
Intracellular Sources of ROS/H2O2 in Health and Neurodegeneration: Spotlight on Endoplasmic Reticulum



Abstract
1. Introduction
2. ER Sources of ROS/H2O2
H2O2 Transport Across the ER Membrane
3. Non-ER Sources of ROS/H2O2
3.1. Mitochondrial ROS/H2O2
3.2. Peroxisomal ROS/H2O2
3.3. Nuclear ROS/H2O2
3.4. Golgi ROS/H2O2
4. ROS/H2O2 and Neurodegeneration
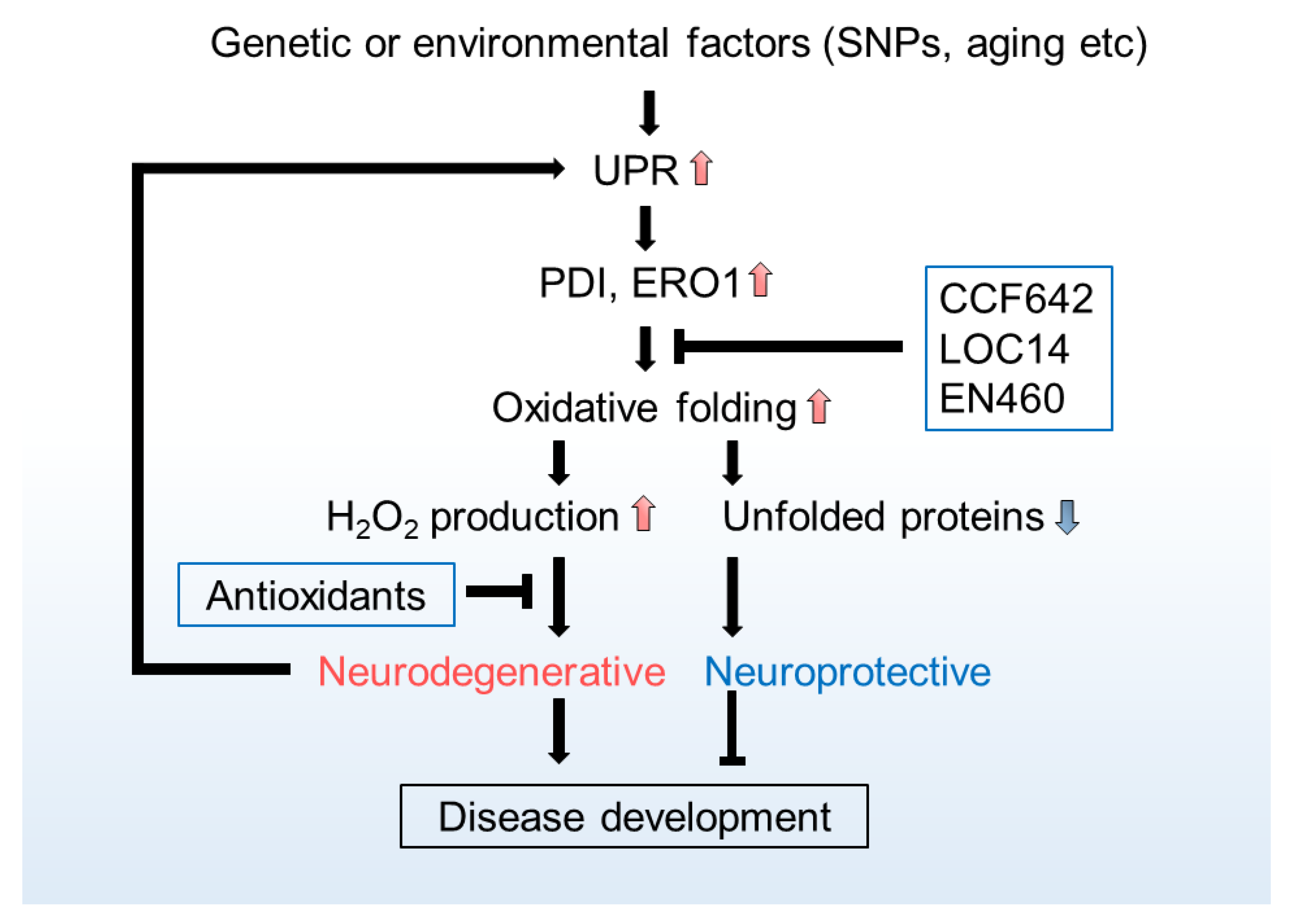
ER H2O2 and Neurodegeneration
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Glossary
| Superoxide (O2−) | A radical ROS produced by the one-electron reduction of molecular oxygen. It is converted to hydrogen peroxide and molecular oxygen by spontaneous and/or SOD-catalysing dismutation. |
| Hydrogen peroxide (H2O2) | A non-radical, relatively stable and functional ROS, hazardous in excess. |
| Hydroxyl radical (·OH) | Highly reactive and short-lived radical. Its production is driven by Fenton reaction, between ferrous ion (Fe2+) and hydrogen peroxide. |
| Redox potential | Expressed in millivolts (mV), a measure of redox state that reflects the tendency of a chemical to receive/donate electrons, and concentrations of its reduced and oxidised species, given by the Nernst equation |
| Superoxide dismutase (SOD) | An antioxidant enzyme catalysing dismutation of superoxide to hydrogen peroxide and molecular oxygen. SOD family consists of Cu/Zn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3). Mutations in the SOD1 gene affect approximately 20% of familial amyotrophic lateral sclerosis (ALS). |
| Peroxiredoxin (PRDX) | An antioxidant enzyme scavenging peroxides including H2O2. PRDX family consists of six isoforms (PRDX1-6). PRDX4 is localised in the ER. |
| Glutathione peroxidase (GPX) | An antioxidant enzyme scavenging H2O2 as well as lipid peroxides by utilising reduced glutathione. GPX family consists of eight isoforms (GPX1-8). GPX1-4 and GPX6 contain an active site selenocysteine (selenoproteins). GPX7 and GPX8 are localised in the ER. GSH peroxidase activity of GPX7 and GPX8 is low. |
| Glutathione | An abundant antioxidant tripeptide consisting of alanine–cysteine–glutamate (with cysteine and glutamate linked by gamma–glutamyl bond). Reduced glutathione (GSH) drives GPXs reactions and as a result GSH is converted to the oxidised form (GSSG), in which two molecules are bound by disulfide bond. GSSG is recycled by reducing by glutathione reductase (GR). |
| Catalase | An antioxidant enzyme that scavenges H2O2. The iron-bound heme group in the active site catalyses the conversion of H2O2 to water (H2O) and molecular oxygen (O2). It mainly localises in the peroxisomes. |
| Protein disulfide isomerase (PDI) | An ER chaperone possessing oxidoreductase activity via CXXC catalytic motif-containing thioredoxin domains. Catalyses protein disulphide bond formation and isomerisation during folding of ER client proteins. |
| ER oxidoreductin 1 (ERO1) | A sulfhydryl oxidase that supplies oxidising equivalents to recycle PDI, in the process ERO1 produces H2O2 by reducing O2 with its active site (CXXCXXC motif/flavin adenine dinucleotide (FAD) prosthetic group). |
| NADPH (nicotinamide adenine dinucleotide phosphate) oxidase (NOX) | An enzyme catalysing the production of a superoxide by transferring one electron to oxygen from NADPH. The NOX family consists of NOX1~NOX5, Dual oxidase 1 (DUOX1) and DUOX2. Among NOXs, NOX4 preferentially produces H2O2 rather than superoxide. |
References
- Paravicini, T.M.; Touyz, R.M. NADPH oxidases, reactive oxygen species, and hypertension: Clinical implications and therapeutic possibilities. Diabetes Care 2008, 31 (Suppl. 2), S170–S180. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Jones, D.P.; Go, Y.M. Redox compartmentalization and cellular stress. Diabetes Obes. Metab. 2010, 12, 116–125. [Google Scholar] [CrossRef]
- Zheng, M.; Åslund, F.; Storz, G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science 1998, 279, 1718–1721. [Google Scholar] [CrossRef]
- Konno, T.; Melo, E.P.; Lopes, C.; Mehmeti, I.; Lenzen, S.; Ron, D.; Avezov, E. ERO1-independent production of H2O2 within the endoplasmic reticulum fuels Prdx4-mediated oxidative protein folding. J. Cell Biol. 2015, 211, 253–259. [Google Scholar] [CrossRef]
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 2006, 3, 281–286. [Google Scholar] [CrossRef]
- Giannoni, E.; Buricchi, F.; Grimaldi, G.; Parri, M.; Cialdai, F.; Taddei, M.L.; Raugei, G.; Ramponi, G.; Chiarugi, P. Redox regulation of anoikis: Reactive oxygen species as essential mediators of cell survival. Cell Death Differ. 2008, 15, 867–878. [Google Scholar] [CrossRef]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen peroxide activates NF-κB through tyrosine phosphorylation of IκBα and serine phosphorylation of p65. Evidence for the involvement of IκBα kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003, 278, 24233–24241. [Google Scholar] [CrossRef]
- Fourquet, S.; Guerois, R.; Biard, D.; Toledano, M.B. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J. Biol. Chem. 2010, 285, 8463–8471. [Google Scholar] [CrossRef]
- Birk, J.; Meyer, M.; Aller, I.; Hansen, H.G.; Odermatt, A.; Dick, T.P.; Meyer, A.J.; Appenzeller-Herzog, C. Endoplasmic reticulum: Reduced and oxidized glutathione revisited. J. Cell Sci. 2013, 126, 1604–1617. [Google Scholar] [CrossRef]
- Kojer, K.; Bien, M.; Gangel, H.; Morgan, B.; Dick, T.P.; Riemer, J. Glutathione redox potential in the mitochondrial intermembrane space is linked to the cytosol and impacts the Mia40 redox state. EMBO J. 2012, 31, 3169–3182. [Google Scholar] [CrossRef]
- Melo, E.P.; Lopes, C.; Gollwitzer, P.; Lortz, S.; Lenzen, S.; Mehmeti, I.; Kaminski, C.F.; Ron, D.; Avezov, E. TriPer, an optical probe tuned to the endoplasmic reticulum tracks changes in luminal H2O2. BMC Biol. 2017, 15. [Google Scholar] [CrossRef]
- Ramming, T.; Hansen, H.G.; Nagata, K.; Ellgaard, L.; Appenzeller-Herzog, C. GPx8 peroxidase prevents leakage of H2O2 from the endoplasmic reticulum. Free Radic. Biol. Med. 2014, 70, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. The biological chemistry of hydrogen peroxide. In Methods in Enzymology; Academic Press Inc.: Cambridge, MA, USA, 2013; Volume 528, pp. 3–25. ISBN 9780124058811. [Google Scholar]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Ellgaard, L. The human PDI family: Versatility packed into a single fold. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.; Sevier, C.S.; Heldman, N.; Vitu, E.; Bentzur, M.; Kaiser, C.A.; Thorpe, C.; Fass, D. Generating disulfides enzymatically: Reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Shergalis, A.G.; Hu, S.; Bankhead, A.; Neamati, N. Role of the ERO1-PDI interaction in oxidative protein folding and disease. Pharmacol. Ther. 2020, 210, 107525. [Google Scholar] [CrossRef]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Princiotta, M.F.; Finzi, D.; Qian, S.B.; Gibbs, J.; Schuchmann, S.; Buttgereit, F.; Bennink, J.R.; Yewdell, J.W. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 2003, 18, 343–354. [Google Scholar] [CrossRef]
- Gess, B.; Hofbauer, K.H.; Wenger, R.H.; Lohaus, C.; Meyer, H.E.; Kurtz, A. The cellular oxygen tension regulates expression of the endoplasmic oxidoreductase ERO1-Lα. Eur. J. Biochem. 2003, 270, 2228–2235. [Google Scholar] [CrossRef] [PubMed]
- Takemori, Y.; Sakaguchi, A.; Matsuda, S.; Mizukami, Y.; Sakurai, H. Stress-induced transcription of the endoplasmic reticulum oxidoreductin gene ERO1 in the yeast Saccharomyces cerevisiae. Mol. Genet. Genomics 2006, 275, 89–96. [Google Scholar] [CrossRef]
- Pagani, M.; Fabbri, M.; Benedetti, C.; Fassio, A.; Pilati, S.; Bulleid, N.J.; Cabibbo, A.; Sitia, R. Endoplasmic reticulum oxidoreductin 1-Lβ (ERO1-Lβ), a human gene induced in the course of the unfolded protein response. J. Biol. Chem. 2000, 275, 23685–23692. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef]
- Ramming, T.; Okumura, M.; Kanemura, S.; Baday, S.; Birk, J.; Moes, S.; Spiess, M.; Jenö, P.; Bernèche, S.; Inaba, K.; et al. A PDI-catalyzed thiol-disulfide switch regulates the production of hydrogen peroxide by human Ero1. Free Radic. Biol. Med. 2015, 83, 361–372. [Google Scholar] [CrossRef]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef]
- Curran, S.P.; Ruvkun, G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007, 3, 479–487. [Google Scholar] [CrossRef]
- Frand, A.R.; Kaiser, C.A. The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol. Cell 1998, 1, 161–170. [Google Scholar] [CrossRef]
- Pollard, M.G.; Travers, K.J.; Weissman, J.S. Ero1p: A novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol. Cell 1998, 1, 171–182. [Google Scholar] [CrossRef]
- Zito, E.; Chin, K.T.; Blais, J.; Harding, H.P.; Ron, D. ERO1-β, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J. Cell Biol. 2010, 188, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010, 29, 4185–4197. [Google Scholar] [CrossRef] [PubMed]
- Zito, E.; Melo, E.P.; Yang, Y.; Wahlander, Å.; Neubert, T.A.; Ron, D. Oxidative Protein Folding by an Endoplasmic Reticulum-Localized Peroxiredoxin. Mol. Cell 2010, 40, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Zito, E.; Hansen, H.G.; Yeo, G.S.H.; Fujii, J.; Ron, D. Endoplasmic Reticulum Thiol Oxidase Deficiency Leads to Ascorbic Acid Depletion and Noncanonical Scurvy in Mice. Mol. Cell 2012, 48, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Kim, M.H.; Lee, H.J.; Huh, J.W.; Lee, H.S.; Lee, D.S. Peroxiredoxin 4 attenuates glutamate-induced neuronal cell death through inhibition of endoplasmic reticulum stress. Free Radic. Res. 2020, 54, 207–220. [Google Scholar] [CrossRef]
- Mehmeti, I.; Lortz, S.; Elsner, M.; Lenzen, S. Peroxiredoxin 4 improves insulin biosynthesis and glucoseinduced insulin secretion in insulin-secreting INS-1E cells. J. Biol. Chem. 2014, 289, 26904–26913. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, M.H.; Lee, H.J.; Huh, J.W.; Lee, S.R.; Lee, H.S.; Lee, D.S. Peroxiredoxin 4 inhibits insulin-induced adipogenesis through regulation of ER stress in 3T3-L1 cells. Mol. Cell. Biochem. 2020, 468, 97–109. [Google Scholar] [CrossRef]
- Iuchi, Y.; Okada, F.; Tsunoda, S.; Kibe, N.; Shirasawa, N.; Ikawa, M.; Okabe, M.; Ikeda, Y.; Fujii, J. Peroxiredoxin 4 knockout results in elevated spermatogenic cell death via oxidative stress. Biochem. J. 2009, 419, 149–158. [Google Scholar] [CrossRef]
- Takagi, T.; Homma, T.; Fujii, J.; Shirasawa, N.; Yoriki, H.; Hotta, Y.; Higashimura, Y.; Mizushima, K.; Hirai, Y.; Katada, K.; et al. Elevated ER stress exacerbates dextran sulfate sodium-induced colitis in PRDX4-knockout mice. Free Radic. Biol. Med. 2019, 134, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, K.; Guo, X.; Shioya, A.; Zhang, J.; Zheng, J.; Kurose, N.; Ishibashi, H.; Motono, N.; Uramoto, H.; Yamada, S. The impact of PRDX4 and the EGFR mutation status on cellular proliferation in lung adenocarcinoma. Int. J. Med. Sci. 2019, 16, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.D.; Saaranen, M.J.; Karala, A.R.; Lappi, A.K.; Wang, L.; Raykhel, I.B.; Alanen, H.I.; Salo, K.E.H.; Wang, C.C.; Ruddock, L.W. Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bond formation. J. Mol. Biol. 2011, 406, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Mehmeti, I.; Lortz, S.; Avezov, E.; Jörns, A.; Lenzen, S. ER-resident antioxidative GPx7 and GPx8 enzyme isoforms protect insulin-secreting INS-1E β-cells against lipotoxicity by improving the ER antioxidative capacity. Free Radic. Biol. Med. 2017, 112, 121–130. [Google Scholar] [CrossRef]
- Fang, J.; Yang, J.; Wu, X.; Zhang, G.; Li, T.; Wang, X.; Zhang, H.; Wang, C.C.; Liu, G.H.; Wang, L. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 2018, 17. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, L.; Miao, Y.; Yang, J.; Wang, X.; Wang, C.C.; Feng, J.; Wang, L. Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 2019, 20, 46–59. [Google Scholar] [CrossRef]
- Wei, P.C.; Hsieh, Y.H.; Su, M.I.; Jiang, X.; Hsu, P.H.; Lo, W.T.; Weng, J.Y.; Jeng, Y.M.; Wang, J.M.; Chen, P.L.; et al. Loss of the Oxidative Stress Sensor NPGPx Compromises GRP78 Chaperone Activity and Induces Systemic Disease. Mol. Cell 2012, 48, 747–759. [Google Scholar] [CrossRef]
- Peng, D.F.; Belkhiri, A.; Hu, T.L.; Chaturvedi, R.; Asim, M.; Wilson, K.T.; Zaika, A.; El-Rifai, W. Glutathione peroxidase 7 protects against oxidative DNA damage in oesophageal cells. Gut 2012, 61, 1250–1260. [Google Scholar] [CrossRef]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef]
- Wu, R.-F.; Ma, Z.; Liu, Z.; Terada, L.S. Nox4-Derived H2O2 Mediates Endoplasmic Reticulum Signaling through Local Ras Activation. Mol. Cell. Biol. 2010, 30, 3553–3568. [Google Scholar] [CrossRef]
- Wu, R.F.; Liao, C.; Hatoum, H.; Fu, G.; Ochoa, C.D.; Terada, L.S. RasGRF couples Nox4-dependent endoplasmic reticulum signaling to Ras. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Auer, S.; Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Breitenbach-Koller, H.; Geisberger, R.; Aigner, E.; Cadamuro, J.; Richter, K.; Sopjani, M.; et al. The human NADPH oxidase, Nox4, regulates cytoskeletal organization in two cancer cell lines, HepG2 and SH-SY5Y. Front. Oncol. 2017, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Prior, K.K.; Wittig, I.; Leisegang, M.S.; Groenendyk, J.; Weissmann, N.; Michalak, M.; Jansen-Dürr, P.; Shah, A.M.; Brandes, R.P. The endoplasmic reticulum chaperone calnexin is a NADPH oxidase NOX4 interacting protein. J. Biol. Chem. 2016, 291, 7045–7059. [Google Scholar] [CrossRef] [PubMed]
- Janiszewski, M.; Lopes, L.R.; Carmo, A.O.; Pedro, M.A.; Brandes, R.P.; Santos, C.X.C.; Laurindo, F.R.M. Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. J. Biol. Chem. 2005, 280, 40813–40819. [Google Scholar] [CrossRef]
- Lee, H.Y.; Zeeshan, H.M.A.; Kim, H.R.; Chae, H.J. Nox4 regulates the eNOS uncoupling process in aging endothelial cells. Free Radic. Biol. Med. 2017, 113, 26–35. [Google Scholar] [CrossRef]
- Santos, C.X.; Hafstad, A.D.; Beretta, M.; Zhang, M.; Molenaar, C.; Kopec, J.; Fotinou, D.; Murray, T.V.; Cobb, A.M.; Martin, D.; et al. Targeted redox inhibition of protein phosphatase 1 by Nox4 regulates eIF 2α-mediated stress signaling. EMBO J. 2016, 35, 319–334. [Google Scholar] [CrossRef]
- Bass, R.; Ruddock, L.W.; Klappa, P.; Freedman, R.B. A Major Fraction of Endoplasmic Reticulum-located Glutathione Is Present as Mixed Disulfides with Protein. J. Biol. Chem. 2004, 279, 5257–5262. [Google Scholar] [CrossRef]
- Montero, D.; Tachibana, C.; Rahr Winther, J.; Appenzeller-Herzog, C. Intracellular glutathione pools are heterogeneously concentrated. Redox Biol. 2013, 1, 508–513. [Google Scholar] [CrossRef]
- Lizák, B.; Birk, J.; Zana, M.; Kosztyi, G.; Kratschmar, D.V.; Odermatt, A.; Zimmermann, R.; Geiszt, M.; Appenzeller-Herzog, C.; Bánhegyi, G. Ca2+ mobilization-dependent reduction of the endoplasmic reticulum lumen is due to influx of cytosolic glutathione. BMC Biol. 2020, 18. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Bánhegyi, G.; Bogeski, I.; Davies, K.J.A.; Delaunay-Moisan, A.; Forman, H.J.; Görlach, A.; Kietzmann, T.; Laurindo, F.; Margittai, E.; et al. Transit of H2O2 across the endoplasmic reticulum membrane is not sluggish. Free Radic. Biol. Med. 2016, 94, 157–160. [Google Scholar] [CrossRef]
- Bienert, G.P.; Møller, A.L.B.; Kristiansen, K.A.; Schulz, A.; Møller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Dynowski, M.; Schaaf, G.; Loque, D.; Moran, O.; Ludewig, U. Plant plasma membrane water channels conduct the signalling molecule H2O2. Biochem. J. 2008, 414, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta Biomembr. 2006, 1758, 994–1003. [Google Scholar] [CrossRef]
- Hooijmaijers, C.; Rhee, J.Y.; Kwak, K.J.; Chung, G.C.; Horie, T.; Katsuhara, M.; Kang, H. Hydrogen peroxide permeability of plasma membrane aquaporins of Arabidopsis thaliana. J. Plant Res. 2012, 125, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.S.; Preston, G.M.; Smith, B.L.; Guggino, W.B.; Agre, P. Molecular structure of the water channel through aquaporin CHIP. The hourglass model—PubMed. J. Biol. Chem. 1994, 269, 14648–14654. [Google Scholar] [CrossRef]
- Bertolotti, M.; Bestetti, S.; García-Manteiga, J.M.; Medraño-Fernandez, I.; Dal Mas, A.; Malosio, M.L.; Sitia, R. Tyrosine Kinase signal modulation: A matter of H2O2 membrane permeability? Antioxid. Redox Signal. 2013, 19, 1447–1451. [Google Scholar] [CrossRef]
- Cavazzin, C.; Ferrari, D.; Facchetti, F.; Russignan, A.; Vescovi, A.L.; La Porta, C.A.M.; Gritti, A. Unique expression and localization of aquaporin- 4 and aquaporin-9 in murine and human neural stem cells and in their glial progeny. Glia 2006, 53, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Almasalmeh, A.; Krenc, D.; Wu, B.; Beitz, E. Structural determinants of the hydrogen peroxide permeability of aquaporins. FEBS J. 2014, 281, 647–656. [Google Scholar] [CrossRef]
- Bestetti, S.; Galli, M.; Sorrentino, I.; Pinton, P.; Rimessi, A.; Sitia, R.; Medraño-Fernandez, I. Human aquaporin-11 guarantees efficient transport of H2O2 across the endoplasmic reticulum membrane. Redox Biol. 2020, 28, 101326. [Google Scholar] [CrossRef]
- Morishita, Y.; Matsuzaki, T.; Hara-chikuma, M.; Andoo, A.; Shimono, M.; Matsuki, A.; Kobayashi, K.; Ikeda, M.; Yamamoto, T.; Verkman, A.; et al. Disruption of Aquaporin-11 Produces Polycystic Kidneys following Vacuolization of the Proximal Tubule. Mol. Cell. Biol. 2005, 25, 7770–7779. [Google Scholar] [CrossRef]
- Hoshino, Y.; Sonoda, H.; Nishimura, R.; Mori, K.; Ishibashi, K.; Ikeda, M. Involvement of the NADPH oxidase 2 pathway in renal oxidative stress in Aqp11-/- mice. Biochem. Biophys. Rep. 2019, 17, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Bestetti, S.; Medraño-Fernandez, I.; Galli, M.; Ghitti, M.; Bienert, G.P.; Musco, G.; Orsi, A.; Rubartelli, A.; Sitia, R. A persulfidation-based mechanism controls aquaporin-8 conductance. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed]
- Kohler, J.J.; Cucoranu, I.; Fields, E.; Green, E.; He, S.; Hoying, A.; Russ, R.; Abuin, A.; Johnson, D.; Hosseini, S.H.; et al. Transgenic mitochondrial superoxide dismutase and mitochondrially targeted catalase prevent antiretroviral-induced oxidative stress and cardiomyopathy. Lab. Investig. 2009, 89, 782–790. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.L.S.; Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Brand, M.D. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 2015, 290, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Goncalves, R.L.S.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef]
- Pak, V.V.; Ezeriņa, D.; Lyublinskaya, O.G.; Pedre, B.; Tyurin-Kuzmin, P.A.; Mishina, N.M.; Thauvin, M.; Young, D.; Wahni, K.; Martínez Gache, S.A.; et al. Ultrasensitive Genetically Encoded Indicator for Hydrogen Peroxide Identifies Roles for the Oxidant in Cell Migration and Mitochondrial Function. Cell Metab. 2020, 31, 642–653.e6. [Google Scholar] [CrossRef]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Das, R.; Xu, S.; Quan, X.; Nguyen, T.T.; Kong, I.D.; Chung, C.H.; Lee, E.Y.; Cha, S.K.; Park, K.S. Upregulation of mitochondrial Nox4 mediates TGF-β-induced apoptosis in cultured mouse podocytes. Am. J. Physiol. Ren. Physiol. 2014, 306, F155–F167. [Google Scholar] [CrossRef] [PubMed]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Stevenson, M.D.; Vendrov, A.E.; Hayami, T.; Robidoux, J.; Xiao, H.; Zhang, Y.Y.; Eitzman, D.T.; Runge, M.S.; Madamanchi, N.R. Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol. 2019, 26. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rapti, K.; Diokmetzidou, A.; Kloukina, I.; Milner, D.J.; Varela, A.; Davos, C.H.; Capetanaki, Y. Opposite effects of catalase and MnSOD ectopic expression on stress induced defects and mortality in the desmin deficient cardiomyopathy model. Free Radic. Biol. Med. 2017, 110, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009, 21, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef]
- Waypa, G.B.; Guzy, R.; Mungai, P.T.; Mack, M.M.; Marks, J.D.; Roe, M.W.; Schumacker, P.T. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ. Res. 2006, 99, 970–978. [Google Scholar] [CrossRef]
- Waypa, G.B.; Chandel, N.S.; Schumacker, P.T. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ. Res. 2001, 88, 1259–1266. [Google Scholar] [CrossRef]
- Backes, S.; Herrmann, J.M. Protein translocation into the intermembrane space and matrix of mitochondria: Mechanisms and driving forces. Front. Mol. Biosci. 2017, 4, 83. [Google Scholar] [CrossRef]
- Bihlmaier, K.; Mesecke, N.; Terziyska, N.; Bien, M.; Hell, K.; Herrmann, J.M. The disulfide relay system of mitochondria is connected to the respiratory chain. J. Cell Biol. 2007, 179, 389–395. [Google Scholar] [CrossRef]
- Dlasková, A.; Clarke, K.J.; Porter, R.K. The role of UCP 1 in production of reactive oxygen species by mitochondria isolated from brown adipose tissue. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yu, M.S.; Huang, Y.; Xiang, Z.; Chen, Y.P. MiR-30e-UCP2 pathway regulates alcoholic hepatitis progress by influencing ATP and hydrogen peroxide expression. Oncotarget 2017, 8, 64294–64302. [Google Scholar] [CrossRef] [PubMed]
- Nègre-Salvayre, A.; Hirtz, C.; Carrera, G.; Cazenave, R.; Troly, M.; Salvayre, R.; Pénicaud, L.; Casteilla, L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation—PubMed. FASEB J. 1997, 11, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Angermüller, S.; Islinger, M.; Völkl, A. Peroxisomes and reactive oxygen species, a lasting challenge. Histochem. Cell Biol. 2009, 131, 459–463. [Google Scholar] [CrossRef]
- Tripathi, D.N.; Walker, C.L. The peroxisome as a cell signaling organelle. Curr. Opin. Cell Biol. 2016, 39, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 1755–1766. [Google Scholar] [CrossRef]
- De Duve, C.; Baudhuin, P. Peroxisomes (microbodies and related particles). Physiol. Rev. 1966, 46, 323–357. [Google Scholar] [CrossRef]
- Zhang, J.; Kim, J.; Alexander, A.; Cai, S.; Tripathi, D.N.; Dere, R.; Tee, A.R.; Tait-Mulder, J.; Di Nardo, A.; Han, J.M.; et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat. Cell Biol. 2013, 15, 1186–1196. [Google Scholar] [CrossRef]
- Lee, J.N.; Dutta, R.K.; Maharjan, Y.; Liu, Z.Q.; Lim, J.Y.; Kim, S.J.; Cho, D.H.; So, H.S.; Choe, S.K.; Park, R. Catalase inhibition induces pexophagy through ROS accumulation. Biochem. Biophys. Res. Commun. 2018, 501, 696–702. [Google Scholar] [CrossRef]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kawałek, A.; van der Klei, I.J. Peroxisomal quality control mechanisms. Curr. Opin. Microbiol. 2014, 22, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Mehmeti, I.; Lortz, S.; Lenzen, S. The H2O2-sensitive HyPer protein targeted to the endoplasmic reticulum as a mirror of the oxidizing thiol-disulfide milieu. Free Radic. Biol. Med. 2012, 53, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Dhaunsi, G.S.; Gupta, M.P.; Orak, J.K.; Asayama, K.; Singh, I. Demonstration of glutathione peroxidase in rat liver peroxisomes and its intraorganellar distribution. Arch. Biochem. Biophys. 1994, 315, 331–338. [Google Scholar] [CrossRef]
- Ohdate, T.; Inoue, Y. Involvement of glutathione peroxidase 1 in growth and peroxisome formation in Saccharomyces cerevisiae in oleic acid medium. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 1295–1305. [Google Scholar] [CrossRef]
- Bottelbergs, A.; Verheijden, S.; Van Veldhoven, P.P.; Just, W.; Devos, R.; Baes, M. Peroxisome deficiency but not the defect in ether lipid synthesis causes activation of the innate immune system and axonal loss in the central nervous system. J. Neuroinflamm. 2012, 9, 61. [Google Scholar] [CrossRef]
- Dubreuil, M.M.; Morgens, D.W.; Okumoto, K.; Honsho, M.; Contrepois, K.; Lee-McMullen, B.; Traber, G.M.A.; Sood, R.S.; Dixon, S.J.; Snyder, M.P.; et al. Systematic Identification of Regulators of Oxidative Stress Reveals Non-canonical Roles for Peroxisomal Import and the Pentose Phosphate Pathway. Cell Rep. 2020, 30, 1417–1433. [Google Scholar] [CrossRef]
- Malinouski, M.; Zhou, Y.; Belousov, V.V.; Hatfield, D.L.; Gladyshev, V.N. Hydrogen peroxide probes directed to different cellular compartments. PLoS ONE 2011, 6, e14564. [Google Scholar] [CrossRef]
- Watson, W.H.; Jones, D.P. Oxidation of nuclear thioredoxin during oxidative stress. FEBS Lett. 2003, 543, 144–147. [Google Scholar] [CrossRef]
- Shelar, S.B.; Kaminska, K.K.; Reddy, S.A.; Kumar, D.; Tan, C.T.; Yu, V.C.; Lu, J.; Holmgren, A.; Hagen, T.; Chew, E.H. Thioredoxin-dependent regulation of AIF-mediated DNA damage. Free Radic. Biol. Med. 2015, 87, 125–136. [Google Scholar] [CrossRef]
- Pallardó, F.V.; Markovic, J.; García, J.L.; Viña, J. Role of nuclear glutathione as a key regulator of cell proliferation. Mol. Asp. Med. 2009, 30, 77–85. [Google Scholar] [CrossRef]
- Söderdahl, T.; Enoksson, M.; Lundberg, M.; Holmgren, A.; Ottersen, O.P.; Orrenius, S.; Bolcsfoldi, G.; Cotgreave, I.A. Visualization of the compartmentalization of glutathione and protein-glutathione mixed disulfides in cultured cells. FASEB J. 2003, 17, 124–126. [Google Scholar] [CrossRef]
- Patterson, J.C.; Joughin, B.A.; van de Kooij, B.; Lim, D.C.; Lauffenburger, D.A.; Yaffe, M.B. ROS and Oxidative Stress Are Elevated in Mitosis during Asynchronous Cell Cycle Progression and Are Exacerbated by Mitotic Arrest. Cell Syst. 2019, 8, 163–167.e2. [Google Scholar] [CrossRef] [PubMed]
- Markovic, J.; Mora, N.J.; Broseta, A.M.; Gimeno, A.; de-la-Concepción, N.; Viña, J.; Pallardó, F.V. The depletion of nuclear glutathione impairs cell proliferation in 3t3 fibroblasts. PLoS ONE 2009, 4, e6413. [Google Scholar] [CrossRef] [PubMed]
- García-Giménez, J.L.; Olaso, G.; Hake, S.B.; Bönisch, C.; Wiedemann, S.M.; Markovic, J.; Dasí, F.; Gimeno, A.; Pérez-Quilis, C.; Palacios, Ò.; et al. Histone H3 glutathionylation in proliferating mammalian cells destabilizes nucleosomal structure. Antioxid. Redox Signal. 2013, 19, 1305–1320. [Google Scholar] [CrossRef] [PubMed]
- Minowa, O.; Arai, T.; Hirano, M.; Monden, Y.; Nakai, S.; Fukuda, M.; Itoh, M.; Takano, H.; Hippou, Y.; Aburatani, H.; et al. Mmh/Ogg1 gene inactivation results in accumulation of 8-hydroxyguanine in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 4156–4161. [Google Scholar] [CrossRef] [PubMed]
- Cardozo-Pelaez, F.; Sanchez-Contreras, M.; Nevin, A.B.C. Ogg1 null mice exhibit age-associated loss of the nigrostriatal pathway and increased sensitivity to MPTP. Neurochem. Int. 2012, 61, 721–730. [Google Scholar] [CrossRef]
- Zhou, X.; Zhuang, Z.; Wang, W.; He, L.; Wu, H.; Cao, Y.; Pan, F.; Zhao, J.; Hu, Z.; Sekhar, C.; et al. OGG1 is essential in oxidative stress induced DNA demethylation. Cell. Signal. 2016, 28, 1163–1171. [Google Scholar] [CrossRef]
- Barciszewska, A.M.; Giel-Pietraszuk, M.; Perrigue, P.M.; Naskręt-Barciszewska, M. Total DNA Methylation Changes Reflect Random Oxidative DNA Damage in Gliomas. Cells 2019, 8, 1065. [Google Scholar] [CrossRef]
- Giorgio, M.; Dellino, I.G.; Gambino, V.; Roda, N.; Pelicci, P.G. On the epigenetic role of guanosine oxidation. Redox Biol. 2020, 29, 101398. [Google Scholar] [CrossRef]
- Redstone, S.C.J.; Fleming, A.M.; Burrows, C.J. Oxidative Modification of the Potential G-Quadruplex Sequence in the PCNA Gene Promoter Can Turn on Transcription. Chem. Res. Toxicol. 2019, 32, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Larsen, E.; Kwon, K.; Coin, F.; Egly, J.M.; Klungland, A. Transcription activities at 8-oxoG lesions in DNA. DNA Repair 2004, 3, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.; Zhang, Y.; Wan, X.; Zhang, C.; Huang, X.; Huang, W.; Pu, H.; Pei, C.; Wu, H.; et al. KDM1A triggers androgen-induced miRNA transcription via H3K4me2 demethylation and DNA oxidation. Prostate 2015, 75, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Amente, S.; Bertoni, A.; Morano, A.; Lania, L.; Avvedimento, E.V.; Majello, B. LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene 2010, 29, 3691–3702. [Google Scholar] [CrossRef]
- Pan, L.; Hao, W.; Zheng, X.; Zeng, X.; Ahmed Abbasi, A.; Boldogh, I.; Ba, X. OGG1-DNA interactions facilitate NF-k B binding to DNA targets. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Guida, M.; Maraldi, T.; Beretti, F.; Follo, M.Y.; Manzoli, L.; De Pol, A. Nuclear Nox4-derived reactive oxygen species in myelodysplastic syndromes. Biomed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- De Mochel, N.S.R.; Seronello, S.; Wang, S.H.; Ito, C.; Zheng, J.X.; Liang, T.J.; Lambeth, J.D.; Choi, J. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 2010, 52, 47–59. [Google Scholar] [CrossRef]
- Lener, B.; Kozieł, R.; Pircher, H.; Hütter, E.; Greussing, R.; Herndler-Brandstetter, D.; Hermann, M.; Unterluggauer, H.; Jansen-Dürr, P. The NADPH oxidase Nox4 restricts the replicative lifespan of human endothelial cells. Biochem. J. 2009, 423, 363–374. [Google Scholar] [CrossRef]
- Matsushima, S.; Kuroda, J.; Zhai, P.; Liu, T.; Ikeda, S.; Nagarajan, N.; Oka, S.I.; Yokota, T.; Kinugawa, S.; Hsu, C.P.; et al. Tyrosine kinase FYN negatively regulates NOX4 in cardiac remodeling. J. Clin. Investig. 2016, 126, 3403–3416. [Google Scholar] [CrossRef]
- Samoylenko, A.; Al Hossain, J.; Mennerich, D.; Kellokumpu, S.; Hiltunen, J.K.; Kietzmann, T. Nutritional countermeasures targeting reactive oxygen species in cancer: From mechanisms to biomarkers and clinical evidence. Antioxid. Redox Signal. 2013, 19, 2157–2196. [Google Scholar] [CrossRef] [PubMed]
- Ilani, T.; Alon, A.; Grossman, I.; Horowitz, B.; Kartvelishvily, E.; Cohen, S.R.; Fass, D. A secreted disulfide catalyst controls extracellular matrix composition and function. Science 2013, 341, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Tury, A.; Mairet-Coello, G.; Poncet, F.; Jacquemard, C.; Risold, P.Y.; Fellmann, D.; Griffond, B. QSOX sulfhydryl oxidase in rat adenohypophysis: Localization and regulation by estrogens. J. Endocrinol. 2004, 183, 353–363. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mairet-Coello, G.; Tury, A.; Esnard-Feve, A.; Fellmann, D.; Risold, P.Y.; Griffond, B. FAD-Linked Sulfhydryl Oxidase QSOX: Topographic, Cellular, and Subcellular Immunolocalization in Adult Rat Central Nervous System. J. Comp. Neurol. 2004, 473, 334–363. [Google Scholar] [CrossRef]
- Chakravarthi, S.; Jessop, C.E.; Willer, M.; Stirling, C.J.; Bulleid, N.J. Intracellular catalysis of disulfide bond formation by the human sulfhydryl oxidase, QSOX1. Biochem. J. 2007, 404, 403–411. [Google Scholar] [CrossRef]
- Raje, S.; Thorpe, C. Inter-domain redox communication in flavoenzymes of the quiescin/sulfhydryl oxidase family: Role of a thioredoxin domain in disulfide bond formation. Biochemistry 2003, 42, 4560–4568. [Google Scholar] [CrossRef]
- Li, J.; Tong, C.; Xu, P.; Wang, L.; Han, T.L.; Wen, L.; Luo, X.; Tan, B.; Zhu, F.; Gui, S.; et al. QSOX1 regulates trophoblastic apoptosis in preeclampsia through hydrogen peroxide production. J. Matern. Neonatal Med. 2019, 32, 3708–3715. [Google Scholar] [CrossRef]
- Morel, C.; Adami, P.; Musard, J.F.; Duval, D.; Radom, J.; Jouvenot, M. Involvement of sulfhydryl oxidase QSOX1 in the protection of cells against oxidative stress-induced apoptosis. Exp. Cell Res. 2007, 313, 3971–3982. [Google Scholar] [CrossRef]
- Lake, D.F.; Faigel, D.O. The emerging role of qsox1 in cancer. Antioxid. Redox Signal. 2014, 21, 485–496. [Google Scholar] [CrossRef]
- Katchman, B.A.; Ocal, I.T.; Cunliffe, H.E.; Chang, Y.H.; Hostetter, G.; Watanabe, A.; LoBello, J.; Lake, D.F. Expression of quiescin sulfhydryl oxidase 1 is associated with a highly invasive phenotype and correlates with a poor prognosis in Luminal B breast cancer. Breast Cancer Res. 2013, 15, R28. [Google Scholar] [CrossRef]
- Pernodet, N.; Hermetet, F.; Adami, P.; Vejux, A.; Descotes, F.; Borg, C.; Adams, M.; Pallandre, J.R.; Viennet, G.; Esnard, F.; et al. High expression of QSOX1 reduces tumorogenesis, and is associated with a better outcome for breast cancer patients. Breast Cancer Res. 2012, 14, R136. [Google Scholar] [CrossRef] [PubMed]
- Mesecke, N.; Spang, A.; Deponte, M.; Herrmann, J.M. A novel group of glutaredoxins in the cis-Golgi critical for oxidative stress resistance. Mol. Biol. Cell 2008, 19, 2673–2680. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Craig Ayre, D.; Christian, S.L. Induction of mitochondrial reactive oxygen species production by GSH mediated S-glutathionylation of 2-oxoglutarate dehydrogenase. Redox Biol. 2016, 8, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, A.; Casas, C.; Mühlenhoff, U.; Lillig, C.H.; Herrero, E. Saccharomyces cerevisiae Grx6 and Grx7 are monothiol glutaredoxins associated with the early secretory pathway. Eukaryot. Cell 2008, 7, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Sessa, W.C.; Garcia-Cardena, G.; Liu, J.; Keh, A.; Pollock, J.S.; Bradley, J.; Thiru, S.; Braverman, I.M.; Desai, K.M. The Golgi association of endothelial nitric oxide synthase is necessary for the efficient synthesis of nitric oxide. J. Biol. Chem. 1995, 270, 17641–17644. [Google Scholar] [CrossRef] [PubMed]
- Sangwung, P.; Greco, T.M.; Wang, Y.; Ischiropoulos, H.; Sessa, W.C.; Iwakiri, Y. Proteomic identification of S-nitrosylated Golgi proteins: New insights into endothelial cell regulation by eNOS-derived NO. PLoS ONE 2012, 7, e31564. [Google Scholar] [CrossRef] [PubMed]
- García-Cardeña, G.; Oh, P.; Liu, J.; Schnitzer, J.E.; Sessa, W.C. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: Implications for nitric oxide signaling. Proc. Natl. Acad. Sci. USA 1996, 93, 6448–6453. [Google Scholar] [CrossRef]
- Druhan, L.J.; Forbes, S.P.; Pope, A.J.; Chen, C.A.; Zweier, J.L.; Cardounel, A.J. Regulation of eNOS-derived superoxide by endogenous methylarginines. Biochemistry 2008, 47, 7256–7263. [Google Scholar] [CrossRef]
- Sindler, A.L.; Delp, M.D.; Reyes, R.; Wu, G.; Muller-Delp, J.M. Effects of ageing and exercise training on eNOS uncoupling in skeletal muscle resistance arterioles. J. Physiol. 2009, 587, 3885–3897. [Google Scholar] [CrossRef]
- Wang, H.; He, Z.; Yang, Y.; Zhang, J.; Zhang, W.; Zhang, W.; Li, P.; Tang, B. Ratiometric fluorescence imaging of Golgi H2O2 reveals a correlation between Golgi oxidative stress and hypertension. Chem. Sci. 2019, 10, 10876–10880. [Google Scholar] [CrossRef]
- Tabner, B.J.; El-Agnaf, O.M.A.; Turnbull, S.; German, M.J.; Paleologou, K.E.; Hayashi, Y.; Cooper, L.J.; Fullwood, N.J.; Allsop, D. Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J. Biol. Chem. 2005, 280, 35789–35792. [Google Scholar] [CrossRef] [PubMed]
- Behl, C.; Davis, J.B.; Lesley, R.; Schubert, D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell 1994, 77, 817–827. [Google Scholar] [CrossRef]
- Tabner, B.J.; Turnbull, S.; El-Agnaf, O.M.A.; Allsop, D. Formation of hydrogen peroxide and hydroxyl radicals from Aβ and α-synuclein as a possible mechanism of cell death in Alzheimer’s disease and Parkinson’s disease. Free Radic. Biol. Med. 2002, 32, 1076–1083. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Boca, M.; Girotto, S.; Martinelli, M.; Valentine, J.S.; Vieru, M. SOD1 and amyotrophic lateral sclerosis: Mutations and oligomerization. PLoS ONE 2008, 3, e1677. [Google Scholar] [CrossRef]
- Deng, H.X.; Shi, Y.; Furukawa, Y.; Zhai, H.; Fu, R.; Liu, E.; Gorrie, G.H.; Khan, M.S.; Hung, W.Y.; Bigio, E.H.; et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7142–7147. [Google Scholar] [CrossRef]
- Sinet, P.M. Metabolism of Oxygen Derivatives in Down’s Syndrome. Ann. N. Y. Acad. Sci. 1982, 396, 83–94. [Google Scholar] [CrossRef]
- Kim, S.H.; Fountoulakis, M.; Cairns, N.; Lubec, G. Protein levels of human peroxiredoxin subtypes in brains of patients with Alzheimer’s disease and Down Syndrome. J. Neural Transm. Suppl. 2001, 223–235. [Google Scholar] [CrossRef]
- Massaad, C.A. Neuronal and Vascular Oxidative Stress in Alzheimers Disease. Curr. Neuropharmacol. 2011, 9, 662–673. [Google Scholar] [CrossRef]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1499. [Google Scholar] [CrossRef]
- Paul, B.D.; Sbodio, J.I.; Xu, R.; Vandiver, M.S.; Cha, J.Y.; Snowman, A.M.; Snyder, S.H. Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature 2014, 508, 96–100. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. Neurodegeneration in Huntington’s disease involves loss of cystathionine γ-lyase. Cell Cycle 2014, 13, 2491–2493. [Google Scholar] [CrossRef] [PubMed]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Transcriptional control of amino acid homeostasis is disrupted in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2016, 113, 8843–8848. [Google Scholar] [CrossRef] [PubMed]
- Acuña, A.I.; Esparza, M.; Kramm, C.; Beltrán, F.A.; Parra, A.V.; Cepeda, C.; Toro, C.A.; Vidal, R.L.; Hetz, C.; Concha, I.I.; et al. A failure in energy metabolism and antioxidant uptake precede symptoms of Huntington’s disease in mice. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias-Pinto, A.; Moll, P.; Solís-Maldonado, M.; Acuña, A.I.; Riveros, A.; Miró, M.P.; Papic, E.; Beltrán, F.A.; Cepeda, C.; Concha, I.I.; et al. Beyond the redox imbalance: Oxidative stress contributes to an impaired GLUT3 modulation in Huntington’s disease. Free Radic. Biol. Med. 2015, 89, 1085–1096. [Google Scholar] [CrossRef] [PubMed]
- Sonati, T.; Reimann, R.R.; Falsig, J.; Baral, P.K.; O’Connor, T.; Hornemann, S.; Yaganoglu, S.; Li, B.; Herrmann, U.S.; Wieland, B.; et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 2013, 501, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Matus, S.; Lopez, E.; Valenzuela, V.; Nassif, M.; Hetz, C. Functional Contribution of the Transcription Factor ATF4 to the Pathogenesis of Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e66672. [Google Scholar] [CrossRef] [PubMed]
- Monacelli, F.; Acquarone, E.; Giannotti, C.; Borghi, R.; Nencioni, A. Vitamin C, aging and Alzheimer’s disease. Nutrients 2017, 9, 670. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, K.; Samanta, S.; Bagoband, V.; Murugan, N.A.; Govindaraju, T. Antioxidant Berberine-Derivative Inhibits Multifaceted Amyloid Toxicity. iScience 2020, 23, 101005. [Google Scholar] [CrossRef]
- Amato, A.; Terzo, S.; Mulè, F. Natural compounds as beneficial antioxidant agents in neurodegenerative disorders: A focus on Alzheimer’s disease. Antioxidants 2019, 8, 608. [Google Scholar] [CrossRef]
- Hase, T.; Shishido, S.; Yamamoto, S.; Yamashita, R.; Nukima, H.; Taira, S.; Toyoda, T.; Abe, K.; Hamaguchi, T.; Ono, K.; et al. Rosmarinic acid suppresses Alzheimer’s disease development by reducing amyloid β aggregation by increasing monoamine secretion. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Park, H.W.; Park, C.G.; Park, M.; Lee, S.H.; Park, H.R.; Lim, J.; Paek, S.H.; Choy, Y. Bin Intrastriatal administration of coenzyme Q10 enhances neuroprotection in a Parkinson’s disease rat model. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Lockrow, J.; Prakasam, A.; Huang, P.; Bimonte-Nelson, H.; Sambamurti, K.; Granholm, A.C. Cholinergic degeneration and memory loss delayed by vitamin E in a Down syndrome mouse model. Exp. Neurol. 2009, 216, 278–289. [Google Scholar] [CrossRef]
- Danino, O.; Grossman, S.; Fischer, B. ATP-γ-S-(α,β-CH2) protects against oxidative stress and amyloid beta toxicity in neuronal culture. Biochem. Biophys. Res. Commun. 2015, 460, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, H.; Speckmann, B.; Sies, H. Toward understanding success and failures in the use of selenium for cancer prevention. Antioxid. Redox Signal. 2013, 19, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Moosmann, B.; Behl, C. Antioxidants as treatment for neurodegenerative disorders. Expert Opin. Investig. Drugs 2002, 11, 1407–1435. [Google Scholar] [CrossRef]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975. [Google Scholar] [CrossRef]
- Sies, H. Findings in redox biology: From H2O2 to oxidative stress. J. Biol. Chem. 2020, 295, 13458–13473. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Maiorino, M.; Ursini, F. Signaling functions of reactive oxygen species. Biochemistry 2010, 49, 835–842. [Google Scholar] [CrossRef]
- Smith, R.A.J.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef]
- Tauskela, J.S. MitoQ—A mitochondria-targeted antioxidant—PubMed. IDrugs 2007, 10, 399–412. [Google Scholar]
- Dringen, R.; Kussmaul, L.; Gutterer, J.M.; Hirrlinger, J.; Hamprecht, B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J. Neurochem. 1999, 72, 2523–2530. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, P.D.; Pérez-De La Cruz, V.; Torres-Ramos, M.; Silva-Islas, C.; Lecona-Vargas, R.; Lugo-Huitrón, R.; Blanco-Ayala, T.; Ugalde-Muñiz, P.; Vázquez-Cervantes, G.I.; Fortoul, T.I.; et al. Selenium-induced antioxidant protection recruits modulation of thioredoxin reductase during excitotoxic/pro-oxidant events in the rat striatum. Neurochem. Int. 2012, 61, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, H.; Alili, L.; Bilgic, E.; Sies, H.; Brenneisen, P. Involvement of selenoprotein P in protection of human astrocytes from oxidative damage. Free Radic. Biol. Med. 2006, 40, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Casas, A.I.; Dao, V.T.V.; Daiber, A.; Maghzal, G.J.; Di Lisa, F.; Kaludercic, N.; Leach, S.; Cuadrado, A.; Jaquet, V.; Seredenina, T.; et al. Reactive Oxygen-Related Diseases: Therapeutic Targets and Emerging Clinical Indications. Antioxid. Redox Signal. 2015, 23, 1171–1185. [Google Scholar] [CrossRef]
- Choi, J.; Sullards, M.C.; Olzmann, J.A.; Rees, H.D.; Weintraub, S.T.; Bostwick, D.E.; Gearing, M.; Levey, A.I.; Chin, L.S.; Li, L. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J. Biol. Chem. 2006, 281, 10816–10824. [Google Scholar] [CrossRef]
- Wang, C.; Ko, H.S.; Thomas, B.; Tsang, F.; Chew, K.C.M.; Tay, S.P.; Ho, M.W.L.; Lim, T.M.; Soong, T.W.; Pletnikova, O.; et al. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin’s protective function. Hum. Mol. Genet. 2005, 14, 3885–3897. [Google Scholar] [CrossRef]
- Dryanovski, D.I.; Guzman, J.N.; Xie, Z.; Galteri, D.J.; Volpicelli-Daley, L.A.; Lee, V.M.Y.; Miller, R.J.; Schumacker, P.T.; James Surmeier, D. Calcium entry and α-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J. Neurosci. 2013, 33, 10154–10164. [Google Scholar] [CrossRef]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef]
- Cristóvão, A.C.; Guhathakurta, S.; Bok, E.; Je, G.; Yoo, S.D.; Choi, D.H.; Kim, Y.S. Nadph oxidase 1 mediates α-synucleinopathy in Parkinson’s disease. J. Neurosci. 2012, 32, 14465–14477. [Google Scholar] [CrossRef]
- Uehara, T.; Nakamura, T.; Yao, D.; Shi, Z.Q.; Gu, Z.; Ma, Y.; Masliah, E.; Nomura, Y.; Lipton, S.A. S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 2006, 441, 513–517. [Google Scholar] [CrossRef]
- Pearce, R.K.B.; Owen, A.; Daniel, S.; Jenner, P.; Marsden, C.D. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J. Neural Transm. 1997, 104, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.L.; Yong, V.W. Idiopathic Parkinson’s disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci. Lett. 1986, 67, 269–274. [Google Scholar] [CrossRef]
- Vural, G.; Gumusyayla, S.; Bektas, H.; Deniz, O.; Alisik, M.; Erel, O. Impairment of dynamic thiol–disulphide homeostasis in patients with idiopathic Parkinson’s disease and its relationship with clinical stage of disease. Clin. Neurol. Neurosurg. 2017, 153, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Hastings, T.G.; Lewis, D.A.; Zigmond, M.J. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc. Natl. Acad. Sci. USA 1996, 93, 1956–1961. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.H.; Zeevalk, G.D. Characterization of reduced and oxidized dopamine and 3,4- dihydrophenylacetic acid, on brain mitochondrial electron transport chain activities. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 819–828. [Google Scholar] [CrossRef]
- Liu, X.; Yamada, N.; Maruyama, W.; Osawa, T. Formation of dopamine adducts derived from brain polyunsaturated fatty acids: Mechanism for Parkinson disease. J. Biol. Chem. 2008, 283, 34887–34895. [Google Scholar] [CrossRef]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.R.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein: A possible molecular link between parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef]
- Martin-Bastida, A.; Lao-Kaim, N.P.; Loane, C.; Politis, M.; Roussakis, A.A.; Valle-Guzman, N.; Kefalopoulou, Z.; Paul-Visse, G.; Widner, H.; Xing, Y.; et al. Motor associations of iron accumulation in deep grey matter nuclei in Parkinson’s disease: A cross-sectional study of iron-related magnetic resonance imaging susceptibility. Eur. J. Neurol. 2017, 24, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Zhang, F.; Zhou, H.; Kam, W.; Wilson, B.; Hong, J.S. Neuroinflammation and α-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environ. Health Perspect. 2011, 119, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.; et al. Aggregated α-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Vandiver, M.S.; Paul, B.D.; Xu, R.; Karuppagounder, S.; Rao, F.; Snowman, A.M.; Seok Ko, H.; Il Lee, Y.; Dawson, V.L.; Dawson, T.M.; et al. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sunico, C.R.; Sultan, A.; Nakamura, T.; Dolatabadi, N.; Parker, J.; Shan, B.; Han, X.; Yates, J.R.; Masliah, E.; Ambasudhan, R.; et al. Role of sulfiredoxin as a peroxiredoxin-2 denitrosylase in human iPSC-derived dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2016, 113, E7564–E7571. [Google Scholar] [CrossRef]
- Xie, H.; Hou, S.; Jiang, J.; Sekutowicz, M.; Kelly, J.; Bacskai, B.J. Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 7904–7909. [Google Scholar] [CrossRef]
- Zhou, W.W.; Lu, S.; Su, Y.J.; Xue, D.; Yu, X.L.; Wang, S.W.; Zhang, H.; Xu, P.X.; Xie, X.X.; Liu, R.T. Decreasing oxidative stress and neuroinflammation with a multifunctional peptide rescues memory deficits in mice with Alzheimer disease. Free Radic. Biol. Med. 2014, 74, 50–63. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef]
- Bradley-Whitman, M.A.; Timmons, M.D.; Beckett, T.L.; Murphy, M.P.; Lynn, B.C.; Lovell, M.A. Nucleic acid oxidation: An early feature of Alzheimer’s disease. J. Neurochem. 2014, 128, 294–304. [Google Scholar] [CrossRef]
- Borghi, R.; Patriarca, S.; Traverso, N.; Piccini, A.; Storace, D.; Garuti, A.; Cirmena, G.; Odetti, P.; Tabaton, M. The increased activity of BACE1 correlates with oxidative stress in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1009–1014. [Google Scholar] [CrossRef]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and redistribution of c-Jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, N.; Trivedi, M.; Muratore, C.; Li, S.; Deth, R. Soluble oligomers of amyloid-β cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimer Dis. 2013, 36, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Sang, W.S.; Hamby, A.M.; Liu, J.; Wai, Y.C.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, X.; Lee, H.G.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. 2010, 468, 267–271. [Google Scholar] [CrossRef]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; McKee, A.C.; Beal, M.F.; Graham, B.H.; Wallace, D.C. Marked changes in mitochondrial dna deletion levels in alzheimer brains. Genomics 1994, 23, 471–476. [Google Scholar] [CrossRef]
- Lu, T.; Aron, L.; Zullo, J.; Pan, Y.; Kim, H.; Chen, Y.; Yang, T.H.; Kim, H.M.; Drake, D.; Liu, X.S.; et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 2014, 507, 448–454. [Google Scholar] [CrossRef]
- Molokanova, E.; Akhtar, M.W.; Sanz-Blasco, S.; Tu, S.; Piña-Crespo, J.C.; Mckercher, S.R.; Lipton, S.A. Differential effects of synaptic and extrasynaptic NMDA receptors on Aβ-induced nitric oxide production in cerebrocortical neurons. J. Neurosci. 2014, 34, 5023–5028. [Google Scholar] [CrossRef]
- Acevedo-Torres, K.; Berríos, L.; Rosario, N.; Dufault, V.; Skatchkov, S.; Eaton, M.J.; Torres-Ramos, C.A.; Ayala-Torres, S. Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington’s disease. DNA Repair 2009, 8, 126–136. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; MacGarvey, U.; Baik, M.J.; Berger, S.C.; Muqit, M.M.K.; Bird, E.D.; Beal, M.F. Oxidative damage and metabolic dysfunction in huntington’s disease: Selective vulnerability of the basal ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef]
- Chen, C.M.; Wu, Y.R.; Cheng, M.L.; Liu, J.L.; Lee, Y.M.; Lee, P.W.; Soong, B.W.; Chiu, D.T.Y. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem. Biophys. Res. Commun. 2007, 359, 335–340. [Google Scholar] [CrossRef]
- Klepac, N.; Relja, M.; Klepac, R.; Hećimović, S.; Babić, T.; Trkulja, V. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects: A cross-sectional study. J. Neurol. 2007, 254, 1676–1683. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Chen, Q.; Wang, X.; Wang, Q.C.; Wang, Y.; Cheng, H.P.; Guo, C.; Sun, Q.; Chen, Q.; Tang, T.S. Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. J. Biol. Chem. 2013, 288, 3070–3084. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G.; Cummings, J.; Perlman, S.; Hance, D.B.; Mintz, J. Increased basal ganglia iron levels in Huntington disease. Arch. Neurol. 1999, 56, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, I.V.; Liu, Y.; Bjoras, M.; Klungland, A.; Wilson, S.H.; McMurray, C.T. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 2007, 447, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.N.; Yu, Y.V.; Gundemir, S.; Jo, C.; Cui, M.; Tieu, K.; Johnson, G.V.W. Impaired Mitochondrial Dynamics and Nrf2 Signaling Contribute to Compromised Responses to Oxidative Stress in Striatal Cells Expressing Full-Length Mutant Huntingtin. PLoS ONE 2013, 8, e57932. [Google Scholar] [CrossRef]
- Johri, A.; Chandra, A.; Beal, M.F. PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic. Biol. Med. 2013, 62, 37–46. [Google Scholar] [CrossRef]
- Weiduschat, N.; Mao, X.; Hupf, J.; Armstrong, N.; Kang, G.; Lange, D.J.; Mitsumoto, H.; Shungu, D.C. Motor cortex glutathione deficit in ALS measured in vivo with the J-editing technique. Neurosci. Lett. 2014, 570, 102–107. [Google Scholar] [CrossRef]
- Beretta, S.; Sala, G.; Mattavelli, L.; Ceresa, C.; Casciati, A.; Ferri, A.; Carrì, M.T.; Ferrarese, C. Mitochondrial dysfunction due to mutant copper/zinc superoxide dismutase associated with amyotrophic lateral sclerosis is reversed by N-acetylcysteine. Neurobiol. Dis. 2003, 13, 213–221. [Google Scholar] [CrossRef]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Flint Beal, M.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef]
- Duran-Aniotz, C.; Cornejo, V.H.; Espinoza, S.; Ardiles, Á.O.; Medinas, D.B.; Salazar, C.; Foley, A.; Gajardo, I.; Thielen, P.; Iwawaki, T.; et al. IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. 2017, 134, 489–506. [Google Scholar] [CrossRef]
- Baleriola, J.; Walker, C.A.; Jean, Y.Y.; Crary, J.F.; Troy, C.M.; Nagy, P.L.; Hengst, U. Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 2014, 158, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C.; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 2013, 33, 9498–9507. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.Y.; Cho, S.B.; Jung, H.Y.; Kim, W.; Lee, K.Y.; Kim, J.W.; Moon, S.M.; Won, M.H.; Choi, J.H.; Yoon, Y.S.; et al. Protein disulfide-isomerase A3 significantly reduces ischemia-induced damage by reducing oxidative and endoplasmic reticulum stress. Neurochem. Int. 2019, 122, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Perri, E.R.; Thomas, C.J.; Parakh, S.; Spencer, D.M.; Atkin, J.D. The unfolded protein response and the role of protein disulfide isomerase in neurodegeneration. Front. Cell Dev. Biol. 2016, 3, 80. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Won, S.M.; Suh, J.; Son, S.J.; Moon, G.J.; Park, U.J.; Gwag, B.J. Induction of the unfolded protein response and cell death pathway in alzheimer’s disease, but not in aged Tg2576 mice. Exp. Mol. Med. 2010, 42, 386–394. [Google Scholar] [CrossRef]
- Conn, K.J.; Gao, W.; McKee, A.; Lan, M.S.; Ullman, M.D.; Eisenhauer, P.B.; Fine, R.E.; Wells, J.M. Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Res. 2004, 1022, 164–172. [Google Scholar] [CrossRef]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407. [Google Scholar] [CrossRef]
- Safren, N.; El Ayadi, A.; Chang, L.; Terrillion, C.E.; Gould, T.D.; Boehning, D.F.; Monteiro, M.J. Ubiquilin-1 overexpression increases the lifespan and delays accumulation of huntingtin aggregates in the R6/2 mouse model of huntington’s disease. PLoS ONE 2014, 9, e87513. [Google Scholar] [CrossRef]
- Hoffstrom, B.G.; Kaplan, A.; Letso, R.; Schmid, R.S.; Turmel, G.J.; Lo, D.C.; Stockwell, B.R. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat. Chem. Biol. 2010, 6, 900–906. [Google Scholar] [CrossRef]
- Kaplan, A.; Gaschler, M.M.; Dunn, D.E.; Colligan, R.; Brown, L.M.; Iii, A.G.P.; Lo, D.C.; Stockwell, B.R. Small molecule-induced oxidation of protein disulfide isomerase is neuroprotective. Proc. Natl. Acad. Sci. USA 2015, 112, E2245–E2252. [Google Scholar] [CrossRef]
- Kamarehei, M.; Pejman, S.; Kaboudanian Ardestani, S.; Zahednasab, H.; Firouzi, M.; Harirchian, M.H. Inhibition of protein disulfide isomerase has neuroprotective effects in a mouse model of experimental autoimmune encephalomyelitis. Int. Immunopharmacol. 2020, 82, 106286. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, G.; Kaplan, A.; Gaschler, M.M.; Zhang, X.; Hou, Z.; Jiang, M.; Zott, R.; Cremers, S.; Stockwell, B.R.; et al. Small molecule modulator of protein disulfide isomerase attenuates mutant huntingtin toxicity and inhibits endoplasmic reticulum stress in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2018, 27, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, Š.; Sonninen, T.M.; Wojciechowski, S.; Goldsteins, G.; Koistinaho, J. Dysfunction of cellular proteostasis in Parkinson’s disease. Front. Neurosci. 2019, 13, 457. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.L.; Su, F.Y.; Tsai, L.K.; Huang, C.C.; Ko, Y.L.; Su, L.W.; Chen, K.Y.; Shih, H.M.; Hu, C.M.; Lee, W.H. NPGPx-Mediated Adaptation to Oxidative Stress Protects Motor Neurons from Degeneration in Aging by Directly Modulating O-GlcNAcase. Cell Rep. 2019, 29, 2134–2143.e7. [Google Scholar] [CrossRef]
- Skladnev, N.V.; Ganeshan, V.; Kim, J.Y.; Burton, T.J.; Mitrofanis, J.; Stone, J.; Johnstone, D.M. Widespread brain transcriptome alterations underlie the neuroprotective actions of dietary saffron. J. Neurochem. 2016, 139, 858–871. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA. 2008, 105, 18525–18530. [Google Scholar] [CrossRef]
- Kam, M.K.; Lee, D.G.; Kim, B.; Lee, H.S.; Lee, S.R.; Bae, Y.C.; Lee, D.S. Peroxiredoxin 4 ameliorates amyloid beta oligomer-mediated apoptosis by inhibiting ER-stress in HT-22 hippocampal neuron cells. Cell Biol. Toxicol. 2019, 35, 573–588. [Google Scholar] [CrossRef]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef]
- Galvan, D.L.; Badal, S.S.; Long, J.; Chang, B.H.; Schumacker, P.T.; Overbeek, P.A.; Danesh, F.R. Real-time in vivo mitochondrial redox assessment confirms enhanced mitochondrial reactive oxygen species in diabetic nephropathy. Kidney Int. 2017, 92, 1282–1287. [Google Scholar] [CrossRef]
- Van Hameren, G.; Campbell, G.; Deck, M.; Berthelot, J.; Gautier, B.; Quintana, P.; Chrast, R.; Tricaud, N. In vivo real-time dynamics of ATP and ROS production in axonal mitochondria show decoupling in mouse models of peripheral neuropathies. Acta Neuropathol. Commun. 2019, 7, 86. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Neurodegenerative Disorder | Possible Source of Oxidative Stress | Cell Component Affected | Reference |
|---|---|---|---|
| Parkinson disease | Not determined | Oxidized DJ-1 1 protein | [186] |
| H2O2/rotenone/6-hydroxydopamine | Parkin | [187] | |
| Ca2+ influx through L-type channels/α-synuclein intracellular inclusions | Mitochondrial oxidant stress | [188,189] | |
| NADPH oxidase | α-synuclein | [190] | |
| NOS 2 | S-nitrosylation of PDI 3 | [191] | |
| Not determined | Depletion of GSH | [192,193,194] | |
| Intrastriatal dopamine injections | Protein-bound cysteinyl catechols/Loss of tyrosine hydroxylase | [195] | |
| Loss of dopamine homeostasis | Inhibition of NADH oxidase/mitochondrial dysfunction | [196] | |
| Formation of the hexanoyl dopamine adduct | Mitochondrial abnormality/apoptosis | [197] | |
| α-synuclein oligomer-induced ROS production | Depletion of GSH | [198] | |
| α-synuclein oligomers | Mitochondrial dysfunction | [199] | |
| Not determined | Mitochondrial respiratory-chain Complex I | [200] | |
| Iron deposition in deep brain nuclei | Fibrillation of α-synuclein/dopaminergenic neurodegeneration | [201,202] | |
| Inflammogen lipopolysaccharide/ NOS 2 and NADPH oxidase | Aggregated nitrated α-synuclein/upregulation of inflammatory markers | [203] | |
| Extracellular aggregated α-synuclein | Activation of NADPH oxidase/microglial activation | [204] | |
| Activation of NOS 2 | Decrease in parkin sulfhydration/increase in parkin nitrosylation | [205] | |
| Nitric oxide | S-nitrosylation of Prx2 4 | [206] | |
| Alzheimer’s disease | Not determined | Oxidized DJ-1 1 protein | [186] |
| Aβ plaques | Neurites | [207] | |
| Aβ plaques | Reduced GSH levels/Decreased SOD activity | [208] | |
| Aβ plaques | Mitochondrial protein import channels | [209] | |
| Not determined | Nuclear and mitochondrial DNA damage | [210] | |
| Increase in BACE1 5 activity | Lipid peroxidation | [211] | |
| Not determined | JNK/SAPK 6 dysregulation | [212] | |
| Aβ oligomers | Inhibition of EAAT3 7-mediated cysteine uptake/decreased GSH levels/decreased DNA methylation | [213,214] | |
| Inhibition of GSH synthesis | Increased levels of tau phosphorylated | [215] | |
| Age-dependent | Mitochondrial DNA deletion | [216] | |
| Not determined | Loss of REST 8 | [217] | |
| NOS 2 | S-nitrosylation of PDI 3 | [191] | |
| Aβ(1-42) oligomers | Activation of NMDARs 9/increase in Nitric oxide | [218] | |
| Huntington’s disease | Age-dependent | Mitochondrial DNA damage | [219] |
| Not determined | Cytosolic SOD 10 activity/nuclear DNA/oxidative phosphorylation enzymes | [220] | |
| Not determined | Cu/Zn-SOD 10 activity/glutathione peroxidase activity/mitochondrial DNA | [221] | |
| Not determined | Reduced GSH levels/plasma lipid peroxidation | [222] | |
| Mitochondrial Ca2+ signalling and superoxide generation | Mitochondrial DNA damage | [223] | |
| Depletion of cystathione γ-lyase 11 | Low levels of GSH/protein carbonylation/protein nitration/lipid peroxidation | [161] | |
| Impaired cysteine biosynthesis and transport | Failure of activating transcriptional factor 4 (ATF4) induction | [163] | |
| Uptake of vitamin C | Impaired trafficking of vitamin C transporters/impaired glucose transporter GLUT3 to the membrane | [164,165] | |
| Iron metabolism | Not determined | [224] | |
| Age-dependent | Oxidative DNA damage (CAG trinucleotide expansion) | [225] | |
| Reduced activity of Nrf2 12 | Impaired mitochondrial dynamics/decreased mitochondrial fusion | [226] | |
| Impaired function of PGC1α 13 | Mitochondrial dysfunction | [227] | |
| Not determined | Reduced levels of GSH | [228] | |
| Amyotrophic lateral sclerosis | Mutations in Cu/Zn SOD 10 (familiar cases) | Increased cytosolic and mitochondrial ROS/impaired mitochondrial respiration | [229,230] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konno, T.; Melo, E.P.; Chambers, J.E.; Avezov, E. Intracellular Sources of ROS/H2O2 in Health and Neurodegeneration: Spotlight on Endoplasmic Reticulum. Cells 2021, 10, 233. https://doi.org/10.3390/cells10020233
Konno T, Melo EP, Chambers JE, Avezov E. Intracellular Sources of ROS/H2O2 in Health and Neurodegeneration: Spotlight on Endoplasmic Reticulum. Cells. 2021; 10(2):233. https://doi.org/10.3390/cells10020233
Chicago/Turabian StyleKonno, Tasuku, Eduardo Pinho Melo, Joseph E. Chambers, and Edward Avezov. 2021. "Intracellular Sources of ROS/H2O2 in Health and Neurodegeneration: Spotlight on Endoplasmic Reticulum" Cells 10, no. 2: 233. https://doi.org/10.3390/cells10020233
APA StyleKonno, T., Melo, E. P., Chambers, J. E., & Avezov, E. (2021). Intracellular Sources of ROS/H2O2 in Health and Neurodegeneration: Spotlight on Endoplasmic Reticulum. Cells, 10(2), 233. https://doi.org/10.3390/cells10020233