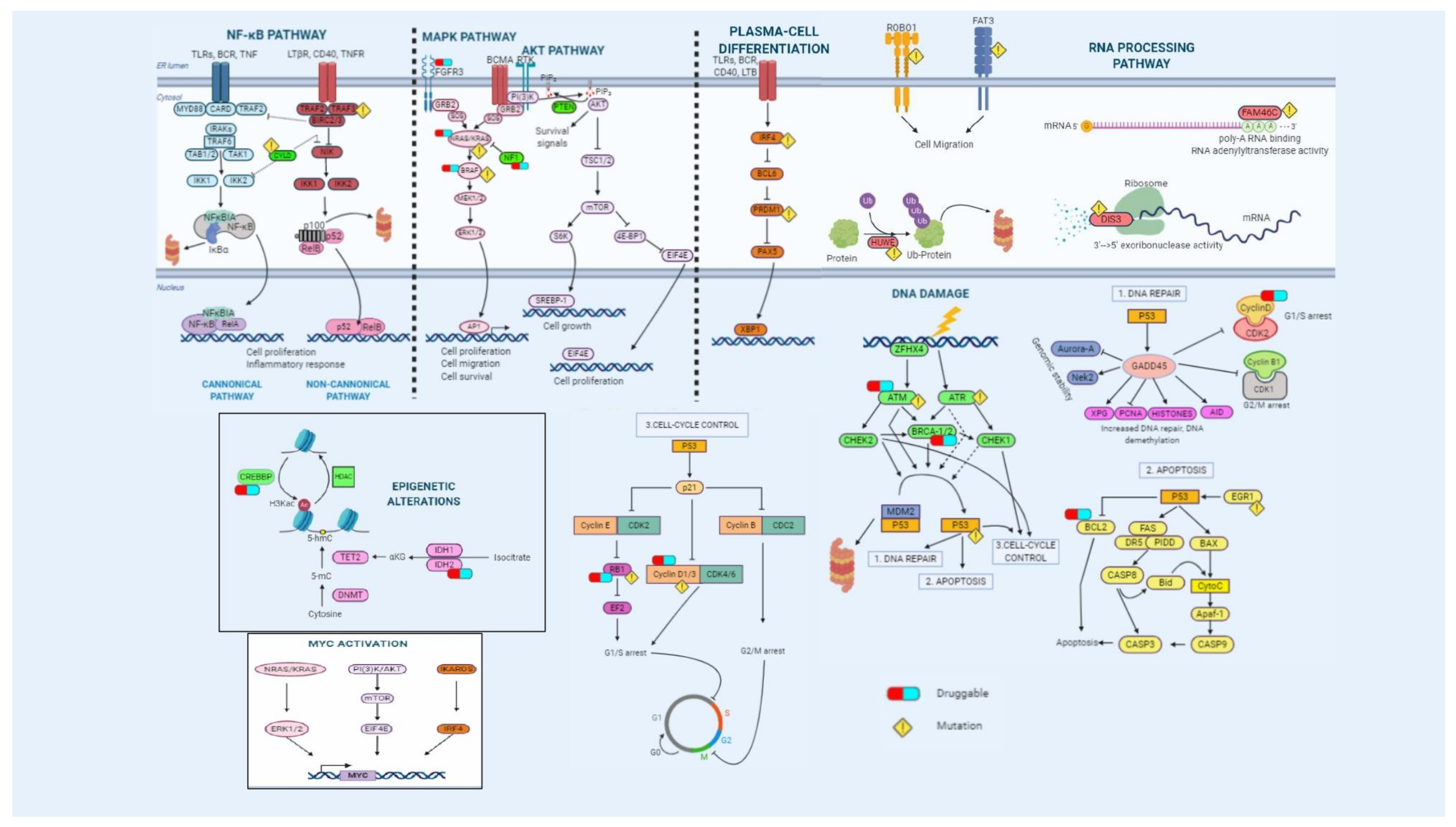
2.3. Mutations
2.3.1. Prognosis
2.3.2. Therapeutic Implications
3. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Pawlyn, C.; Davies, F.E. Toward personalized treatment in multiple myeloma based on molecular characteristics. Blood 2019, 133, 660–675. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, N.C.; García-Sanz, R.; San Miguel, J.F. Molecular biology of myeloma. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2007, 9, 618–624. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised international staging system for multiple myeloma: A report from International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef]
- Chesi, M.; Bergsagel, P.L. Advances in the pathogenesis and diagnosis of multiple myeloma. Int. J. Lab. Hematol. 2015, 37 (Suppl. 1), 108–114. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Melchor, L.; Pawlyn, C.; Kaiser, M.F.; et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 2015, 6, 6997. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.; Walker, B.; Wardell, C.; Johnson, D.; Kaiser, M.; Begum, D.; Dahir, N.; Ross, F.; Davies, F.; Gonzalez, D. Characterization of IgH breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pre-germinal center B Cells. Blood 2013, 121, 3413–3420. [Google Scholar] [CrossRef]
- Barwick, B.G.; Gupta, V.A.; Vertino, P.M.; Boise, L.H. Cell of origin and genetic alterations in the pathogenesis of multiple myeloma. Front Immunol. 2019, 10, 1121. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Mikhael, J.R.; Dingli, D.; Roy, V.; Reeder, C.B.; Buadi, F.K.; Hayman, S.R.; Dispenzieri, A.; Fonseca, R.; Sher, T.; Kyle, R.A.; et al. Management of newly diagnosed symptomatic multiple myeloma: Updated Mayo stratification of myeloma and risk-adapted therapy (mSMART) consensus guidelines 2013. Mayo Clin. Proc. 2013, 88, 360–376. [Google Scholar] [CrossRef]
- Kaufman, G.P.; Gertz, M.A.; Dispenzieri, A.; Lacy, M.Q.; Buadi, F.K.; Dingli, D.; Hayman, S.R.; Kapoor, P.; Lust, J.A.; Russell, S.; et al. Impact of cytogenetic classification on outcomes following early high-dose therapy in multiple myeloma. Leukemia 2016, 30, 633–639. [Google Scholar] [CrossRef]
- Lakshman, A.; Alhaj Moustafa, M.; Rajkumar, S.V.; Dispenzieri, A.; Gertz, M.A.; Buadi, F.K.; Lacy, M.Q.; Dingli, D.; Fonder, A.L.; Hayman, S.R.; et al. Natural history of t(11;14) multiple myeloma. Leukemia 2018, 32, 131–138. [Google Scholar] [CrossRef]
- Gran, C.; Uttervall, K.; Borg Bruchfeld, J.; Wallblom, A.; Alici, E.; Gahrton, G.; Nahi, H. Translocation (11;14) in newly diagnosed multiple myeloma, time to reclassify this standard risk chromosomal aberration? Eur. J. Haematol. 2019, ejh.13325. [Google Scholar] [CrossRef]
- Paner, A.; Patel, P.; Dhakal, B. The evolving role of translocation t(11;14) in the biology, prognosis, and management of multiple myeloma. Blood Rev. 2020, 41, 100643. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Cavo, M.; Cellini, C.; Terragna, C.; Zamagni, E.; Ruggeri, D.; Testoni, N.; Tosi, P.; De Vivo, A.; Amabile, M.; et al. Cyclin D1 overexpression is a favorable prognostic variable for newly diagnosed multiple myeloma patients treated with high-dose chemotherapy and single or double autologous transplantation. Blood 2003, 102, 1588–1594. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.J.; Rajkumar, S.V.; Therneau, T.M.; Singh, P.P.; Dispenzieri, A.; Kumar, S.K. Relationship between initial clinical presentation and the molecular cytogenetic classification of myeloma. Leukemia 2014, 28, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vij, R.; Pegourie, B.; Benboubker, L.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist venetoclax in multiple myeloma models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef]
- Moreau, P.; Chanan-Khan, A.; Roberts, A.W.; Agarwal, A.B.; Facon, T.; Kumar, S.; Touzeau, C.; Punnoose, E.A.; Cordero, J.; Munasinghe, W.; et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood 2017, 130, 2392–2400. [Google Scholar] [CrossRef]
- Ghobrial, I. BELLINI: A renaissance for an era of precision therapy in multiple myeloma. Lancet Oncol. 2020, 21, 1547–1549. [Google Scholar] [CrossRef]
- Kumar, S.K.; Harrison, S.J.; Cavo, M.; de la Rubia, J.; Popat, R.; Gasparetto, C.; Hungria, V.; Salwender, H.; Suzuki, K.; Kim, I.; et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020, 21, 1630–1642. [Google Scholar] [CrossRef]
- Venkata, J.K.; An, N.; Stuart, R.; Costa, L.J.; Cai, H.; Coker, W.; Song, J.H.; Gibbs, K.; Matson, T.; Garrett-Mayer, E.; et al. Inhibition of sphingosine kinase 2 downregulates the expression of c-myc and Mcl-1 and induces apoptosis in multiple myeloma. Blood 2014, 124, 1915–1925. [Google Scholar] [CrossRef]
- Sundaramoorthy, P.; Gasparetto, C.; Kang, Y. The combination of a sphingosine kinase 2 inhibitor (ABC294640) and a Bcl-2 inhibitor (ABT-199) displays synergistic anti-myeloma effects in myeloma cells without a t(11;14) translocation. Cancer Med. 2018, 7, 3257–3268. [Google Scholar] [CrossRef] [PubMed]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.T.; Huang, C.; Panaroni, C.; Mukaihara, K.; Fulzele, K.; Soucy, R.; Thorburn, C.; Cidado, J.; Drew, L.; Chattopadhyay, S.; et al. BCL2 blockade overcomes MCL1 resistance in multiple myeloma. Leukemia 2019, 33, 2098–2102. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Hulin, C.; Campion, L.; Rodon, P.; Marit, G.; Attal, M.; Royer, B.; Dib, M.; Voillat, L.; Bouscary, D.; et al. Chromosomal abnormalities are major prognostic factors in elderly patients with multiple myeloma: The Intergroupe Francophone Du Myélome experience. J. Clin. Oncol. 2013, 31, 2806–2809. [Google Scholar] [CrossRef]
- Pawlyn, C.; Morgan, G.J. Evolutionary biology of high-risk multiple myeloma. Nat. Rev. Cancer 2017, 17, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Brito, J.L.R.; Walker, B.; Jenner, M.; Dickens, N.J.; Brown, N.J.M.; Ross, F.M.; Avramidou, A.; Irving, J.A.E.; Gonzalez, D.; Davies, F.E.; et al. MMSET deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica 2009, 94, 78–86. [Google Scholar] [CrossRef]
- Misiewicz-Krzeminska, I.; Sarasquete, M.E.; Vicente-Dueñas, C.; Krzeminski, P.; Wiktorska, K.; Corchete, L.A.; Quwaider, D.; Rojas, E.A.; Corral, R.; Martín, A.A.; et al. Post-transcriptional modifications contribute to the upregulation of cyclin D2 in multiple myeloma. Clin. Cancer Res. 2016, 22, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Bergsagel, P.L.; Kuehl, W.M.; Zhan, F.; Sawyer, J.; Barlogie, B.; Shaughnessy, J., Jr. Cyclin D dysregulation: An early and unifying pathogenic event in multiple myeloma. Blood 2005, 106, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; Van Ness, B.; Chesi, M.; Minvielle, S.; et al. International Myeloma Working Group molecular classification of multiple myeloma: Spotlight review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Durie, B.G.M.; Cavo, M.; Attal, M.; Gutierrez, N.; Haessler, J.; Goldschmidt, H.; Hajek, R.; Lee, J.H.; Sezer, O.; et al. Combining fluorescent in situ hybridization data with ISS staging improves risk assessment in myeloma: An International Myeloma Working Group collaborative project. Leukemia 2013, 27, 711–717. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Leleu, X.; Roussel, M.; Moreau, P.; Guerin-Charbonnel, C.; Caillot, D.; Marit, G.; Benboubker, L.; Voillat, L.; Mathiot, C.; et al. Bortezomib plus dexamethasone induction improves outcome of patients with t(4;14) myeloma but not outcome of patients with del(17p). J. Clin. Oncol. 2010, 28, 4630–4634. [Google Scholar] [CrossRef] [PubMed]
- Tacchetti, P.; Pantani, L.; Patriarca, F.; Petrucci, M.T.; Zamagni, E.; Dozza, L.; Galli, M.; Di Raimondo, F.; Crippa, C.; Boccadoro, M.; et al. Bortezomib, thalidomide, and dexamethasone followed by double autologous haematopoietic stem-cell transplantation for newly diagnosed multiple myeloma (GIMEMA-MMY-3006): Long-term follow-up analysis of a randomised phase 3, open-label study. Lancet Haematol. 2020, 7, e861–e873. [Google Scholar] [CrossRef]
- Cavo, M.; Tacchetti, P.; Patriarca, F.; Petrucci, M.T.; Pantani, L.; Galli, M.; Di Raimondo, F.; Crippa, C.; Zamagni, E.; Palumbo, A.; et al. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: A randomised phase 3 study. Lancet Lond. Engl. 2010, 376, 2075–2085. [Google Scholar] [CrossRef]
- Sonneveld, P.; Schmidt-Wolf, I.G.H.; van der Holt, B.; El Jarari, L.; Bertsch, U.; Salwender, H.; Zweegman, S.; Vellenga, E.; Broyl, A.; Blau, I.W.; et al. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: Results of the randomized phase III HOVON-65/GMMG-HD4 trial. J. Clin. Oncol. 2012, 30, 2946–2955. [Google Scholar] [CrossRef]
- Neben, K.; Lokhorst, H.M.; Jauch, A.; Bertsch, U.; Hielscher, T.; van der Holt, B.; Salwender, H.; Blau, I.W.; Weisel, K.; Pfreundschuh, M.; et al. Administration of bortezomib before and after autologous stem cell transplantation improves outcome in multiple myeloma patients with deletion 17p. Blood 2012, 119, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Reece, D.; Song, K.W.; Fu, T.; Roland, B.; Chang, H.; Horsman, D.E.; Mansoor, A.; Chen, C.; Masih-Khan, E.; Trieu, Y.; et al. Influence of cytogenetics in patients with relapsed or refractory multiple myeloma treated with lenalidomide plus dexamethasone: Adverse effect of deletion 17p13. Blood 2009, 114, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Soulier, J.; Fermand, J.-P.; Yakoub-Agha, I.; Attal, M.; Hulin, C.; Garderet, L.; Belhadj, K.; Dorvaux, V.; Minvielle, S.; et al. Impact of high-risk cytogenetics and prior therapy on outcomes in patients with advanced relapsed or refractory multiple myeloma treated with lenalidomide plus dexaméthasone. Leukemia 2010, 24, 623–628. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Weisel, K.C.; Song, K.W.; Delforge, M.; Karlin, L.; Goldschmidt, H.; Moreau, P.; Banos, A.; Oriol, A.; Garderet, L.; et al. Cytogenetics and long-term survival of patients with refractory or relapsed and refractory multiple myeloma treated with pomalidomide and low-dose dexamethasone. Haematologica 2015, 100, 1327–1333. [Google Scholar] [CrossRef]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.-V.; Magen, H.; et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Jakubowiak, A.J.; Siegel, D.S.; Martin, T.; Wang, M.; Vij, R.; Lonial, S.; Trudel, S.; Kukreti, V.; Bahlis, N.; Alsina, M.; et al. Treatment outcomes in patients with relapsed and refractory multiple myeloma and high-risk cytogenetics receiving single-agent carfilzomib in the PX-171-003-A1 study. Leukemia 2013, 27, 2351–2356. [Google Scholar] [CrossRef]
- Hadari, Y.; Schlessinger, J. FGFR3-targeted MAb therapy for bladder cancer and multiple myeloma. J. Clin. Invest. 2009, 119, 1077–1079. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kamath, A.V.; Lu, D.; Gupta, P.; Jin, D.; Xin, Y.; Brady, A.; Stephan, J.P.; Li, H.; Tien, J.; Qing, J.; et al. Preclinical pharmacokinetics of MFGR1877A, a human monoclonal antibody to FGFR3, and prediction of its efficacious clinical dose for the treatment of t(4;14)-positive multiple myeloma. Cancer Chemother. Pharmacol. 2012, 69, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Trudel, S.; Stewart, A.K.; Rom, E.; Wei, E.; Zhi, H.L.; Kotzer, S.; Chumakov, I.; Singer, Y.; Chang, H.; Liang, S.B.; et al. The inhibitory anti-FGFR3 antibody, PRO-001, is cytotoxic to t(4;14) multiple myeloma Cells. Blood 2006, 107, 4039–4046. [Google Scholar] [CrossRef]
- Kalff, A.; Spencer, A. The t(4;14) translocation and FGFR3 overexpression in multiple myeloma: Prognostic implications and current clinical strategies. Blood Cancer J. 2012, 2, e89-8. [Google Scholar] [CrossRef]
- Scheid, C.; Reece, D.; Beksac, M.; Spencer, A.; Callander, N.; Sonneveld, P.; Kalimi, G.; Cai, C.; Shi, M.; Scott, J.W.; et al. Phase 2 study of dovitinib in patients with relapsed or refractory multiple myeloma with or without t(4;14) translocation. Eur. J. Haematol. 2015, 95, 316–324. [Google Scholar] [CrossRef]
- Majumder, M.M.; Silvennoinen, R.; Anttila, P.; Tamborero, D.; Eldfors, S.; Yadav, B.; Karjalainen, R.; Kuusanmäki, H.; Lievonen, J.; Parsons, A.; et al. Identification of precision treatment strategies for relapsed/refractory multiple myeloma by functional drug sensitivity testing. Oncotarget 2017, 8, 56338–56350. [Google Scholar] [CrossRef]
- Xie, Z.; Bi, C.; Chooi, J.Y.; Chan, Z.L.; Mustafa, N.; Chng, W.J. MMSET regulates expression of IRF4 in t(4;14) myeloma and its silencing potentiates the effect of bortezomib. Leukemia 2015, 29, 2347–2354. [Google Scholar] [CrossRef]
- Shah, M.Y.; Martinez-Garcia, E.; Phillip, J.M.; Chambliss, A.B.; Popovic, R.; Ezponda, T.; Small, E.C.; Will, C.; Phillip, M.P.; Neri, P.; et al. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene 2016, 35, 5905–5915. [Google Scholar] [CrossRef]
- Tonon, G.; Anderson, K.C. Multiple myeloma. Mol. Basis Cancer 2015, 455–466.e4. [Google Scholar] [CrossRef]
- Lauring, J.; Abukhdeir, A.M.; Konishi, H.; Garay, J.P.; Gustin, J.P.; Wang, Q.; Arceci, R.J.; Matsui, W.; Park, B.H. The multiple myeloma associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood 2008, 111, 856–864. [Google Scholar] [CrossRef] [PubMed]
- International myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: A report of the International Myeloma Working Group. Br. J. Haematol. 2003, 121, 749–757. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: 2018 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2018, 93, 1091–1110. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.; Blood, E.; Rue, M.; Harrington, D.; Oken, M.M.; Kyle, R.A.; Dewald, G.W.; Van Ness, B.; Van Wier, S.A.; Henderson, K.J.; et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood 2003, 101, 4569–4575. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Malard, F.; Campion, L.; Magrangeas, F.; Sebban, C.; Lioure, B.; Decaux, O.; Lamy, T.; Legros, L.; Fuzibet, J.G.; et al. Translocation t(14;16) and multiple myeloma: Is it really an independent prognostic factor? Blood 2011, 117, 2009–2011. [Google Scholar] [CrossRef]
- Tai, Y.T.; Fulciniti, M.; Hideshima, T.; Song, W.; Leiba, M.; Li, X.F.; Rumizen, M.; Burger, P.; Morrison, A.; Podar, K.; et al. Targeting MEK induces myeloma-cell cytotoxicity and inhibits osteoclastogenesis. Blood 2007, 110, 1656–1663. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Hernandez, L.; Davis, R.E.; Zingone, A.; Lamy, L.; Lam, L.T.; Hurt, E.M.; Shaffer, A.L.; Kuehl, W.M.; Staudt, L.M. A mechanistic rationale for MEK inhibitor therapy in myeloma based on blockade of MAF oncogene expression. Blood 2011, 117, 2396–2404. [Google Scholar] [CrossRef]
- Suzuki, R.; Kikuchi, S.; Harada, T.; Mimura, N.; Minami, J.; Ohguchi, H.; Yoshida, Y.; Sagawa, M.; Gorgun, G.; Cirstea, D.; et al. Combination of a selective HSP90α/β inhibitor and a RAS-RAF-MEK-ERK signaling pathway inhibitor triggers synergistic cytotoxicity in multiple myeloma cells. PLoS ONE 2015, 10, 1–20. [Google Scholar] [CrossRef]
- Ragon, B.K.; Odenike, O.; Baer, M.R.; Stock, W.; Borthakur, G.; Patel, K.; Han, L.; Chen, H.; Ma, H.; Joseph, L.; et al. Oral MEK 1/2 inhibitor trametinib in combination with AKT inhibitor GSK2141795 in patients with acute myeloid leukemia with RAS mutations: A phase II study. Clin. Lymphoma Myeloma Leuk. 2019, 19, 431–440.e13. [Google Scholar] [CrossRef]
- Qiang, Y.-W.; Ye, S.; Chen, Y.; Buros, A.F.; Edmonson, R.; van Rhee, F.; Barlogie, B.; Epstein, J.; Morgan, G.J.; Davies, F.E. MAF protein mediates innate resistance to proteasome inhibition therapy in multiple myeloma. Blood 2016, 128, 2919–2930. [Google Scholar] [CrossRef]
- Qiang, Y.-W.; Ye, S.; Huang, Y.; Chen, Y.; Van Rhee, F.; Epstein, J.; Walker, B.A.; Morgan, G.J.; Davies, F.E. MAFb protein confers intrinsic resistance to proteasome inhibitors in multiple myeloma. BMC Cancer 2018, 18, 724. [Google Scholar] [CrossRef]
- Herath, N.I.; Rocques, N.; Garancher, A.; Eychène, A.; Pouponnot, C. GSK3-mediated MAF phosphorylation in multiple myeloma as a potential therapeutic target. Blood Cancer J. 2014, 4, e175. [Google Scholar] [CrossRef] [PubMed]
- Misund, K.; Keane, N.; Stein, C.K.; Asmann, Y.W.; Day, G.; Welsh, S.; Van Wier, S.A.; Riggs, D.L.; Ahmann, G.; Chesi, M.; et al. MYC dysregulation in the progression of multiple myeloma. Leukemia 2019, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Affer, M.; Chesi, M.; Chen, W.-D.G.; Keats, J.J.; Demchenko, Y.N.; Roschke, A.V.; Van Wier, S.; Fonseca, R.; Bergsagel, P.L.; Kuehl, W.M. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia 2014, 28, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, W.M.; Bergsagel, P.L. MYC addiction: A potential therapeutic target in MM. Blood 2012, 120, 2351–2352. [Google Scholar] [CrossRef]
- Cottini, F.; Hideshima, T.; Suzuki, R.; Tai, Y.-T.; Bianchini, G.; Richardson, P.G.; Anderson, K.C.; Tonon, G. Synthetic lethal approaches exploiting DNA damage in aggressive myeloma. Cancer Discov. 2015, 5, 972–987. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting MYC in multiple myeloma. Leukemia 2018, 32, 1295–1306. [Google Scholar] [CrossRef]
- Abdallah, N.; Baughn, L.B.; Rajkumar, S.V.; Kapoor, P.; Gertz, M.A.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; et al. Implications of MYC rearrangements in newly diagnosed multiple myeloma. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Chng, W.J.; Huang, G.F.; Chung, T.H.; Ng, S.B.; Gonzalez-Paz, N.; Troska-Price, T.; Mulligan, G.; Chesi, M.; Bergsagel, P.L.; Fonseca, R. Clinical and biological implications of MYC activation: A common difference between MGUS and newly diagnosed multiple myeloma. Leukemia 2011, 25, 1026–1035. [Google Scholar] [CrossRef]
- Brioli, A.; Melchor, L.; Walker, B.A.; Davies, F.E.; Morgan, G.J. Biology and treatment of myeloma. Clin. Lymphoma Myeloma Leuk. 2014, 14, S65–S70. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Attal, M.; Moreau, P.; Charbonnel, C.; Garban, F.; Hulin, C.; Leyvraz, S.; Michallet, M.; Yakoub-Agha, I.; Garderet, L.; et al. Genetic abnormalities and survival in multiple myeloma: The experience of the Intergroupe Francophone Du Myélome. Blood 2007, 109, 3489–3495. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.P.; Brioli, A.; Boyle, E.; Kaiser, M.F.; Begum, D.B.; Dahir, N.B.; Johnson, D.C.; Ross, F.M.; Davies, F.E.; et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J. 2014, 4, e191-7. [Google Scholar] [CrossRef]
- Glitza, I.C.; Lu, G.; Shah, R.; Bashir, Q.; Shah, N.; Champlin, R.E.; Shah, J.; Orlowski, R.Z.; Qazilbash, M.H. Chromosome 8q24.1/c-MYC abnormality: A marker for high-risk myeloma. Leuk. Lymphoma 2015, 56, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Kenneth, C. Anderson Progress and paradigms in multiple myeloma. Clin. Cancer Res. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Stubbs, M.C.; Burn, T.C.; Sparks, R.; Maduskuie, T.; Diamond, S.; Rupar, M.; Wen, X.; Volgina, A.; Zolotarjova, N.; Waeltz, P.; et al. The novel bromodomain and extraterminal domain inhibitor INCB054329 induces vulnerabilities in myeloma cells that inform rational combination strategies. Clin. Cancer Res. 2019, 25, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.T.; Ramachandran, J.; Yee, A.J.; Eda, H.; Santo, L.; Panaroni, C.; Mertz, J.A.; Sims Iii, R.J.; Cooper, M.R.; Raje, N. Preclinical activity of CPI-0610, a novel small-molecule bromodomain and extra-terminal protein inhibitor in the therapy of multiple myeloma. Leukemia 2017, 31, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.L.; Damnernsawad, A.; Shyamsunder, P.; Chng, W.J.; Han, B.C.; Xu, L.; Pan, J.; Pravin, D.P.; Alkan, S.; Tyner, J.W.; et al. Proteolysis targeting chimeric molecules as therapy for multiple myeloma: Efficacy, biomarker and drug combinations. Haematologica 2019, 104, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Buettner, R.; Morales, C.; Caserta, E.; Troadec, E.; Gunes, E.G.; Viola, D.; Khalife, J.; Li, H.; Keats, J.J.; Christofferson, A.; et al. Leflunomide regulates c-Myc expression in myeloma cells through PIM targeting. Blood Adv. 2019, 3, 1027–1032. [Google Scholar] [CrossRef]
- Caracciolo, D.; Scionti, F.; Juli, G.; Altomare, E.; Golino, G.; Todoerti, K.; Grillone, K.; Riillo, C.; Arbitrio, M.; Iannone, M.; et al. Exploiting MYC-induced PARPness to target genomic instability in multiple myeloma. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Ketterling, R.P.; Fonseca, R. Analysis of genetic abnormalities provides insights into genetic evolution of hyperdiploid myeloma. Genes Chromosomes Cancer 2006, 45, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Van Wier, S.A.; Ahmann, G.J.; Winkler, J.M.; Jalal, S.M.; Bergsagel, P.L.; Chesi, M.; Trendle, M.C.; Oken, M.M.; Blood, E.; et al. A validated FISH trisomy index demonstrates the hyperdiploid and nonhyperdiploid dichotomy in MGUS. Blood 2005, 106, 2156–2161. [Google Scholar] [CrossRef]
- Hoctor, V.T.; Campbell, L.J. Hyperhaploid plasma cell myeloma. Cancer Genet 2012, 205, 414–418. [Google Scholar] [CrossRef]
- Sawyer, J.R.; Tian, E.; Shaughnessy, J.D.; Epstein, J.; Swanson, C.M.; Stangeby, C.; Hale, C.L.; Parr, L.; Lynn, M.; Sammartino, G.; et al. Hyperhaploidy is a novel high-risk cytogenetic subgroup in multiple myeloma. Leukemia 2017, 31, 637–644. [Google Scholar] [CrossRef]
- Magrangeas, F.; Avet-Loiseau, H.; Munshi, N.C.; Minvielle, S. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood 2011, 118, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Santana-Dávila, R.; Van Wier, S.A.; Ahmann, G.J.; Jalal, S.M.; Bergsagel, P.L.; Chesi, M.; Trendle, M.C.; Jacobus, S.; Blood, E.; et al. Prognostic factors for hyperdiploid-myeloma: Effects of chromosome 13 deletions and IgH translocations. Leukemia 2006, 20, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Smadja, N.V.; Bastard, C.; Brigaudeau, C.; Leroux, D.; Fruchart, C. Hypodiploidy is a major prognostic factor in multiple myeloma. Blood 2001, 98. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Rajkumar, S.V.; Kapoor, P.; Ketterling, R.P.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.; Dingli, D.; Hayman, S.R.; Dispenzieri, A.; et al. Impact of FISH abnormalities on response to lenalidomide in patients with multiple myeloma. Blood 2013, 122, 3210. [Google Scholar] [CrossRef]
- Mei, J.; Zhai, Y.; Li, H.; Li, F.; Zhou, X.; Song, P.; Zhao, Q.; Yu, Y.; An, Z.; Wang, L. Prognostic impact of hyperdiploidy in multiple myeloma patients with high-risk cytogenetics: A pilot study in China. J. Cancer Res. Clin. Oncol. 2018, 144, 2263–2273. [Google Scholar] [CrossRef]
- Chretien, M.-L.; Corre, J.; Lauwers-Cances, V.; Magrangeas, F.; Cleynen, A.; Yon, E.; Hulin, C.; Leleu, X.; Orsini-Piocelle, F.; Blade, J.-S.; et al. Understanding the role of hyperdiploidy in myeloma prognosis: Which trisomies really matter? Blood 2015, 126, 2713–2719. [Google Scholar] [CrossRef]
- Walker, B.A.; Leone, P.E.; Chiecchio, L.; Dickens, N.J.; Jenner, M.W.; Boyd, K.D.; Johnson, D.C.; Gonzalez, D.; Dagrada, G.P.; Protheroe, R.K.M.; et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood 2010, 116, e56–e65. [Google Scholar] [CrossRef]
- Fassas, A.B.-T.; Spencer, T.; Sawyer, J.; Zangari, M.; Lee, C.-K.; Anaissie, E.; Muwalla, F.; Morris, C.; Barlogie, B.; Tricot, G. Both hypodiploidy and deletion of chromosome 13 independently confer poor prognosis in multiple myeloma. Br. J. Haematol. 2002, 118, 1041–1047. [Google Scholar] [CrossRef]
- Kaur, G.; Gupta, R.; Mathur, N.; Rani, L.; Kumar, L.; Sharma, A.; Singh, V.; Gupta, A.; Sharma, O.D. Clinical impact of chromothriptic complex chromosomal rearrangements in newly diagnosed multiple myeloma. Leuk. Res. 2019, 76, 58–64. [Google Scholar] [CrossRef]
- Boyd, K.D.; Ross, F.M.; Walker, B.A.; Wardell, C.P.; Tapper, W.J.; Chiecchio, L.; Dagrada, G.; Konn, Z.J.; Gregory, W.M.; Jackson, G.H.; et al. Mapping of chromosome 1p deletions in myeloma identifies FAM46C at 1p12 and CDKN2C at 1p32.3 as being genes in regions associated with adverse survival. Clin. Cancer Res. 2011, 17, 7776–7784. [Google Scholar] [CrossRef]
- Qazilbash, M.H.; Saliba, R.M.; Ahmed, B.; Parikh, G.; Mendoza, F.; Ashraf, N.; Hosing, C.; Flosser, T.; Weber, D.M.; Wang, M.; et al. Deletion of the short arm of chromosome 1 (Del 1p) is a strong predictor of poor outcome in myeloma patients undergoing an autotransplant. Biol. Blood Marrow Transpl. 2007, 13, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- López-Corral, L.; Sarasquete, M.E.; Beà, S.; García-Sanz, R.; Mateos, M.V.; Corchete, L.A.; Sayagués, J.M.; García, E.M.; Bladé, J.; Oriol, A.; et al. SNP-based mapping arrays reveal high genomic complexity in monoclonal gammopathies, from MGUS to myeloma status. Leukemia 2012, 26, 2521–2529. [Google Scholar] [CrossRef]
- Li, F.; Hu, L.; Xu, Y.; Li, Z.; Yi, S.; Gu, Z.; Li, C.; Hao, M.; Ru, K.; Zhan, F.; et al. Identification of characteristic and prognostic values of chromosome 1p abnormality by multi-gene fluorescence in situ hybridization in multiple myeloma. Leukemia 2016, 30, 1197–1201. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.E.; Walker, B.A.; Jenner, M.W.; Chiecchio, L.; Dagrada, G.; Protheroe, R.K.M.; Johnson, D.C.; Dickens, N.J.; Brito, J.L.; Else, M.; et al. Deletions of CDKN2C in multiple myeloma: Biological and clinical implications. Clin. Cancer Res. 2008, 14, 6033–6041. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.K.; Chen, Y.; Lian, L.; Ha, P.C.; Petersen, K.; Laity, K.; Carillo, A.; Emerson, M.; Heichman, K.; Gupte, J.; et al. Genomic structure, chromosomal location, and mutation analysis of the human CDC14A gene. Genomics 1999, 59, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Hebraud, B.; Leleu, X.; Lauwers-Cances, V.; Roussel, M.; Caillot, D.; Marit, G.; Karlin, L.; Hulin, C.; Gentil, C.; Guilhot, F.; et al. Deletion of the 1p32 region is a major independent prognostic factor in young patients with myeloma: The IFM Experience on 1195 patients. Leukemia 2014, 28, 675–679. [Google Scholar] [CrossRef]
- Chang, H.; Ning, Y.; Qi, X.; Yeung, J.; Xu, W. Chromosome 1p21 deletion is a novel prognostic marker in patients with multiple myeloma. Br. J. Haematol. 2007, 139, 51–54. [Google Scholar] [CrossRef]
- Chang, H.; Jiang, A.; Qi, C.; Trieu, Y.; Chen, C.; Reece, D. Impact of genomic aberrations including chromosome 1 abnormalities on the outcome of patients with relapsed or refractory multiple myeloma treated with lenalidomide and dexamethasone. Leuk. Lymphoma 2010, 51, 2084–2091. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Shi, C.-X.; Bruins, L.A.; Jedlowski, P.; Wang, X.; Kortüm, K.M.; Luo, M.; Ahmann, J.M.; Braggio, E.; Stewart, A.K. Loss of FAM46C promotes cell survival in myeloma. Cancer Res. 2017, 77, 4317–4327. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, double-hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019, 33, 159–170. [Google Scholar] [CrossRef]
- Herrero, A.B.; Quwaider, D.; Corchete, L.A.; Mateos, M.V.; García-Sanz, R.; Gutiérrez, N.C. FAM46C controls antibody production by the polyadenylation of immunoglobulin mRNAs and inhibits cell migration in multiple myeloma. J. Cell Mol. Med. 2020, 24, 4171–4182. [Google Scholar] [CrossRef] [PubMed]
- An, G.; Xu, Y.; Shi, L.; Shizhen, Z.; Deng, S.; Xie, Z.; Sui, W.; Zhan, F.; Qiu, L. Chromosome 1q21 gains confer inferior outcomes in multiple myeloma treated with bortezomib but copy number variation and percentage of plasma cells involved have no additional prognostic value. Haematologica 2014, 99, 353–359. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Attal, M.; Campion, L.; Caillot, D.; Hulin, C.; Marit, G.; Stoppa, A.-M.; Voillat, L.; Wetterwald, M.; Pegourie, B.; et al. Long-term analysis of the IFM 99 trials for myeloma: Cytogenetic abnormalities [t(4;14), del(17p), 1q gains] play a major role in defining long-term survival. J. Clin. Oncol. 2012, 30, 1949–1952. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational spectrum, copy number changes, and outcome: Results of a sequencing study of patients with newly diagnosed myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef] [PubMed]
- Neben, K.; Jauch, A.; Hielscher, T.; Hillengass, J.; Lehners, N.; Seckinger, A.; Granzow, M.; Raab, M.S.; Ho, A.D.; Goldschmidt, H.; et al. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J. Clin. Oncol. 2013, 31, 4325–4332. [Google Scholar] [CrossRef]
- Chang, H.; Yeung, J.; Xu, W.; Ning, Y.; Patterson, B. Significant increase of CKS1B amplification from monoclonal gammopathy of undetermined significance to multiple myeloma and plasma cell leukaemia as demonstrated by interphase fluorescence in situ hybridisation. Br. J. Haematol. 2006, 134, 613–615. [Google Scholar] [CrossRef]
- Hanamura, I.; Stewart, J.P.; Huang, Y.; Zhan, F.; Santra, M.; Sawyer, J.R.; Hollmig, K.; Zangarri, M.; Pineda-Roman, M.; van Rhee, F.; et al. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: Incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantat. Blood 2006, 108, 1724–1732. [Google Scholar] [CrossRef]
- Fonseca, R.; Van Wier, S.A.; Chng, W.J.; Ketterling, R.; Lacy, M.Q.; Dispenzieri, A.; Bergsagel, P.L.; Rajkumar, S.V.; Greipp, P.R.; Litzow, M.R.; et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia 2006, 20, 2034–2040. [Google Scholar] [CrossRef]
- Zhan, F.; Colla, S.; Wu, X.; Chen, B.; Stewart, J.P.; Kuehl, W.M.; Barlogie, B.; Shaughnessy, J.D. CKS1B, Overexpressed in aggressive disease, regulates multiple myeloma growth and survival through SKP2- and p27Kip1-dependent and -independent mechanisms. Blood 2007, 109, 4995–5001. [Google Scholar] [CrossRef]
- Shi, L.; Wang, S.; Zangari, M.; Xu, H.; Cao, T.M.; Xu, C.; Wu, Y.; Xiao, F.; Liu, Y.; Yang, Y.; et al. Over-expression of CKS1B activates both MEK/ERK and JAK/STAT3 signaling pathways and promotes myeloma cell drug-resistance. Oncotarget 2010, 1, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P.; Maimonis, P.; Bua, D.; Young, G.; Raje, N.; Mollick, J.; Chauhan, D.; Tai, Y.T.; Hideshima, T.; Shima, Y.; et al. Elevated soluble MUC1 levels and decreased anti-MUC1 antibody levels in patients with multiple myeloma. Blood 2000, 96, 3147–3153. [Google Scholar] [CrossRef]
- Legartova, S.; Krejci, J.; Harnicarova, A.; Hajek, R.; Kozubek, S.; Bartova, E. Nuclear topography of the 1q21 genomic region and Mcl-1 protein levels associated with pathophysiology of multiple myeloma. Neoplasma 2009, 56, 404–413. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sawyer, J.R.; Tricot, G.; Lukacs, J.L.; Binz, R.L.; Tian, E.; Barlogie, B.; Shaughnessy, J. Genomic instability in multiple myeloma: Evidence for jumping segmental duplications of chromosome arm 1q. Genes Chromosomes Cancer 2005, 42, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, J.D.; Qu, P.; Usmani, S.; Heuck, C.J.; Zhang, Q.; Zhou, Y.; Tian, E.; Hanamura, I.; van Rhee, F.; Anaissie, E.; et al. Pharmacogenomics of bortezomib test-dosing identifies hyperexpression of proteasome genes, especially PSMD4, as novel high-risk feature in myeloma treated with total therapy 3. Blood 2011, 118, 3512–3524. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.; Otsuki, T.; Hirasawa, A.; Imoto, I.; Matsuo, Y.; Shimizu, S.; Taniwaki, M.; Inazawa, J. Overexpression of PDZK1 within the 1q12-Q22 amplicon is likely to be associated with drug-resistance phenotype in multiple myeloma. Am. J. Pathol. 2004, 165, 71–81. [Google Scholar] [CrossRef]
- Schmidt, T.M.; Barwick, B.G.; Joseph, N.; Heffner, L.T.; Hofmeister, C.C.; Bernal, L.; Dhodapkar, M.V.; Gupta, V.A.; Jaye, D.L.; Wu, J.; et al. Gain of chromosome 1q is associated with early progression in multiple myeloma patients treated with lenalidomide, bortezomib, and dexamethasone. Blood Cancer J. 2019, 9, 94. [Google Scholar] [CrossRef] [PubMed]
- Shah, G.L.; Landau, H.; Londono, D.; Devlin, S.M.; Kosuri, S.; Lesokhin, A.M.; Lendvai, N.; Hassoun, H.; Chung, D.J.; Koehne, G.; et al. Gain of chromosome 1q portends worse prognosis in multiple myeloma despite novel agent-based induction regimens and autologous transplantation. Leuk. Lymphoma 2017, 58, 1823–1831. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.; Lu, G.; Srour, S.A.; Gaballa, S.; Lin, H.Y.; Baladandayuthapani, V.; Honhar, M.; Stich, M.; Das Shah, N.; Bashir, Q.; et al. Outcome of patients with multiple myeloma and CKS1B gene amplification after autologous hematopoietic stem cell transplantation. Biol. Blood Marrow Transpl. 2016, 22, 2159–2164. [Google Scholar] [CrossRef]
- Jackson, G.H.; Davies, F.E.; Pawlyn, C.; Cairns, D.A.; Striha, A.; Collett, C.; Hockaday, A.; Jones, J.R.; Kishore, B.; Garg, M.; et al. Lenalidomide maintenance versus observation for patients with newly diagnosed multiple myeloma (myeloma XI): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 57–73. [Google Scholar] [CrossRef]
- Walker, B.A.; Morgan, G.J. The genomic features associated with high-risk multiple myeloma. Oncotarget 2018, 9, 35478–35479. [Google Scholar] [CrossRef]
- Sherbenou, D.W.; Aftab, B.T.; Su, Y.; Behrens, C.R.; Wiita, A.; Logan, A.C.; Acosta-Alvear, D.; Hann, B.C.; Walter, P.; Shuman, M.A.; et al. Antibody-drug conjugate targeting CD46 eliminates multiple myeloma cells. J. Clin. Invest. 2016, 126, 4640–4653. [Google Scholar] [CrossRef]
- Slomp, A.; Moesbergen, L.M.; Gong, J.-N.; Cuenca, M.; von dem Borne, P.A.; Sonneveld, P.; Huang, D.C.S.; Minnema, M.C.; Peperzak, V. Multiple myeloma with 1q21 amplification is highly sensitive to MCL-1 targeting. Blood Adv. 2019, 3, 4202–4214. [Google Scholar] [CrossRef]
- Avet-Louseau, H.; Daviet, A.; Sauner, S.; Bataille, R.; Intergroupe Francophone du Myélome. Chromosome 13 abnormalities in multiple myeloma are mostly monosomy 13. Br. J. Haematol. 2000, 111, 1116–1117. [Google Scholar]
- Fonseca, R.; Oken, M.M.; Harrington, D.; Bailey, R.J.; Van Wier, S.A.; Henderson, K.J.; Kay, N.E.; Van Ness, B.; Greipp, P.R.; Dewald, G.W. Deletions of chromosome 13 in multiple myeloma identified by interphase FISH usually denote large deletions of the q arm or monosomy. Leukemia 2001, 15, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Zojer, N.; Königsberg, R.; Ackermann, J.; Fritz, E.; Dallinger, S.; Krömer, E.; Kaufmann, H.; Riedl, L.; Gisslinger, H.; Schreiber, S.; et al. Deletion of 13q14 remains an independent adverse prognostic variable in multiple myeloma despite its frequent detection by interphase fluorescence in situ hybridization. Blood 2000, 95, 1925–1930. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, J.; Tian, E.; Sawyer, J.; Bumm, K.; Landes, R.; Badros, A.; Morris, C.; Tricot, G.; Epstein, J.; Barlogie, B. High incidence of chromosome 13 deletion in multiple myeloma detected by multiprobe interphase FISH. Blood 2000, 96, 1505–1511. [Google Scholar] [CrossRef]
- Tricot, G.; Barlogie, B.; Jagannath, S.; Bracy, D.; Mattox, S.; Vesole, D.H.; Naucke, S.; Sawyer, J.R. Poor prognosis in multiple myeloma is associated only with partial or complete deletions of chromosome 13 or abnormalities involving 11q and not with other karyotype abnormalities. Blood 1995, 86, 4250–4256. [Google Scholar] [CrossRef] [PubMed]
- Kiyota, M.; Kobayashi, T.; Fuchida, S.; Yamamoto-Sugitani, M.; Ohshiro, M.; Shimura, Y.; Mizutani, S.; Nagoshi, H.; Sasaki, N.; Nakayama, R.; et al. Monosomy 13 in metaphase spreads is a predictor of poor long-term outcome after bortezomib plus dexamethasone treatment for relapsed/refractory multiple myeloma. Int. J. Hematol. 2012, 95, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, N.C.; Castellanos, M.V.; Martín, M.L.; Mateos, M.V.; Hernández, J.M.; Fernández, M.; Carrera, D.; Rosiñol, L.; Ribera, J.M.; Ojanguren, J.M.; et al. Prognostic and biological implications of genetic abnormalities in multiple myeloma undergoing autologous stem cell transplantation: t(4;14) is the most relevant adverse prognostic factor, whereas RB deletion as a unique abnormality is not associated with adverse prognosis. Leukemia 2007, 21, 143–150. [Google Scholar] [CrossRef]
- Binder, M.; Rajkumar, S.V.; Ketterling, R.P.; Greipp, P.T.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Hayman, S.R.; Hwa, Y.L.; et al. Prognostic implications of abnormalities of chromosome 13 and the presence of multiple cytogenetic high-risk abnormalities in newly diagnosed multiple myeloma. Blood Cancer J. 2017, 7, e600. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB promotes cancer immunity by inhibiting NF-ΚB activation and PD-L1 expression. Mol. Cell 2019, 73, 22–35.e6. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Qi, C.; Yi, Q.-L.; Reece, D.; Stewart, A.K. P53 gene deletion detected by fluorescence in situ hybridization is an adverse prognostic factor for patients with multiple myeloma following autologous stem cell transplantation. Blood 2005, 105, 358–360. [Google Scholar] [CrossRef]
- Hu, B.; Thall, P.; Milton, D.R.; Sasaki, K.; Bashir, Q.; Shah, N.; Patel, K.; Popat, U.; Hosing, C.; Nieto, Y.; et al. High-risk myeloma and minimal residual disease postautologous-HSCT predict worse outcomes. Leuk. Lymphoma 2019, 60, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Tiedemann, R.E.; Gonzalez-Paz, N.; Kyle, R.A.; Santana-Davila, R.; Price-Troska, T.; Van Wier, S.A.; Chng, W.J.; Ketterling, R.P.; Gertz, M.A.; Henderson, K.; et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia 2008, 22, 1044–1052. [Google Scholar] [CrossRef]
- Merz, M.; Hielscher, T.; Seckinger, A.; Hose, D.; Mai, E.K.; Raab, M.S.; Goldschmidt, H.; Jauch, A.; Hillengass, J. Baseline Characteristics, chromosomal alterations, and treatment affecting prognosis of deletion 17p in newly diagnosed myeloma. Am. J. Hematol. 2016, 91, E473–E477. [Google Scholar] [CrossRef]
- An, G.; Li, Z.; Tai, Y.-T.; Acharya, C.; Li, Q.; Qin, X.; Yi, S.; Xu, Y.; Feng, X.; Li, C.; et al. The impact of clone size on the prognostic value of chromosome aberrations by fluorescence in situ hybridization in multiple myeloma. Clin. Cancer Res. 2015, 21, 2148–2156. [Google Scholar] [CrossRef]
- Lakshman, A.; Painuly, U.; Rajkumar, S.V.; Ketterling, R.P.; Kapoor, P.; Greipp, P.T.; Gertz, M.A.; Buadi, F.K.; Lacy, M.Q.; Dingli, D.; et al. Natural history of multiple myeloma with de novo del(17p). Blood Cancer J. 2019, 9, 32. [Google Scholar] [CrossRef]
- Ross, F.M.; Avet-Loiseau, H.; Ameye, G.; Gutiérrez, N.C.; Liebisch, P.; O’Connor, S.; Dalva, K.; Fabris, S.; Testi, A.M.; Jarosova, M.; et al. Report from the European Myeloma Network on interphase FISH in multiple myeloma and related disorders. Haematologica 2012, 97, 1272–1277. [Google Scholar] [CrossRef]
- Herrero, A.B.; Rojas, E.A.; Misiewicz-Krzeminska, I.; Krzeminski, P.; Gutiérrez, N.C. Molecular mechanisms of P53 deregulation in cancer: An overview in multiple myeloma. Int. J. Mol. Sci. 2016, 17, 2003. [Google Scholar] [CrossRef]
- Lodé, L.; Eveillard, M.; Trichet, V.; Soussi, T.; Wuillème, S.; Richebourg, S.; Magrangeas, F.; Ifrah, N.; Campion, L.; Traullé, C.; et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica 2010, 95, 1973–1976. [Google Scholar] [CrossRef]
- Thakurta, A.; Ortiz, M.; Blecua, P.; Towfic, F.; Corre, J.; Serbina, N.V.; Flynt, E.; Yu, Z.; Yang, Z.; Palumbo, A.; et al. High subclonal fraction of 17p deletion is associated with poor prognosis in multiple myeloma. Blood 2019, 133, 1217–1221. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Mateos, M.-V.; Gutierrez, N.C.; Rajkumar, S.V.; San Miguel, J.F. Improving overall survival and overcoming adverse prognosis in the treatment of cytogenetically high-risk multiple myeloma. Blood 2013, 121, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, J.D.; Zhou, Y.; Haessler, J.; van Rhee, F.; Anaissie, E.; Nair, B.; Waheed, S.; Alsayed, Y.; Epstein, J.; Crowley, J.; et al. TP53 deletion is not an adverse feature in multiple myeloma treated with total therapy 3. Br. J. Haematol. 2009, 147, 347–351. [Google Scholar] [CrossRef]
- Chng, W.-J.; Goldschmidt, H.; Dimopoulos, M.A.; Moreau, P.; Joshua, D.; Palumbo, A.; Facon, T.; Ludwig, H.; Pour, L.; Niesvizky, R.; et al. Carfilzomib-dexamethasone vs. bortezomib-dexamethasone in relapsed or refractory multiple myeloma by cytogenetic risk in the phase 3 study ENDEAVOR. Leukemia 2017, 31, 1368–1374. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Fonseca, R.; Siegel, D.; Dimopoulos, M.A.; Špička, I.; Masszi, T.; Hájek, R.; Rosiñol, L.; Goranova-Marinova, V.; Mihaylov, G.; et al. Carfilzomib significantly improves the progression-free survival of high-risk patients in multiple myeloma. Blood 2016, 128, 1174–1180. [Google Scholar] [CrossRef]
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N. Engl. J. Med. 2016, 374, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Leleu, X.; Karlin, L.; Macro, M.; Hulin, C.; Garderet, L.; Roussel, M.; Arnulf, B.; Pegourie, B.; Kolb, B.; Stoppa, A.M.; et al. Pomalidomide plus low-dose dexamethasone in multiple myeloma with deletion 17p and/or translocation (4;14): IFM 2010-02 trial results. Blood 2015, 125, 1411–1417. [Google Scholar] [CrossRef]
- Mikhael, J.; Richter, J.; Vij, R.; Cole, C.; Zonder, J.; Kaufman, J.L.; Bensinger, W.; Dimopoulos, M.; Lendvai, N.; Hari, P.; et al. A dose-finding phase 2 study of single agent isatuximab (anti-CD38 MAb) in relapsed/refractory multiple myeloma. Leukemia 2020, 34, 3298–3309. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Dispenzieri, A.; Chim, C.-S.; Fonseca, R.; Goldschmidt, H.; Lentzsch, S.; Munshi, N.; Palumbo, A.; Miguel, J.S.; Sonneveld, P.; et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia 2014, 28, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, N.; Ashby, C.; Rasche, L.; Chavan, S.S.; Stein, C.; Stephens, O.W.; Tytarenko, R.; Bauer, M.A.; Meissner, T.; Deshpande, S.; et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016, 128, 1735–1744. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Han, C.; Wan, G.; Huang, X.; Ivan, C.; Jiang, D.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Rao, P.H.; et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 2015, 520, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Pahl, A.; Lutz, C.; Hechler, T. Amanitins and their development as a payload for antibody-drug conjugates. Drug Discov. Today Technol. 2018, 30, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Vazquez, V.; Ko, J.; Breunig, C.; Baumann, A.; Giesen, N.; Pálfi, A.; Müller, C.; Lutz, C.; Hechler, T.; Kulke, M.; et al. HDP-101, anti-BCMA antibody-drug conjugate, safely delivers amanitin to induce cell death in proliferating and resting multiple myeloma cells. Mol. Cancer Ther. 2020. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Xu, H.; Jiang, G.; der Jeught, K.; Fang, Y.; Zhou, Z.; Zhang, L.; Frieden, M.; Wang, L.; et al. Heterozygous deletion of chromosome 17p renders prostate cancer vulnerable to inhibition of RNA polymerase II. Nat. Commun. 2018, 9, 4394. [Google Scholar] [CrossRef]
- Moreau, P.; Cavo, M.; Sonneveld, P.; Rosinol, L.; Attal, M.; Pezzi, A.; Goldschmidt, H.; Lahuerta, J.J.; Marit, G.; Palumbo, A.; et al. Combination of international scoring system 3, high lactate dehydrogenase, and t(4;14) and/or del(17p) identifies patients with multiple myeloma (MM) treated with front-line autologous stem-cell transplantation at high risk of early MM progression-related death. J. Clin. Oncol. 2014, 32, 2173–2180. [Google Scholar] [CrossRef]
- Lancman, G.; Tremblay, D.; Barley, K.; Barlogie, B.; Cho, H.J.; Jagannath, S.; Madduri, D.; Moshier, E.; Parekh, S.; Chari, A. The effect of novel therapies in high-molecular-risk multiple myeloma. Clin. Adv. Hematol. Oncol. 2017, 15, 870–879. [Google Scholar]
- Avet-Loiseau, H.; Fonseca, R.; Siegel, D.; Dimopoulos, M.A.; Spicka, I.; Masszi, T.; Hájek, R.; Rosiñol, L.; Goranova-Marinova, V.; Mihaylov, G.; et al. Efficacy and safety of carfilzomib, lenalidomide, and dexamethasone vs. lenalidomide and dexamethasone in patients with relapsed multiple myeloma based on cytogenetic risk status: Subgroup analysis from the phase 3 study aspire (NCT01080391). Blood 2015, 126, 731. [Google Scholar] [CrossRef]
- Larocca, A.; Mina, R.; Offidani, M.; Liberati, A.M.; Ledda, A.; Patriarca, F.; Evangelista, A.; Spada, S.; Benevolo, G.; Oddolo, D.; et al. First-line therapy with either bortezomib-melphalan-prednisone or lenalidomide-dexamethasone followed by lenalidomide for transplant-ineligible multiple myeloma patients: A pooled analysis of two randomized trials. Haematologica 2019. [Google Scholar] [CrossRef]
- Shah, V.; Sherborne, A.L.; Walker, B.A.; Johnson, D.C.; Boyle, E.M.; Ellis, S.; Begum, D.B.; Proszek, P.Z.; Jones, J.R.; Pawlyn, C.; et al. Prediction of outcome in newly diagnosed myeloma: A meta-analysis of the molecular profiles of 1905 trial patients. Leukemia 2018, 32, 102–110. [Google Scholar] [CrossRef]
- Cook, G.; Royle, K.; O’Connor, S.; Cairns, D.A.; Ashcroft, A.J.; Williams, C.D.; Hockaday, A.; Cavenagh, J.D.; Snowden, J.A.; Ademokun, D.; et al. The impact of cytogenetics on duration of response and overall survival in patients with relapsed multiple myeloma (long-term follow-up results from BSBMT/UKMF myeloma X relapse [intensive]): A randomised, open-label, phase 3 trial. Br. J. Haematol. 2019, 185, 450–467. [Google Scholar] [CrossRef]
- Corre, J.; Cleynen, A.; Robiou du Pont, S.; Buisson, L.; Bolli, N.; Attal, M.; Munshi, N.; Avet-Loiseau, H. Multiple myeloma clonal evolution in homogeneously treated patients. Leukemia 2018, 32, 2636–2647. [Google Scholar] [CrossRef]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef]
- Jones, J.R.; Weinhold, N.; Ashby, C.; Walker, B.A.; Wardell, C.; Pawlyn, C.; Rasche, L.; Melchor, L.; Cairns, D.A.; Gregory, W.M.; et al. Clonal evolution in myeloma: The impact of maintenance lenalidomide and depth of response on the genetics and sub-clonal structure of relapsed disease in uniformly treated newly diagnosed patients. Haematologica 2019, 104, 1440–1450. [Google Scholar] [CrossRef]
- Lionetti, M.; Neri, A. utilizing next-generation sequencing in the management of multiple myeloma. Expert. Rev. Mol. Diagn. 2017, 17, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Rajkumar, S.V. The multiple myelomas - current concepts in cytogenetic classification and therapy. Nat. Rev. Clin. Oncol. 2018, 15, 409–421. [Google Scholar] [CrossRef]
- Mulligan, G.; Lichter, D.I.; Di Bacco, A.; Blakemore, S.J.; Berger, A.; Koenig, E.; Bernard, H.; Trepicchio, W.; Li, B.; Neuwirth, R.; et al. Mutation of NRAS but not KRAS significantly reduces myeloma sensitivity to single-agent bortezomib therapy. Blood 2014, 123, 632–639. [Google Scholar] [CrossRef]
- Boyle, E.M.; Ashby, C.; Tytarenko, R.; Deshpande, S.; Wang, Y.; Sawyer, J.; Tian, E.; Johnson, S.; Rutherford, M.W.; Wardell, C.P.; et al. BRAF and DIS3 mutations associate with adverse outcome in a long-term follow-up of patients with multiple myeloma. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.-J.; Van Wier, S.; Tiedemann, R.; Shi, C.-X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef]
- Mroczek, S.; Chlebowska, J.; Kuliński, T.M.; Gewartowska, O.; Gruchota, J.; Cysewski, D.; Liudkovska, V.; Borsuk, E.; Nowis, D.; Dziembowski, A. The non-canonical poly(A) polymerase FAM46C acts as an onco-suppressor in multiple myeloma. Nat. Commun. 2017, 8, 619. [Google Scholar] [CrossRef]
- Ruiz-Heredia, Y.; Sánchez-Vega, B.; Onecha, E.; Barrio, S.; Alonso, R.; Martínez-Ávila, J.C.; Cuenca, I.; Agirre, X.; Braggio, E.; Hernández, M.-T.; et al. Mutational screening of newly diagnosed multiple myeloma patients by deep targeted sequencing. Haematologica 2018, 103, e544–e548. [Google Scholar] [CrossRef] [PubMed]
- Kortuem, K.M.; Braggio, E.; Bruins, L.; Barrio, S.; Shi, C.S.; Zhu, Y.X.; Tibes, R.; Viswanatha, D.; Votruba, P.; Ahmann, G.; et al. Panel sequencing for clinically oriented variant screening and copy number detection in 142 untreated multiple myeloma patients. Blood Cancer J. 2016, 6, e397. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Niesvizky, R.; Badros, A.Z.; Costa, L.J.; Ely, S.A.; Singhal, S.B.; Stadtmauer, E.A.; Haideri, N.A.; Yacoub, A.; Hess, G.; Lentzsch, S.; et al. Phase 1/2 study of cyclin-dependent kinase (CDK)4/6 inhibitor palbociclib (PD-0332991) with bortezomib and dexamethasone in relapsed/refractory multiple myeloma. Leuk. Lymphoma 2015, 56, 3320–3328. [Google Scholar] [CrossRef]
- Kumar, S.K.; LaPlant, B.; Chng, W.J.; Zonder, J.; Callander, N.; Fonseca, R.; Fruth, B.; Roy, V.; Erlichman, C.; Stewart, A.K.; et al. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 2015, 125, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Mey, U.J.M.; Renner, C.; von Moos, R. Vemurafenib in combination with cobimetinib in relapsed and refractory extramedullary multiple myeloma harboring the BRAF V600E mutation. Hematol. Oncol. 2017, 35, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Čepulytė, R.; Žučenka, A.; Pečeliūnas, V. Combination of dabrafenib and trametinib for the treatment of relapsed and refractory multiple myeloma harboring BRAF V600E mutation. Case Rep. Hematol. 2020, 2020, 8894031. [Google Scholar] [CrossRef]
- Blanden, A.R.; Yu, X.; Loh, S.N.; Levine, A.J.; Carpizo, D.R. Reactivating mutant P53 using small molecules as zinc metallochaperones: A wakening a sleeping giant in cancer. Drug Discov. Today 2015, 20, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 reactivates mutant P53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Deneberg, S.; Cherif, H.; Lazarevic, V.; Andersson, P.-O.; von Euler, M.; Juliusson, G.; Lehmann, S. An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J. 2016, 6, e447. [Google Scholar] [CrossRef]
- Saha, M.N.; Jiang, H.; Chang, H. Molecular mechanisms of nutlin-induced apoptosis in multiple myeloma: Evidence for P53-transcription-dependent and -independent pathways. Cancer Biol. Ther. 2010, 10, 567–578. [Google Scholar] [CrossRef]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.G.C.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to P53, blocks P53-HDM-2 interaction and activates P53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef]
- Grasberger, B.L.; Lu, T.; Schubert, C.; Parks, D.J.; Carver, T.E.; Koblish, H.K.; Cummings, M.D.; LaFrance, L.V.; Milkiewicz, K.L.; Calvo, R.R.; et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate P53 in cells. J. Med. Chem. 2005, 48, 909–912. [Google Scholar] [CrossRef]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2−p53 interaction. J. Med. Chem. 2006, 49, 3432–3435. [Google Scholar] [CrossRef]
- Rao, B.; Lain, S.; Thompson, A.M. P53-based cyclotherapy: Exploiting the “guardian of the genome” to protect normal cells from cytotoxic therapy. Br. J. Cancer 2013, 109, 2954–2958. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Han, Z.-G.; He, K.-Y. Malignancy of cancers and synthetic lethal interactions associated with mutations of cancer driver genes. Med. Baltim 2016, 95, e2697. [Google Scholar] [CrossRef]
- Pasca, S.; Tomuleasa, C.; Teodorescu, P.; Ghiaur, G.; Dima, D.; Moisoiu, V.; Berce, C.; Stefan, C.; Ciechanover, A.; Einsele, H. KRAS/NRAS/BRAF mutations as potential targets in multiple myeloma. Front. Oncol. 2019, 9, 1137. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.-Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Patnaik, A.; Papadopoulos, K.P.; Rasco, D.W.; Becerra, C.R.; Allred, A.J.; Orford, K.; Aktan, G.; Ferron-Brady, G.; Ibrahim, N.; et al. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother. Pharmacol. 2015, 75, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.K.S.; Li, S.; Macrae, E.R.; Park, J.-I.; Mitchell, E.P.; Zwiebel, J.A.; Chen, H.X.; Gray, R.J.; McShane, L.M.; Rubinstein, L.V.; et al. Dabrafenib and trametinib in patients with tumors with BRAFV600E mutations: Results of the NCI-MATCH trial subprotocol H. J. Clin. Oncol. 2020, 38, 3895–3904. [Google Scholar] [CrossRef]
- Holkova, B.; Zingone, A.; Kmieciak, M.; Bose, P.; Badros, A.Z.; Voorhees, P.M.; Baz, R.; Korde, N.; Lin, H.-Y.; Chen, J.-Q.; et al. A phase II trial of AZD6244 (selumetinib, ARRY-142886), an oral MEK1/2 inhibitor, in relapsed/refractory multiple myeloma. Clin. Cancer Res. 2016, 22, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, V.G.; Miller, K.C.; Macon, E.P.; Kimlinger, T.K.; Haug, J.; Kumar, S.; Gonsalves, W.I.; Rajkumar, S.V.; Kumar, S.K. Histone deacetylase inhibition in combination with MEK or BCL-2 inhibition in multiple myeloma. Haematologica 2019, 104, 2061–2074. [Google Scholar] [CrossRef] [PubMed]
- Díaz, T.; Rodríguez, V.; Lozano, E.; Mena, M.-P.; Calderón, M.; Rosiñol, L.; Martínez, A.; Tovar, N.; Pérez-Galán, P.; Bladé, J.; et al. The BET bromodomain inhibitor CPI203 improves lenalidomide and dexamethasone activity in vitro and in vivo models of multiple myeloma by blockade of ikaros and MYC signaling. Haematologica 2017, 102, 1776–1784. [Google Scholar] [CrossRef]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Corre, J.; Munshi, N.C.; Avet-Loiseau, H. Risk factors in multiple myeloma: Is it time for a revision? Blood 2021, 137, 16–19. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| mSMART [10] | IFM [160] | IMWG [3] | |
|---|---|---|---|
| t(11;14) | Standard risk | ||
| t(4;14) | Intermediate risk | High risk | High risk |
| t(14;16) | High risk | High risk | |
| Del(17p) | High risk | High risk | High risk |
| Del(13q) * | Intermediate risk | ||
| Gain 1q | High risk ** |
| Trial/Reference | Patients/N | Treatment | Survival Outcomes | ||||
|---|---|---|---|---|---|---|---|
| t(4;14) | del(17p) | t(14;16) | 1q gain | t(11;14) | |||
| IFM99-02, 03, 04 [72,108] | NDMM ≤ 65 years/ 1064 | IFM-02: VAD+auto-SCTx2 +/- Pam vs Tm+Pam IFM-03: VAD+auto-SCT+allo-SCT IFM-04: VAD+auto-SCTx2 +/- IL6 Inhibitor | - Negative vs. non- t(4;14) - No differences | - Negative vs. non-del(17p) - No differences | - Neutral - No differences | ||
| IFM2005-01 [33] | NDMM/507 | Vd vs. VAD | - Negative vs. non-t(4;14) - Vd improved EFS and OS | - Negative vs. non-del(17p) - No differences | |||
| TT2 and TT3 [148] | NDMM/668 NDMM/303 | TT2: VAD/CECD+/-T+auto-SCTx2+T+INF TT3:VTD+auto-SCTx2 +VTD+VTD/VRDm | - Negative in TT2 vs. neutral in TT3 | ||||
| HOVON65/ GMMG-HD4 [37] | NDMM/399 | PAD-auto-SCTx2-Vm vs. VAD-auto-SCTx2-Tm | - Negative vs. non-t(4;14) - No differences | - Negative - Bortezomib arm improved PFS and OS | - Neutral | - Negative - No differences | - Neutral - No differences |
| Eloquent-2 [41,161] | RRMM/646 | ERd vs. Rd | - Negative - ERd improved PFS | - Neutral - ERd improved PFS | |||
| Aspire [161,162] | RRMM/792 | KRd vs. Rd | - Negative vs. SR | - Negative vs. SR | |||
| MM-003 [40] | RRMM/455 | Pd vs. HDd | - Negative vs. SR - Pd improved PFS | - Neutral vs. SR - Pd improved PFS | |||
| Tourmaline [151] | RRMM/722 | IRd vs. Rd | - No differences | - No differences | |||
| Endeavor [40,149] | RRMM/929 | Kd vs. Vd | - Kd improved PFS | - No differences | |||
| GIMEMA MM-03-05 + EMN01 [163] | NDMM Ineligible for auto-SCT/474 | VMP vs. Rd+Rm | - VMP improved PFS | - No differences | - No differences | ||
| Myeloma IX + Myeloma XI [124,164] | NDMM/1905 | IX: CVAD/MP/CTD-auto-SCT-Tm XI: CTD/CRD/CVD-auto-SCT-Rm | - Negative vs. non-t(4;14) - Rm improved PFS | - Negative vs. non-del(17p) - Rm had no impact on PFS | - Negative vs. non-t(14;16) - Rm had no impact on PFS | - Negative vs. non-1q gains - Rm improved PFS | |
| Myeloma X [165] | RRMM/297 | Second auto-SCT vs. CFM | - No differences | - Not evaluable | - No differences | ||
| Gene Mutations/ Chromosomal Abnormalities | Signaling Pathways | Mechanisms of Action | * Targeted Drugs | Clinical Trial (Identifier)/ Reference | Trial Design/Phase |
|---|---|---|---|---|---|
| ATM | DNA damage repair | PARP inhibitor | Olaparib | NCT02693535 | Master protocol (basket)/2 |
| BRCA1/2 | DNA damage repair | PARP inhibitor | Veliparib + Bortezomib | NCT01495351 | Traditional design/1 |
| PARP inhibitor | Olaparib | NCT03297606 | Master protocol (basket)/2 | ||
| PARP inhibitor | Talazoparib | NCT02693535 | Master protocol (basket)/2 | ||
| WEE1 inhibitor | Adavosertib | NCT02465060 | Master protocol (basket)/2 | ||
| BRAFV600 | MAPK | BRAF inhibitor | Vemurafenib | NCT01524978/[194] | Master protocol (basket)/2 |
| MEK inhibitor BRAF inhibitor | Cobimetinib + Vemurafenib | NCT03297606 | Master protocol (basket)/2 | ||
| MEK inhibitor AKT inhibitor | Trametinib + Afuresertib | NCT01476137/[195] | Traditional design/1–2 | ||
| MEK inhibitor BRAF inhibitor | Trametinib +/- Dabrafenib | NCT02465060/[196] NCT03091257 | Master protocol (basket)/2 Traditional design/1 | ||
| BRAF inhibitor MEK inhibitor | Encorafenib + Binimetinib | NCT02834364 | Traditional design/2 | ||
| BRAF non-V600 | MAPK | ERK1/ERK2 inhibitor | Ulixertinib | NCT02465060 | Master protocol (basket)/2 |
| MEK inhibitor | Selumetinib | NCT01085214/[197] | Traditional design/2 | ||
| KRAS | MAPK | MEK inhibitor BRAF inhibitor | Trametinib + Afuresertib | NCT01476137/[195] | Traditional design/1–2 |
| MEK inhibitor BRAF inhibitor | Trametinib +/- Dabrafenib | NCT02465060/[196] NCT03091257 | Master protocol (basket)/2 Traditional design/1 | ||
| MEK inhibitor | Cobimetinib + Pomalidomide + Ixazomib + Dexamethasone | NCT03732703 | Master protocol (basket)/1–2 | ||
| MEK inhibitor | Selumetinib + Panobinostat | [198] | Preclinical study (cell lines) | ||
| NRAS | MAPK | MEK inhibitor | Cobimetinib + Pomalidomide + Ixazomib + Dexamethasone | NCT03732703 | Master protocol (basket)/1–2 |
| MEK inhibitor | Binimetinib | NCT02465060 | Master protocol (basket)/2 | ||
| MEK inhibitor BRAF inhibitor | Trametinib +/- Dabrafenib | NCT02465060/[196] NCT03091257 | Master protocol (basket)/2 Traditional design/1 | ||
| MEK inhibitor | Cobimetinib + Pomalidomide + Ixazomib + Dexamethasone | NCT03732703 | Master protocol (basket)/1–2 | ||
| MEK inhibitor | Selumetinib + Panobinostat | [198] | Preclinical study (cell lines) | ||
| FGFR3 | MAPK | FGFR inhibitor | Erdafitinib | NCT02465060 | Master protocol (basket)/2 |
| FGFR inhibitor | Erdafitinib + Ixazomib + Pomalidomide + Dexamethasone | NCT03732703 | Master protocol (basket)/1–2 | ||
| VEGFR/PDGFR/ CSFR inhibitor | Sunitinib | NCT02693535 | Master protocol (basket)/2 | ||
| FGFR inhibitor | AZD4547 | NCT02465060 | Master protocol (basket)/2 | ||
| CCND1 CCND3 | Cell cycle | CDK4/6 inhibitor | Palbociclib + Bortezomib + Dexamethasone | NCT00555906/[180] | Traditional design/2 |
| CDK4/6 inhibitor | Palbociclib | NCT02465060 | Master protocol (basket)/2 | ||
| IDH2 | Epigenetics | IDH2 inhibitor | Enasidenib + Ixazomib + Pomalidomide + Dexamethasone | NCT03732703 | Master protocol (basket)/1–2 |
| NF1 | MAPK | MEK inhibitor | Trametinib | NCT02465060 | Master protocol (basket)/2 |
| t(11;14) | Cell cycle | BCL2 inhibitor | Venetoclax | NCT01794520/[17] | Traditional design/1 |
| BCL2 inhibitor | Venetoclax + Carfilzomib + Dexamethasone | NCT02899052 | Traditional design/2 | ||
| BCL2 inhibitor | Venetoclax + Pomalidomide + Dexamethasone | NCT03567616 NCT03539744 | Traditional design/2 Traditional design/3 | ||
| BCL2 inhibitor | Venetoclax + Bortezomib + Dexamethasone | NCT01794507/[19] NCT02755597/[21] | Traditional design/1 Traditional design/3 | ||
| BCL2 inhibitor | Venetoclax + Daratumumab + Bortezomib + Dexamethasone | NCT03314181 | Traditional design/1–2 | ||
| BCL2 inhibitor | Venetoclax | NCT03878524 | Master protocol (basket)/1 | ||
| BCL2 inhibitor | Venetoclax + Ixazomib + Pomalidomide + Dexamethasone | NCT03732703 | Master protocol (basket)/1–2 | ||
| t(4;14) | VEGFR/FGFR/ PDGFR inhibitor | Dovitinib | NCT01058434/[47] | Traditional design/2 | |
| BET inhibitor | INCB054329 | [77] | Preclinical study (cell lines) | ||
| MYC translocations | MYC activation | BET inhibitor | CPI203 + Lenalidomide + Dexamethasone | [199] | Preclinical study (cell lines and patient samples) |
| BET inhibitor | OTX015 | NCT01713582/[200] | Traditional design/1 | ||
| BET inhibitor | Molibresib | NCT01943851 | Traditional design/2 | ||
| Del13q (RB gene) | Cell cycle | CDK4/6 inhibitor | Palbociclib | NCT02465060 | Master protocol (basket)/2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardona-Benavides, I.J.; de Ramón, C.; Gutiérrez, N.C. Genetic Abnormalities in Multiple Myeloma: Prognostic and Therapeutic Implications. Cells 2021, 10, 336. https://doi.org/10.3390/cells10020336
Cardona-Benavides IJ, de Ramón C, Gutiérrez NC. Genetic Abnormalities in Multiple Myeloma: Prognostic and Therapeutic Implications. Cells. 2021; 10(2):336. https://doi.org/10.3390/cells10020336
Chicago/Turabian StyleCardona-Benavides, Ignacio J., Cristina de Ramón, and Norma C. Gutiérrez. 2021. "Genetic Abnormalities in Multiple Myeloma: Prognostic and Therapeutic Implications" Cells 10, no. 2: 336. https://doi.org/10.3390/cells10020336
APA StyleCardona-Benavides, I. J., de Ramón, C., & Gutiérrez, N. C. (2021). Genetic Abnormalities in Multiple Myeloma: Prognostic and Therapeutic Implications. Cells, 10(2), 336. https://doi.org/10.3390/cells10020336