
Sensitivity of Osteosarcoma Cells to Concentration-Dependent Bioactivities of Lipid Peroxidation Product 4-Hydroxynonenal Depend on Their Level of Differentiation


 , , ,
, , ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. HOS Cell Line
2.2. Alkaline Phosphatase Staining
2.3. 3H-Thymidine Incorporation Assay
2.4. GSH Measurement
2.5. GST Activity
2.6. Cell Viability Assay
2.7. Cell Treatments with HNE for Free HNE Analysis, GSH Analysis, and HNE-ELISA Analysis
2.8. Determination of Free HNE by HPLC Method
2.9. Determination of HNE-Protein Adducts by HNE-ELISA
2.10. Analysis of Nuclear Morphology with 4′,6-diamidino-2-phenylindole
2.11. ROS Measurement
2.12. Immunocytochemical Fluorescence Labeling for c-FOS and c-MYC
2.13. Proteasomal Activity
2.14. Fatty Acid Analysis
2.15. Statistical Analysis
3. Results
3.1. Characterization of Undifferentiated and Differentiated HOS
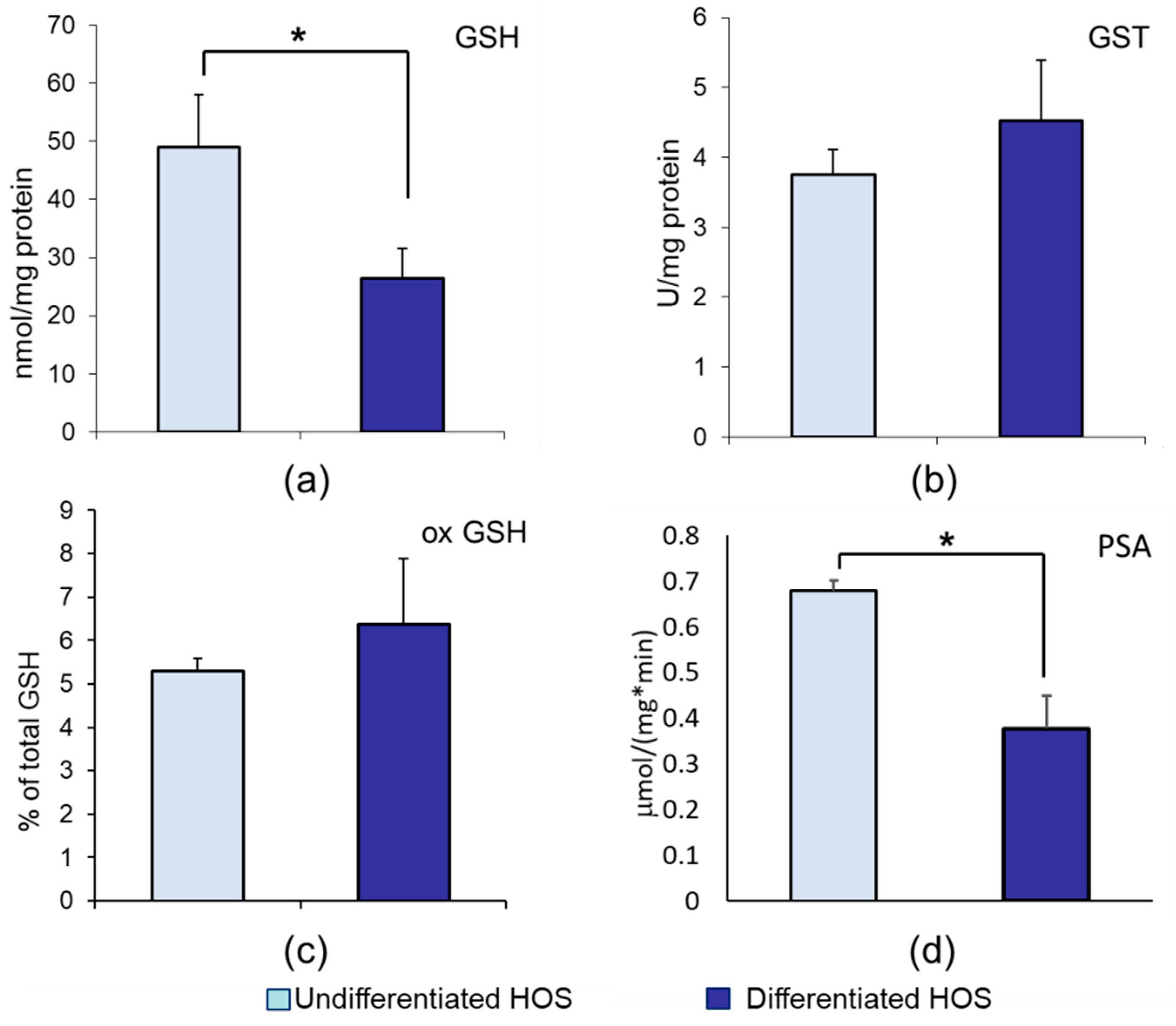
3.2. GSH, ox GSH, GST, and Proteasomal Activity in Undifferentiated and Differentiated HOS
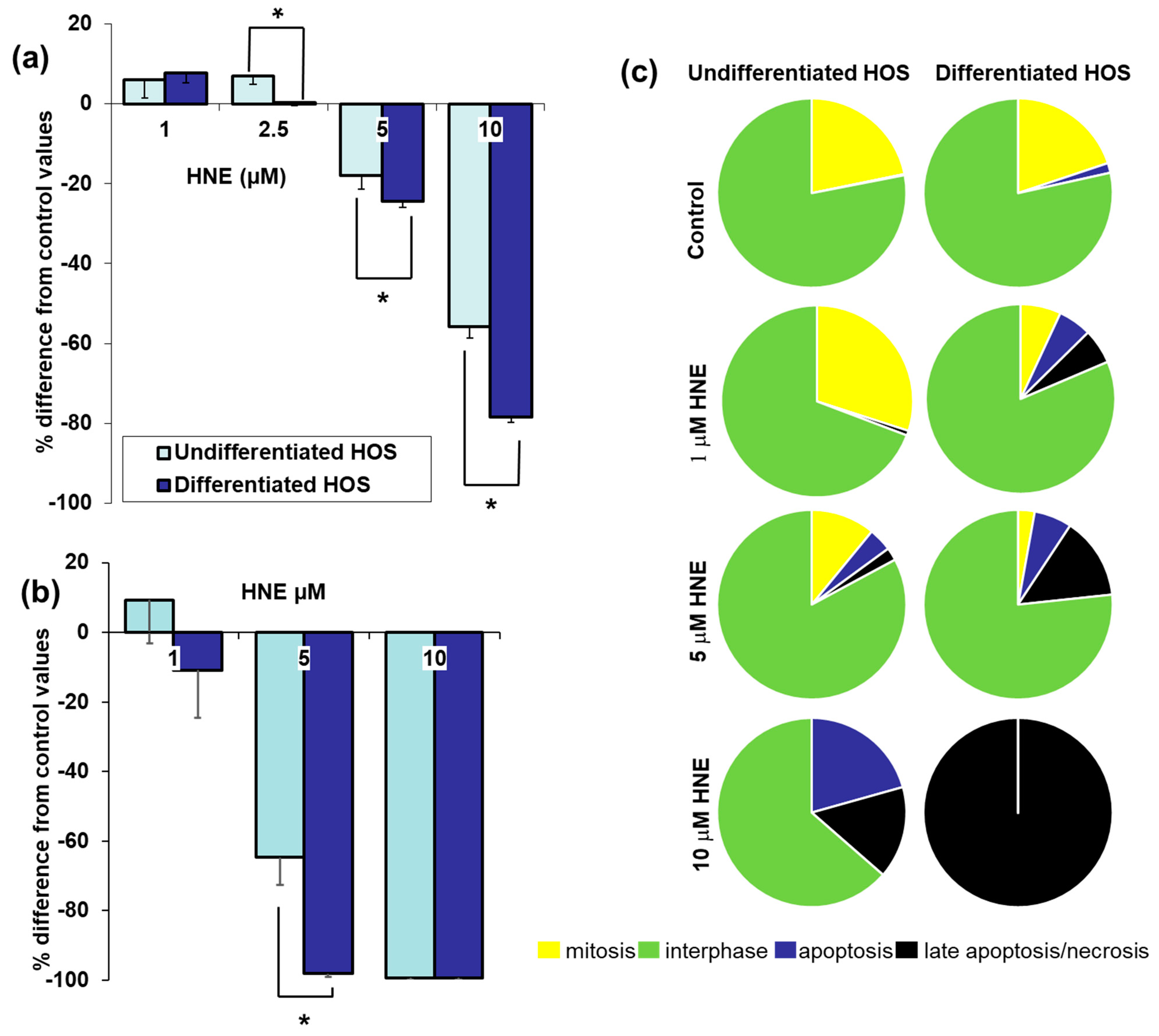
3.3. Cell Viability, Proliferation, Mitosis, Apoptosis, and Necrosis after Treatment with HNE
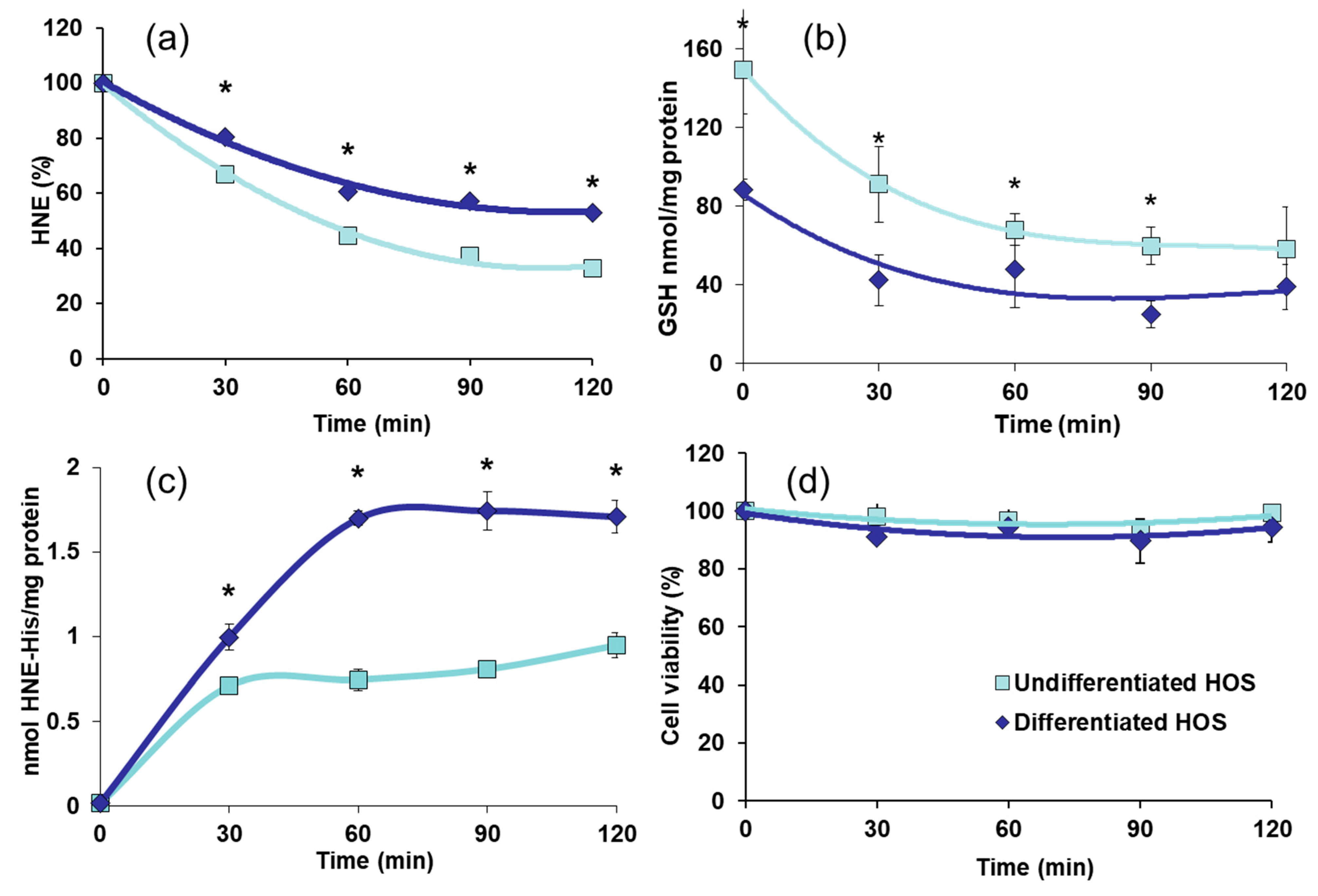
3.4. HNE Metabolism, GSH Content, Formation of HNE-Protein Adducts
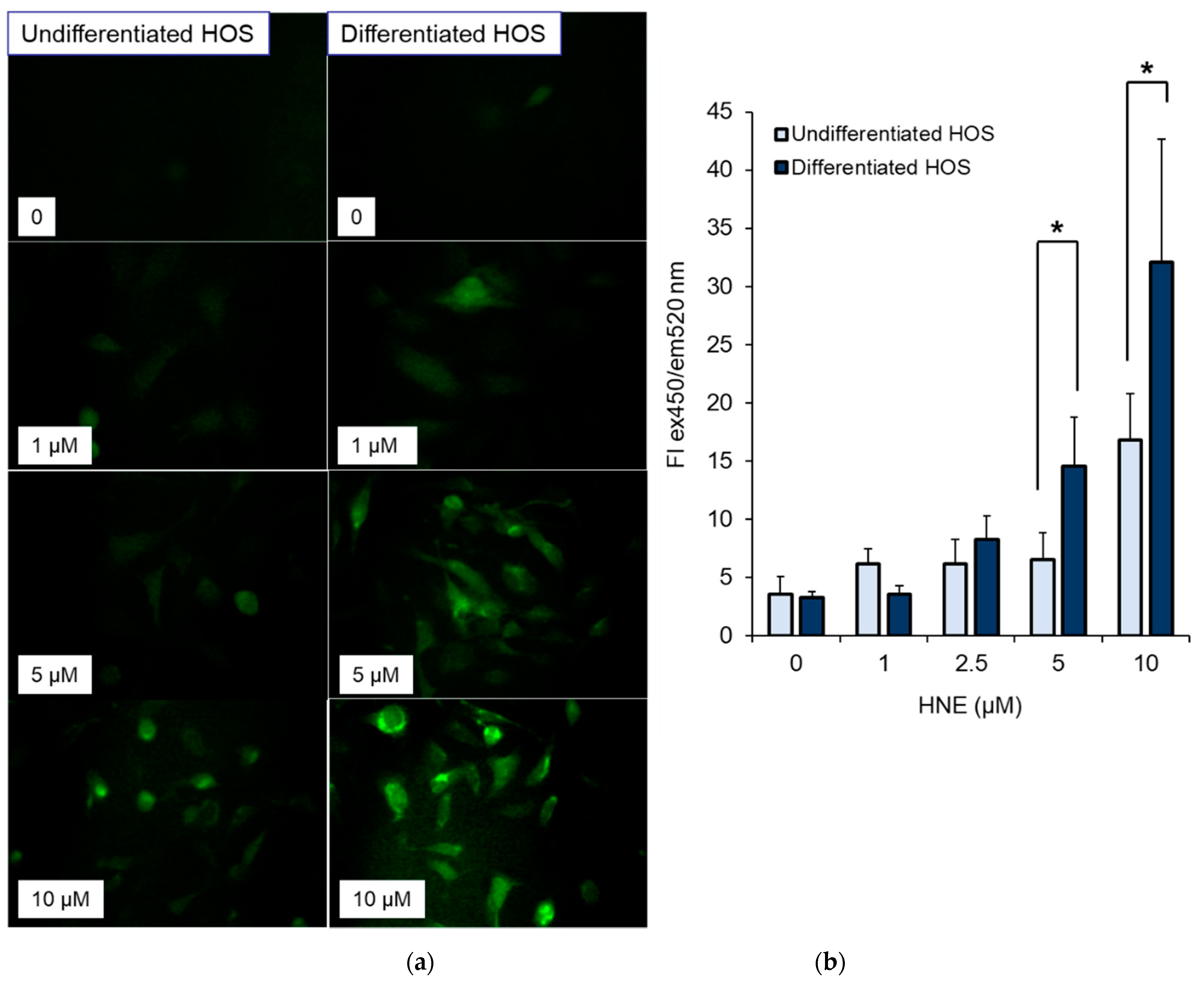
3.5. ROS Production in Cells after Treatment with HNE
3.6. c-FOS and c-MYC in Undifferentiated and Differentiated HOS
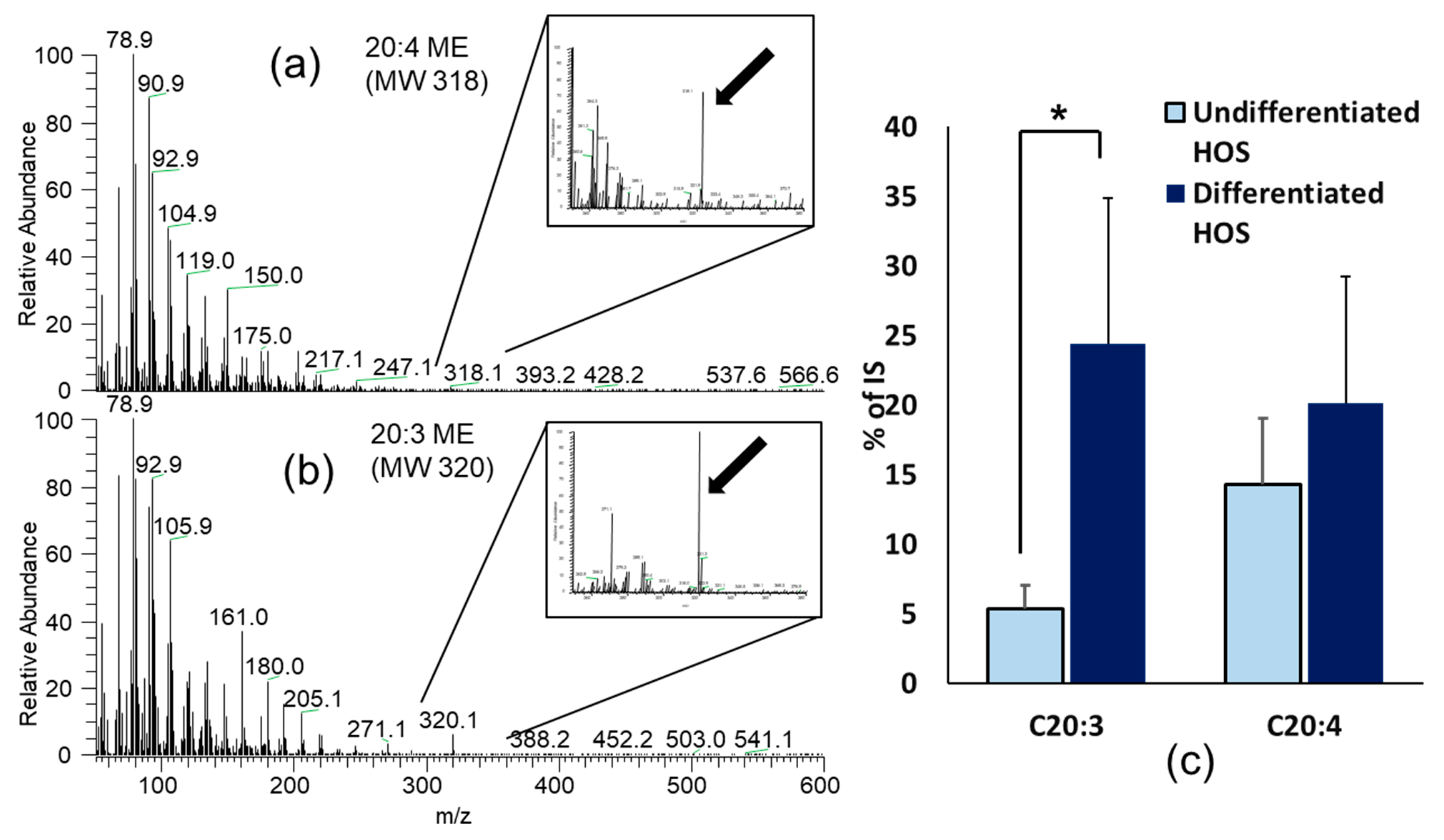
3.7. Fatty Acids Composition in Undifferentiated and Differentiated HOS
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Halliwell, B.; Gutteridge, J.M.C. Oxygen Free Radicals and Iron in Relation to Biology and Medicine: Some Problems and Concepts. Arch. Biochem. Biophys. 1986, 246, 501–514. [Google Scholar] [CrossRef]
- Burcham, P.C. Genotoxic Lipid Peroxidation Products: Their DNA Damaging Properties and Role in Formation of Endogenous DNA Adducts. Mutagenesis 1998, 13, 287–305. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and Biochemistry of 4-Hydroxynonenal, Malonaldehyde and Related Aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Dalleau, S.; Baradat, M.; Guéraud, F.; Huc, L. Cell Death and Diseases Related to Oxidative Stress: 4-Hydroxynonenal (HNE) in the Balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef]
- Toyokuni, S.; Okamoto, K.; Yodoi, J.; Hiai, H. Persistent Oxidative Stress in Cancer. FEBS Lett. 1995, 358, 1–3. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies. Antioxidants 2020, 9, 1151. [Google Scholar] [CrossRef]
- Pennington, J.D.; Wang, T.J.C.; Nguyen, P.; Sun, L.; Bisht, K.; Smart, D.; Gius, D. Redox-Sensitive Signaling Factors as a Novel Molecular Targets for Cancer Therapy. Drug Resist. Updates 2005, 8, 322–330. [Google Scholar] [CrossRef]
- Jaganjac, M.; Borovic Sunjic, S.; Zarkovic, N. Utilizing Iron for Targeted Lipid Peroxidation as Anticancer Option of Integrative Biomedicine: A Short Review of Nanosystems Containing Iron. Antioxidants 2020, 9, 191. [Google Scholar] [CrossRef]
- He, H.; Ni, J.; Huang, J. Molecular Mechanisms of Chemoresistance in Osteosarcoma (Review). Oncol. Lett. 2014, 7, 1352–1362. [Google Scholar] [CrossRef]
- dos Santos Cavalcanti, A.; Meohas, W.; de Ribeiro, G.O.; de Sá Lopes, A.C.; Gholamin, S.; Razavi, M.; Hanae Kasai Brunswick, T.; Avan, A.; Matheus Guimarães, J.A.; Leite Duarte, M.E.; et al. Patient-Derived Osteosarcoma Cells Are Resistant to Methotrexate. PLoS ONE 2017, 12, e0184891. [Google Scholar] [CrossRef]
- Milkovic, L.; Gasparovic, A.C.; Zarkovic, N. Overview on Major Lipid Peroxidation Bioactive Factor 4-Hydroxynonenal as Pluripotent Growth-Regulating Factor. Free Radic. Res. 2015, 49, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Semlitsch, T.; Tillian, H.M.; Zarkovic, N.; Borovic, S.; Purtscher, M.; Hohenwarter, O.; Schaur, R.J. Differential Influence of the Lipid Peroxidation Product 4-Hydroxynonenal on the Growth of Human Lymphatic Leukaemia Cells and Human Periopherial Blood Lymphocytes. Anticancer Res. 2002, 22, 1689–1697. [Google Scholar] [PubMed]
- Borovic, S.; Cipak, A.; Meinitzer, A.; Kejla, Z.; Perovic, D.; Waeg, G.; Zarkovic, N. Differential Sensitivity to 4-Hydroxynonenal for Normal and Malignant Mesenchymal Cells. Redox Rep. 2007, 12, 50–54. [Google Scholar] [PubMed]
- Sunjic, S.B.; Cipak, A.; Rabuzin, F.; Wildburger, R.; Zarkovic, N. The Influence of 4-Hydroxy-2-Nonenal on Proliferation, Differentiation and Apoptosis of Human Osteosarcoma Cells. BioFactors 2005, 24, 141–148. [Google Scholar] [CrossRef]
- Diduch, D.R.; Coe, M.R.; Joyner, O.; Owen, M.E.; Balian, G. Two Cell Lines from Bone Marrow That Differ in Terms of Collagen Synthesis, Osteogenic Characteristics, and Matrix Mineralization. J. Bone Jt. Surg. Ser. A 1993, 75, 92–105. [Google Scholar] [CrossRef]
- Tietze, F. Enzymic Method for Quantitative Determination of Nanogram Amounts of Total and Oxidized Glutathione: Applications to Mammalian Blood and Other Tissues. Anal. Biochem. 1969, 27, 502–522. [Google Scholar] [CrossRef]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Carmagnol, F.; Sinet, P.M.; Rapin, J.; Jerome, H. Glutathione-S-Transferase of Human Red Blood Cells; Assay, Values in Normal Subjects and in Two Pathological Circumstances: Hyperbilirubinemia and Impaired Renal Function. Clin. Chim. Acta 1981, 117, 209–217. [Google Scholar] [CrossRef]
- Mastelić, J.; Jerković, I.; Blažević, I.; Poljak-Blaži, M.; Borović, S.; Ivančić-Baće, I.; Smrěcki, V.; Žarković, N.; Brčić-Kostic, K.; Vikić-Topić, D.; et al. Comparative Study on the Antioxidant and Biological Activities of Carvacrol, Thymol, and Eugenol Derivatives. J. Agric. Food Chem. 2008, 56. [Google Scholar] [CrossRef]
- Borovic, S.; Rabuzin, F.; Waeg, G.; Zarkovic, N. Enzyme-Linked Immunosorbent Assay for 4-Hydroxynonenal-Histidine Conjugates. Free Radic. Res. 2006, 40, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Tarnowski, B.I.; Spinale, F.G.; Nicholson, J.H. DAPI as a Useful Stain for Nuclear Quantitation. Biotech. Histochem. 1991, 66, 296–302. [Google Scholar] [CrossRef]
- Rosenkranz, A.R.; Schmaldienst, S.; Stuhlmeier, K.M.; Chen, W.; Knapp, W.; Zlabinger, G.J. A Microplate Assay for the Detection of Oxidative Products Using 2′,7′-Dichlorofluorescin-Diacetate. J. Immunol. Methods 1992, 156, 39–45. [Google Scholar] [CrossRef]
- McLennan, H.R.; Esposti, M.D. The Contribution of Mitochondrial Respiratory Complexes to the Production of Reactive Oxygen Species. J. Bioenerg. Biomembr. 2000, 32, 153–162. [Google Scholar] [CrossRef]
- Reinheckel, T.; Ullrich, O.; Sitte, N.; Grune, T. Differential Impairment of 20S and 26S Proteasome Activities in Human Hematopoietic K562 Cells during Oxidative Stress. Arch. Biochem. Biophys. 2000, 377, 65–68. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Kang, J.X.; Man, S.F.P.; Brown, N.E.; Labrecque, P.A.; Garg, M.L.; Clandinin, M.T. Essential Fatty Acid Metabolism in Cultured Human Airway Epithelial Cells. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab. 1992, 1128, 267–274. [Google Scholar] [CrossRef]
- Cipak, A.; Hasslacher, M.; Tehlivets, O.; Collinson, E.J.; Zivkovic, M.; Matijevic, T.; Wonisch, W.; Waeg, G.; Dawes, I.W.; Zarkovic, N.; et al. Saccharomyces Cerevisiae Strain Expressing a Plant Fatty Acid Desaturase Produces Polyunsaturated Fatty Acids and Is Susceptible to Oxidative Stress Induced by Lipid Peroxidation. Free Radic. Biol. Med. 2006, 40, 897–906. [Google Scholar] [CrossRef]
- Zhu, D.; Shi, S.; Wang, H.; Liao, K. Growth Arrest Induces Primary-Cilium Formation and Sensitizes IGF-1-Receptor Signaling during Differentiation Induction of 3T3-L1 Preadipocytes. J. Cell Sci. 2009, 122, 2760–2768. [Google Scholar] [CrossRef]
- Barrera, G.; di Mauro, C.; Muraca, R.; Ferrero, D.; Cavalli, G.; Fazio, V.M.; Paradisi, L.; Dianzani, M.U. Induction of Differentiation in Human HL-60 Cells by 4-Hydroxynonenal, a Product of Lipid Peroxidation. Exp. Cell Res. 1991, 197, 148–152. [Google Scholar] [CrossRef]
- Cheng, J.Z.; Singhal, S.S.; Saini, M.; Singhal, J.; Piper, J.T.; van Kuijk, F.J.G.M.; Zimniak, P.; Awasthi, Y.C.; Awasthi, S. Effects of MGST A4 Transfection on 4-Hydroxynonenal-Mediated Apoptosis and Differentiation of K562 Human Erythroleukemia Cells. Arch. Biochem. Biophys. 1999, 372, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G.; Pizzimenti, S.; Serra, A.; Ferretti, C.; Fazio, V.M.; Saglio, G.; Dianzani, M.U. 4-Hydroxynonenal Specifically Inhibits c-Myb but Does Not Affect c-Fos Expressions in HL-60 Cells. Biochem. Biophys. Res. Commun. 1996, 227, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G.; Muraca, R.; Pizzimenti, S.; Serra, A.; Rosso, C.; Saglio, G.; Farce, M.G.; Fazio, V.M.; Danzani, M.U. Inhibition of C-Myc Expression Induced by 4-Hydroxynonenal, a Product of Lipid Peroxidation, in the HL-60 Human Leukemic Cell Line. Biochem. Biophys. Res. Commun. 1994, 203, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Melnik, S.; Werth, N.; Boeuf, S.; Hahn, E.M.; Gotterbarm, T.; Anton, M.; Richter, W. Impact of C-MYC Expression on Proliferation, Differentiation, and Risk of Neoplastic Transformation of Human Mesenchymal Stromal Cells. Stem Cell Res. Ther. 2019, 10, 73. [Google Scholar] [CrossRef]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic Targets of the Human C-Myc Protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef]
- Brandl, A.; Hartmann, A.; Bechmann, V.; Graf, B.; Nerlich, M.; Angele, P. Oxidative Stress Induces Senescence in Chondrocytes. J. Orthop. Res. 2011, 29, 1114–1120. [Google Scholar] [CrossRef]
- Forman, H.J.; Dickinson, D.A.; Iles, K.E. HNE-Signaling Pathways Leading to Its Elimination. In Molecular Aspects of Medicine; Elsevier Ltd.: Amsterdam, The Netherlands, 2003; Volume 24, pp. 189–194. [Google Scholar]
- Söderdahl, T.; Enoksson, M.; Lundberg, M.; Holmgren, A.; Ottersen, O.P.; Orrenius, S.; Bolcsfoldi, G.; Cotgreave, I.A. Visualization of the Compartmentalization of Glutathione and Protein-Glutathione Mixed Disulfides in Cultured Cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 124–126. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Kim, H.G.; Jung, H.I.; Shin, Y.H.; Hong, S.M.; Park, E.H.; Sa, J.H.; Lim, C.J. Activities of Antioxidant and Redox Enzymes in Human Normal Hepatic and Hepatoma Cell Lines. Mol. Cells 2002, 14, 305–311. [Google Scholar]
- Castro, J.P.; Jung, T.; Grune, T.; Siems, W. 4-Hydroxynonenal (HNE) Modified Proteins in Metabolic Diseases. Free Radic. Biol. Med. 2017, 111, 309–315. [Google Scholar] [CrossRef]
- Oberley, T.D.; Toyokuni, S.; Szweda, L.I. Localization of Hydroxynonenal Protein Adducts in Normal Human Kidney and Selected Human Kidney Cancers. Free Radic. Biol. Med. 1999, 27, 695–703. [Google Scholar] [CrossRef]
- Juric-Sekhar, G.; Zarkovic, K.; Waeg, G.; Cipak, A.; Zarkovic, N. Distribution of 4-Hydroxynonenal-Protein Conjugates as a Marker of Lipid Peroxidation and Parameter of Malignancy in Astrocytic and Ependymal Tumors of the Brain. Tumori J. 2009, 95, 762–768. [Google Scholar] [CrossRef]
- Ålin, P.; Danielson, U.H.; Mannervik, B. 4-Hydroxyalk-2-Enals Are Substrates for Glutathione Transferase. FEBS Lett. 1985, 179, 267–270. [Google Scholar] [CrossRef]
- Hubatsch, I.; Ridderström, M.; Mannervik, B. Human Glutathione Transferase A4-4: An Alpha Class Enzyme with High Catalytic Efficiency in the Conjugation of 4-Hydroxynonenal and Other Genotoxic Products of Lipid Peroxidation. Biochem. J. 1998, 330, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.S.; Zimniak, P.; Awasthi, S.; Piper, J.T.; He, N.G.; Teng, J.I.; Petersen, D.R.; Awasthi, Y.C. Several Closely Related Glutathione S-Transferase Isozymes Catalyzing Conjugation of 4-Hydroxynonenal Are Differentially Expressed in Human Tissues. Arch. Biochem. Biophys. 1994, 311, 242–250. [Google Scholar] [CrossRef]
- Awasthi, S.; Sharma, R.; Singhal, S.S.; Zimniak, P.; Awasthi, Y.C. RLIP76, a Novel Transporter Catalyzing ATP-Dependent Efflux of Xenobiotics. Drug Metab. Dispos. 2002, 30, 1300–1310. [Google Scholar] [CrossRef]
- Welters, M.J.P.; Fichtinger-Schepman, A.M.J.; Baan, R.A.; Flens, M.J.; Scheper, R.J.; Braakhuis, B.J.M. Role of Glutathione, Glutathione S-Transferases and Multidrug Resistance-Related Proteins in Cisplatin Sensitivity of Head and Neck Cancer Cell Lines. Br. J. Cancer 1998, 77, 556–561. [Google Scholar] [CrossRef][Green Version]
- Chao, C.C.; Huang, Y.T.; Ma, C.M.; Chou, W.Y.; Lin-Chao, S. Overexpression of Glutathione S-Transferase and Elevation of Thiol Pools in a Multidrug-Resistant Human Colon Cancer Cell Line. Mol. Pharmacol. 1992, 41, 69–75. [Google Scholar]
- Aldini, G.; Granata, P.; Orioli, M.; Santaniello, E.; Carini, M. Detoxification of 4-Hydroxynonenal (HNE) in Keratinocytes: Characterization of Conjugated Metabolites by Liquid Chromatography/Electrospray Ionization Tandem Mass Spectrometry. J. Mass Spectrom. 2003, 38, 1160–1168. [Google Scholar] [CrossRef]
- Srivastava, S.; Dixit, B.L.; Cai, J.; Sharma, S.; Hurst, H.E.; Bhatnagar, A.; Srivastava, S.K. Metabolism of Lipid Peroxidation Product, 4-Hydroxynonenal (HNE) in Rat Erythrocytes: Role of Aldose Reductase. Free Radic. Biol. Med. 2000, 29, 642–651. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. Proteasome Expression and Activity in Cancer and Cancer Stem Cells. Tumor Biol. 2017, 39, 1–17. [Google Scholar] [CrossRef]
- Grune, T.; Davies, K.J.A. The Proteasomal System and HNE-Modified Proteins. In Molecular Aspects of Medicine; Elsevier Ltd.: Amsterdam, The Netherlands, 2003; Volume 24, pp. 195–204. [Google Scholar]
- Chen, X.; Zhong, Z.; Xu, Z.; Chen, L.; Wang, Y. 2′,7′-Dichlorodihydrofluorescein as a Fluorescent Probe for Reactive Oxygen Species Measurement: Forty Years of Application and Controversy. Free Radic. Res. 2010, 44, 587–604. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J.A.; Caldwell, M.D.; Meng, H.C. Essential Fatty Acid Deficiency in Surgical Patients. Ann. Surg. 1977, 185, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, D.S.; Simão, A.C.; Dichi, I. Use of Olive Oil in Patients with Rheumatoid Arthritis. In Olives and Olive Oil in Health and Disease Prevention; Elsevier Inc.: Amsterdam, The Netherlands, 2010; pp. 1057–1064. ISBN 9780123744203. [Google Scholar]
- Ichi, I.; Kono, N.; Arita, Y.; Haga, S.; Arisawa, K.; Yamano, M.; Nagase, M.; Fujiwara, Y.; Arai, H. Identification of Genes and Pathways Involved in the Synthesis of Mead Acid (20:3n-9), an Indicator of Essential Fatty Acid Deficiency. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2014, 1841, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Trugnan, G.; Thomas-Benhamou, G.; Cardot, P.; Rayssiguier, Y.; Bereziat, G. Short Term Essential Fatty Acid Deficiency in Rats. Influence of Dietary Carbohydrates. Lipids 1985, 20, 862–868. [Google Scholar] [CrossRef]
- Adkisson, H.D.; Risener, F.S.; Zarrinkar, P.P.; Walla, M.D.; Christie, W.W.; Wuthier, R.E. Unique Fatty Acid Composition of Normal Cartilage: Discovery of High Levels of N-9 Eicosatrienoic Acid and Low Levels of N-6 Polyunsaturated Fatty Acids. FASEB J. 1991, 5, 344–353. [Google Scholar] [CrossRef]
- Hamazaki, T.; Nagasawa, T.; Hamazaki, K.; Itomura, M. Inhibitory Effect of 5,8,11-Eicosatrienoic Acid on Angiogenesis. Prostaglandins Leukot. Essent. Fat. Acids 2012, 86, 221–224. [Google Scholar] [CrossRef]
- Dołȩgowska, B.; Machoy, Z.; Chlubek, D. Profiles of Fatty Acids in Different Bone Structures of Growing Chicks. Vet. Res. Commun. 2006, 30, 735–747. [Google Scholar] [CrossRef]
- James, M.J.; Gibson, R.A.; Cleland, L.G. Dietary Polyunsaturated Fatty Acids and Inflammatory Mediator Production. Am. J. Clin. Nutr. 2000, 71, 343s–348s. [Google Scholar] [CrossRef]
- Hamazaki, T.; Suzuki, N.; Widyowati, R.; Miyahara, T.; Kadota, S.; Ochiai, H.; Hamazaki, K. The Depressive Effects of 5,8,11-Eicosatrienoic Acid (20:3n-9) on Osteoblasts. Lipids 2009, 44, 97–102. [Google Scholar] [CrossRef]
- Sung Kang, K.; Wang, P.; Yamabe, N.; Fukui, M.; Jay, T.; Ting Zhu, B. Docosahexaenoic Acid Induces Apoptosis in MCF-7 Cells In Vitro and In Vivo via Reactive Oxygen Species Formation and Caspase 8 Activation. PLoS ONE 2010, 5, e10296. [Google Scholar] [CrossRef]
- Guichardant, M.; Chen, P.; Liu, M.; Calzada, C.; Colas, R.; Véricel, E.; Lagarde, M. Functional Lipidomics of Oxidized Products from Polyunsaturated Fatty Acids. Chem. Phys. Lipids 2011, 164, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, J.P.; Church, D.F.; Pryor, W.A. The Kinetics of the Autoxidation of Polyunsaturated Fatty Acids. Lipids 1987, 22, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Astarita, G.; Kendall, A.C.; Dennis, E.A.; Nicolaou, A. Targeted Lipidomic Strategies for Oxygenated Metabolites of Polyunsaturated Fatty Acids. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2015, 1851, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G.; Zarkovic, N. Revealing mechanisms of selective, concentration-dependent potentials of 4-hydroxy-2-nonenal to induce apoptosis in cancer cells through inactivation of membrane-associated catalase. Free Radic. Biol. Med. 2015, 81, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Xiao, M.; Zarkovic, K.; Zhu, M.; Sa, R.; Lu, J.; Tao, Y.; Chen, Q.; Xia, L.; Cheng, S.; et al. Mitochondrial Control of Apoptosis through Modulation of Cardiolipin Oxidation in Hepatocellular Carcinoma: A Novel Link between Oxidative Stress and Cancer. Free Radic. Biol. Med. 2015, 176, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Žarković, K.; Uchida, K.; Kolenc, D.; Hlupić, L.; Žarković, N. Tissue distribution of lipid peroxidation product acrolein in human colon carcinogenesis. Free Radic. Res. 2006, 40, 543–552. [Google Scholar] [CrossRef]
- Jaganjac, M.; Matijevic Glavan, T.; Zarkovic, N. The Role of Acrolein and NADPH Oxidase in the Granulocyte-Mediated Growth-Inhibition of Tumor Cells. Cells 2019, 8, 292. [Google Scholar] [CrossRef]
- Zarkovic, K.; Jakovcevic, A.; Zarkovic, N. Contribution of the HNE-Immunohistochemistry to Modern Pathological Concepts of Major Human Diseases. Free Radic. Biol. Med. 2017, 111, 110–125. [Google Scholar] [CrossRef]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.A.; Zarkovic, N. Short overview of ROS as cell function regulators and their implications in therapy concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mitosis | Apoptosis | ALP Positive Nodules/10 cm2 | 3H-Thymidine | |
|---|---|---|---|---|
| Undifferentiated HOS | 23% | 0% | 0.74 ± 0.64 | 17,698 ± 1230 |
| Differentiated HOS | 3% | 0% | 37.77 ± 4.84 | 9814 ± 920 |
| t-test | p < 0.001 | p < 0.0002 | p < 0.05 |
| Fatty Acid | Undifferentiated HOS | Differentiated HOS | |
|---|---|---|---|
| C14:0 | Myristic acid (Tetradecanoic acid) | 3.03 ± 0.72 | 3.57 ± 1.27 |
| C16:0 | Palmitic acid (Hexadecanoic acid) | 74.01 ± 14.45 | 85.41 ± 32.26 |
| C16:1 | Palmitoleic acid (9-hexadecenoic acid) | 10.00 ± 3.58 | 11.56 ± 5.01 |
| C17:0 1 | Margaric acid (Heptadecanoic acid) | ||
| C18:1 | Oleic acid (9-Octadecenoic acid) | 70.88 ± 21.05 | 108.37 ± 47.25 |
| C18:0 | Stearic acid (Octadecanoic acid) | 40.72 ± 11.56 | 59.75 ± 20.63 |
| C20:4 | Arachidonic acid (5,8,11,14-Eicosatetraenoic acid) | 14.29 ± 4.75 | 20.11 ± 9.15 |
| C20:3 | Mead acid (5,8,11-Eicosatrienoic acid) | 5.40 ± 1.65 | 24.40 ± 10.50 * |
| C22:6 | (4,7,10,13,16,19-Docosahexaenoic acid) | 4.33 ± 1.86 | 6.13 ± 3.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sunjic, S.B.; Gasparovic, A.C.; Jaganjac, M.; Rechberger, G.; Meinitzer, A.; Grune, T.; Kohlwein, S.D.; Mihaljevic, B.; Zarkovic, N. Sensitivity of Osteosarcoma Cells to Concentration-Dependent Bioactivities of Lipid Peroxidation Product 4-Hydroxynonenal Depend on Their Level of Differentiation. Cells 2021, 10, 269. https://doi.org/10.3390/cells10020269
Sunjic SB, Gasparovic AC, Jaganjac M, Rechberger G, Meinitzer A, Grune T, Kohlwein SD, Mihaljevic B, Zarkovic N. Sensitivity of Osteosarcoma Cells to Concentration-Dependent Bioactivities of Lipid Peroxidation Product 4-Hydroxynonenal Depend on Their Level of Differentiation. Cells. 2021; 10(2):269. https://doi.org/10.3390/cells10020269
Chicago/Turabian StyleSunjic, Suzana Borovic, Ana Cipak Gasparovic, Morana Jaganjac, Gerald Rechberger, Andreas Meinitzer, Tilman Grune, Sepp D. Kohlwein, Branka Mihaljevic, and Neven Zarkovic. 2021. "Sensitivity of Osteosarcoma Cells to Concentration-Dependent Bioactivities of Lipid Peroxidation Product 4-Hydroxynonenal Depend on Their Level of Differentiation" Cells 10, no. 2: 269. https://doi.org/10.3390/cells10020269
APA StyleSunjic, S. B., Gasparovic, A. C., Jaganjac, M., Rechberger, G., Meinitzer, A., Grune, T., Kohlwein, S. D., Mihaljevic, B., & Zarkovic, N. (2021). Sensitivity of Osteosarcoma Cells to Concentration-Dependent Bioactivities of Lipid Peroxidation Product 4-Hydroxynonenal Depend on Their Level of Differentiation. Cells, 10(2), 269. https://doi.org/10.3390/cells10020269